

Abstract
Hepatitis C virus (HCV) is highly variable and associated with chronic liver disease. Viral isolates are grouped into seven genotypes (GTs). Accumulating evidence indicates that viral determinants in the core to NS2 proteins modulate the efficiency of virus production. However, the role of the glycoproteins E1 and E2 in this process is currently poorly defined. Therefore, we constructed chimeric viral genomes to explore the role of E1 and E2 in HCV assembly. Comparison of the kinetics and efficiency of particle production by intragenotypic chimeras highlighted core and p7 as crucial determinants for efficient virion release. Glycoprotein sequences, however, had only a minimal impact on this process. In contrast, in the context of intergenotypic HCV chimeras, HCV assembly was profoundly influenced by glycoprotein genes. On the one hand, insertion of GT1a-derived (H77) E1-E2 sequences into a chimeric GT2a virus (Jc1) strongly suppressed virus production. On the other hand, replacement of H77 glycoproteins within the GT1a-GT2a chimeric genome H77/C3 by GT2a-derived (Jc1) E1-E2 increased infectious particle production. Thus, within intergenotypic chimeras, glycoprotein features strongly modulate virus production. Replacement of Jc1 glycoprotein genes by H77-derived E1-E2 did not grossly affect subcellular localization of core, E2, and NS2. However, it caused an accumulation of nonenveloped core protein and increased abundance of nonenveloped core protein structures with slow sedimentation. These findings reveal an important role for the HCV glycoproteins E1 and E2 in membrane envelopment, which likely depends on a genotype-specific interplay with additional viral factors.
INTRODUCTION
Globally, an estimated 160 million people are chronically infected with hepatitis C virus (HCV) (1) and are therefore at a high risk for developing severe liver damage, including hepatic steatosis, fibrosis, cirrhosis, and hepatocellular carcinoma (2). Due to error-prone RNA replication, HCV is a highly variable virus, and based on phylogenetic analyses, viral isolates are grouped into seven genotypes (GTs), which are associated with specific global regions and modes of transmission (3). Importantly, different HCV GTs display differential treatment response rates, and the clinical course of infection also differs, indicating that, in addition to host factors, viral factors play a role in the outcome of infection/treatment. Specifically, infections by HCV GTs 1 and 4 are more difficult to treat with interferons than infections by HCV GTs 2 and 3, and GT3 is associated with liver steatosis (4). The current standard therapy, a combination of pegylated alpha interferon and ribavirin combined with one of two available protease inhibitors, is associated with serious side effects and does not achieve an effective response in every patient (5).
HCV is a hepatotropic RNA virus that establishes a chronic infection in the majority of cases. The HCV genome, 9.6 kb in size, encodes a single polyprotein that is cleaved by cellular and viral proteases into at least 10 different proteins: the structural proteins core, E1, and E2; the ion channel p7; and the nonstructural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B (6). The genetic variability exhibited by HCV facilitates immune evasion and contributes to viral persistence. The development of in vitro cell culture systems, including subgenomic replicons (7), HCV pseudoparticles (8, 9), and, ultimately, the JFH1-based infection system (10–12), have allowed detailed investigation of multiple aspects of the viral life cycle (13). With respect to assembly and release of infectious HCV particles, essential roles of viral and host factors have been defined. On the one hand, cellular factors involved in the secretion of lipoproteins, including the microsomal triglyceride transfer protein (MTP), apolipoprotein B (ApoB), and ApoE, have been implicated in HCV particle production (14–16). On the other hand, it was demonstrated that besides the canonical structural proteins core, E1, and E2, other viral factors, including p7, NS2, NS3, NS4B, NS5A, and NS5B, also play crucial roles in the production of infectious viral progeny (17–24).
The two viral glycoproteins E1 and E2, which show the highest variability among the HCV proteins (3), are embedded in the virus envelope, mediate uptake of HCV into liver cells, and are the main target for neutralizing antibodies. However, their role in the production of infectious particles is incompletely defined. Both are type I membrane proteins with a C-terminal transmembrane domain (TMD) and a large N-terminal extracellular domain. Their biogenesis is an intricate process involving various endoplasmic reticulum (ER)-resident chaperones, including calnexin, calreticulin, and BiP (25–27). Localization of the glycoproteins in the ER and signals for heterodimerization are determined by the TMDs (28–30). E2 folding is independent of other viral proteins, whereas the maturation of E1 requires the coexpression of E2 (31). The two envelope proteins are heavily glycosylated, mostly at highly conserved sites (32), and several glycans are important for HCV assembly and/or infectivity (33).
Chimeric HCV GT2a genomes such as “Fl-J6/JFH1” or “Jc1,” consisting of J6CF and JFH1 segments, produce much higher virus titers than parental JFH1 or genotype 1 viral chimeras, indicating that viral determinants in the core to NS2 proteins modulate efficiency of virus production (10, 34). However, the role of the HCV glycoproteins E1 and E2 in this regard and in assembly and release in general is not well defined. Therefore, we used a genetic approach to investigate the function of different E1 and E2 variants in HCV assembly and release.
MATERIALS AND METHODS
Cell culture.
Huh7.5 cells were grown in Dulbecco's modified minimal essential medium (DMEM; Invitrogen, Karlsruhe, Germany) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 10% fetal calf serum (FCS) (DMEM complete).
Plasmid construction.
The genomes and plasmids pFK-JFH1 (11), H77 (35), Con1 (7), J6CF (36), Jc1, JFH1/p7J6, JFH1/coreJ6, and JFH1/H77/C3 were described previously (23, 34, 37). Further chimeric HCV constructs, JFH1/corep7J6, Jc1/E1E2JFH1, JFH1/E1E2J6, Jc1/E1E2H77, and JFH1/H77/C3/E1E2J6, were generated by a PCR-based strategy. The integrity of these constructs was verified by sequencing. Additional information, including the sequences of these constructs, is available upon request.
In vitro transcription, electroporation of HCV RNA, and quantification of viral core protein.
In vitro transcripts of the individual constructs were generated by linearizing 5 to 10 μg of the respective plasmid by digestion for 1 h with MluI. Plasmid DNA was extracted with phenol and chloroform and, after precipitation with ethanol, dissolved in RNase-free water. In vitro transcription reaction mixtures contained 80 mM HEPES (pH 7.5), 12 mM MgCl2, 2 mM spermidine, 40 mM dithiothreitol (DTT), 3.125 mM each ribonucleoside triphosphate, 1 U of RNasin (Promega, Mannheim, Germany) per μl, 0.1 μg plasmid DNA/μl, and 0.6 U of T7 RNA polymerase (Promega) per μl. After incubation for 2 h at 37°C, an additional 0.3 U of T7 RNA polymerase/μl reaction mixture was added, followed by another 2 h at 37°C. Transcription was terminated by the addition of 1.2 U of RNase-free DNase (Promega) per μg of plasmid DNA and 30 min of incubation at 37°C. The RNA was extracted with acidic phenol and chloroform, precipitated with isopropanol, and dissolved in RNase-free water. The concentration was determined by measurement of the optical density at 260 nm. Denaturing agarose gel electrophoresis was used to check RNA integrity.
For electroporation of HCV RNA into Huh7.5 cells, single-cell suspensions were prepared by trypsinization of monolayers and subsequent resuspension with DMEM complete. Huh7.5 cells were washed with phosphate-buffered saline (PBS), counted, and resuspended at 1.5 × 107 cells per ml in Cytomix containing 2 mM ATP and 5 mM glutathione. Unless otherwise stated, 10 μg of in vitro-transcribed RNA was mixed with 400 μl of cell suspension by pipetting and then electroporated with a Gene Pulser system (Bio-Rad, Munich, Germany) in a cuvette with a gap width of 0.4 cm (Bio-Rad) at 975 μF and 270 V. Cells were immediately transferred into 16 ml complete DMEM, and 2 ml of the cell suspension was seeded per well of a six-well plate.
To quantify HCV core protein, samples were inactivated with 1% (vol/vol) Triton X-100 in PBS, and core protein levels were measured by using the Architect system (Abbott, Wiesbaden, Germany).
Proteolytic digestion protection assay.
HCV-transfected cells seeded into 6-well dishes were scraped into 170 μl proteinase K buffer (50 mM Tris-HCl [pH 8.0] 10 mM CaCl2, 1 mM DTT) at 48 h posttransfection and subjected to five cycles of freezing and thawing. Subsequently, 50 μl of the crude lysate were left untreated, 50 μl was treated with 50 μg/ml proteinase K (Roche, Mannheim, Germany) for 1 h on ice, and another 50 μl was lysed with 5% (vol/vol) Triton X-100 prior to proteinase K treatment. Proteinase K digestion was terminated by addition of PMSF (phenylmethylsulfonyl fluoride; AppliChem) at a final concentration of 5 mM and incubation of the sample on ice for 10 min. Subsequently, 13 μl of 5× SDS sample buffer was added, and the sample was heated to 95°C for 10 min. The amount of residual core protein was determined by SDS-PAGE and by a core-specific enzyme-linked immunosorbent assay (ELISA).
Indirect immunofluorescence analysis.
Indirect immunofluorescence staining was performed as described previously (38). Primary antibodies against HCV core protein (mouse monoclonal antibody C7-50 at a dilution of 1:1,000), E2 (mouse anti-E2 antibody AP33 at a 1:250 dilution) (39), and NS2 (anti-mouse 6H6 antibody at a 1:1,000 dilution) (40), supplemented in PBS with 5% goat serum (Sigma), were incubated for 1 h, followed by a secondary antibody specific for murine IgG conjugated with Alexa Fluor 568 (Invitrogen) at a dilution of 1:1,000. Cell nuclei were stained for 1 min at room temperature (RT) with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) diluted 1:3,000.
Immunohistochemical staining and virus titration.
Virus titers were determined as described previously (41). In brief, Huh7.5 cells were seeded into 96-well plates at a density of 1 × 104 cells per well 24 h prior to inoculation with dilutions of filtered cell culture supernatant (at least six wells were used per dilution). Two to 3 days later, cells were washed with PBS, fixed for 20 min with ice-cold methanol at −20°C, washed three times with PBS, and then permeabilized and blocked for 1 h with PBS containing 0.5% saponin, 1% bovine serum albumin, 0.2% dried skim milk, and 0.02% sodium azide. Endogenous peroxidases were blocked by incubating cells for 5 min with PBS containing 0.3% hydrogen peroxide. After washing three times with PBS and once with PBS containing 0.5% saponin (PBS-saponin), NS5A was detected with a 1:1,000 dilution of hybridoma supernatant 9E10 (10) in PBS-saponin for 1 h at RT or overnight at 4°C. Cells were washed as described above, and bound 9E10 was detected by incubation with peroxidase-conjugated antibodies specific to murine IgG (Sigma-Aldrich, Steinheim, Germany) diluted 1:200 in PBS-saponin. After 1 h of incubation at RT, cells were washed as specified above. Finally, peroxidase activity was detected by using the Vector NovaRED substrate kit (Linaris Biologische Produkte GmbH, Wertheim, Germany).
Metabolic radiolabeling of proteins, immunoprecipitation, and Western blotting.
Twenty-four hours after electroporation, Huh7.5 cells were washed with PBS, starved in methionine-cysteine-free medium for 1 h, and incubated for 6 h in methionine-cysteine-free DMEM supplemented with 100 μCi/ml of Express Protein labeling mix (PerkinElmer, Rodgau-Jügesheim, Germany). Cell lysates were prepared by using 1 ml of ice-cold NPB per well of a 6-well plate (50 mM Tris-Cl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS), supplemented with a complete protease inhibitor cocktail, as recommended by the manufacturer (Roche). Lysates were cleared by centrifugation at 13,800 × g for 15 min at 4°C. The cleared lysates were used for immunoprecipitation using the E2-specific antibody AP33 (42). Immune complexes were resolved by denaturing SDS-PAGE and detected by autoradiography. For immunoprecipitation without radiolabeling, the cells were harvested at 48 h posttransfection by trypsinization, and cell pellets were lysed in 230 μl lysis buffer (PBS supplemented with 1% Triton X-100 and a protease inhibitor cocktail [Complete; Roche]). Out of these, 200 μl was used for immunoprecipitation with AP33, and 20 μl was kept for the inputs (one-fifth of the input). For each immunoprecipitation, 25 μl of protein G-agarose beads (Roche) was washed 3 times in PBS. Subsequently, washed beads were incubated with 3.3 μg antibody diluted in PBS for 2 h at 4°C under rotation. In parallel, cell lysates were precleared on protein G-agarose in the absence of antibody for 2 h at 4°C. Afterwards, precleared cell lysates were added onto the washed antibody-bound beads and incubated overnight at 4°C, under constant rotation, in PBS–1% Triton X-100. Beads were then washed 5 times in PBS–1% Triton X-100 (three times quickly and twice with a 5-min incubation time under rotation) and finally washed once quickly in water before protein elution in Laemmli buffer and SDS-PAGE analysis.
Freeze-and-thaw lysates of HCV-transfected cells.
Huh7.5 cells were transfected with different HCV genomes, cell culture supernatants were harvested at 48 h posttransfection, and virus titers were determined by a 50% tissue culture infective dose (TCID50) assay. Cell-associated infectivity was determined essentially as described previously (23). Briefly, cells were extensively washed with PBS, scraped, and centrifuged for 5 min at 400 × g. Cell pellets were resuspended in 1 ml of DMEM containing 5% FCS and subjected to three freeze-thaw cycles using liquid nitrogen and a heating block set to 37°C. Samples were then centrifuged at 10,000 × g for 10 min at 4°C to remove cell debris, and cell-associated infectivity was determined by a TCID50 assay.
Rate-zonal centrifugation.
HCV-transfected cells seeded into 10-cm dishes were scraped in 250 μl TNE buffer (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA) at 48 h posttransfection and subjected to five cycles of freezing and thawing. Postnuclear supernatants collected after centrifugation of the crude lysate for 5 min at 4°C at 1,700 × g were layered on top of preformed continuous 0 to 30% sucrose-TNE gradients and spun at 270,000 × g for 1 h at 4°C in a Sorvall TH-641 ultracentrifuge rotor. Subsequently, 10 fractions (1 ml each) were harvested from the top, and the respective refraction index was measured by refractometry.
Statistical analysis.
Statistical analysis was done with R (http://www.r-project.org/). Due to small sample sizes, a two-sample Mann-Whitney U test was used to asses the difference between sample distributions. P values were calculated, and differences are reported as significant if the P value was ≤0.05. Differences were considered not significant at P values of >0.05.
RESULTS
Features of HCV core and p7 determine efficiency of particle production by intragenotypic HCV chimeras.
It has been described that the intragenotypic chimeric genome Jc1, composed of the region encoding core up to the first putative transmembrane segment of NS2 of the J6CF isolate and the remainder of the genome of the JFH1 isolate, replicates at levels similar to those of JFH1 but supports much higher levels of production of infectious HCV particles (34). We assumed that the higher titers achieved and faster kinetics of virus production must be due to determinants residing in the J6CF sequence from core to p7. Thus, we constructed chimeric HCV genomes by transferring J6CF-derived core (JFH1/coreJ6), p7 (JFH1/p7J6), and a combination of both (JFH1/corep7J6) into JFH1 (Fig. 1A). These chimeras were transfected into highly permissive Huh7.5 cells, in parallel with Jc1 and JFH1 as reference genomes. The kinetics of release of infectivity from transfected cells were determined with a limiting-dilution assay (23). As previously described, JFH1 titers were comparatively low at 24 h posttransfection and increased to 104 TCID50/ml at later time points. In contrast, peak Jc1 titers were 100-fold higher with 106 TCID50/ml (34) (Fig. 1B). Interchange of J6CF p7 into JFH1 increased viral titers 10-fold over the titers of the parental genome, and core transfer yielded even higher titers, 50-fold over JFH1 titers. Insertion of both J6CF-derived proteins core and p7 enhanced viral titers 100-fold, reaching a level comparable to that of Jc1. To rule out that the differences observed were not caused by changes in RNA replication, translation, or specific infectivity of viral particles, extra- and intracellular core protein levels were determined by an ELISA (Fig. 1C). Intracellular core amounts showed only slight differences for all genomes tested, and the level of released core protein grossly correlated with supernatant infectivity. Thus, efficiency of virus assembly/release was affected by core and p7 exchanges. These results indicate that core and p7 are crucial determinants for viral particle production in the context of JFH1-J6-intergenotypic chimeras.
Fig 1.

Characterization of genotype 2a core and p7 recombinants in production of infectious particles. (A) Schematic representation of the parental Jc1 and JFH1 genomes (top) and the core and p7 chimeras (bottom). JFH1 and J6 sequences are depicted as white and gray boxes, respectively. JFH1-derived 5′ and 3′ nontranslated regions are drawn as thick black lines. Note that the junction site of J6 and JFH1 in the Jc1 chimera resides in the N-terminal membrane domain of NS2 (22). (B) Huh7.5 cells transfected with RNA encoding each chimera. After 24 h, 48 h, and 72 h, cell supernatants were harvested, and infectious virus release was determined by TCID50 titration on Huh7.5 cells. Mean values and standard deviations of three independent experiments are shown. Statistical analysis with a two-sample Mann-Whitney U test was performed for the indicated HCV genomes and time points. (C) Intra- and extracellular core levels from cells transfected with the HCV genomes specified in panel A at 48 posttransfection. Data from a representative experiment conducted with two technical replicates, representative of three independent experiments in total, are shown.
Glycoprotein genes modulate efficiency of virus production within intragenotypic HCV chimeras.
To investigate the relevance of E1 and E2 genes for the efficiency of virus production by intragenotypic GT2a chimeras, we constructed a JFH1 genome with the glycoproteins of J6CF (JFH1/E1E2J6) and a chimeric Jc1 genome with the glycoproteins of JFH1 (Jc1/E1E2JFH1) (Fig. 2A). After transfection of these genomes into Huh7.5 cells, release of infectivity into the cell culture supernatant over time was determined. As depicted in Fig. 2B, the transfer of JFH1-derived E1 and E2 into Jc1 caused a modest decrease in virus titers compared with the titers of the parental Jc1 genome. Exchange of J6CF-derived glycoproteins into JFH1 yielded titers of about 104 TCID50/ml at 72 h posttransfection, which is comparable to the titers of the parental JFH1 genome (Fig. 2B). Levels of intracellular core protein accumulating in transfected cells showed only slight differences between the chimeric genomes, arguing for nearly equivalent levels of RNA replication (Fig. 2C). The differences in virus production between Jc1- and JFH1-based genomes were also comparable, with the differences of the amounts of core protein released from transfected cells indicating that the specific infectivity conferred by the glycoprotein transfer was not affected (Fig. 2C). In summary, these data suggest that within intragenotypic GT2a-GT2a chimeras, glycoprotein genes can be exchanged without causing gross changes in virus production efficiency.
Fig 2.
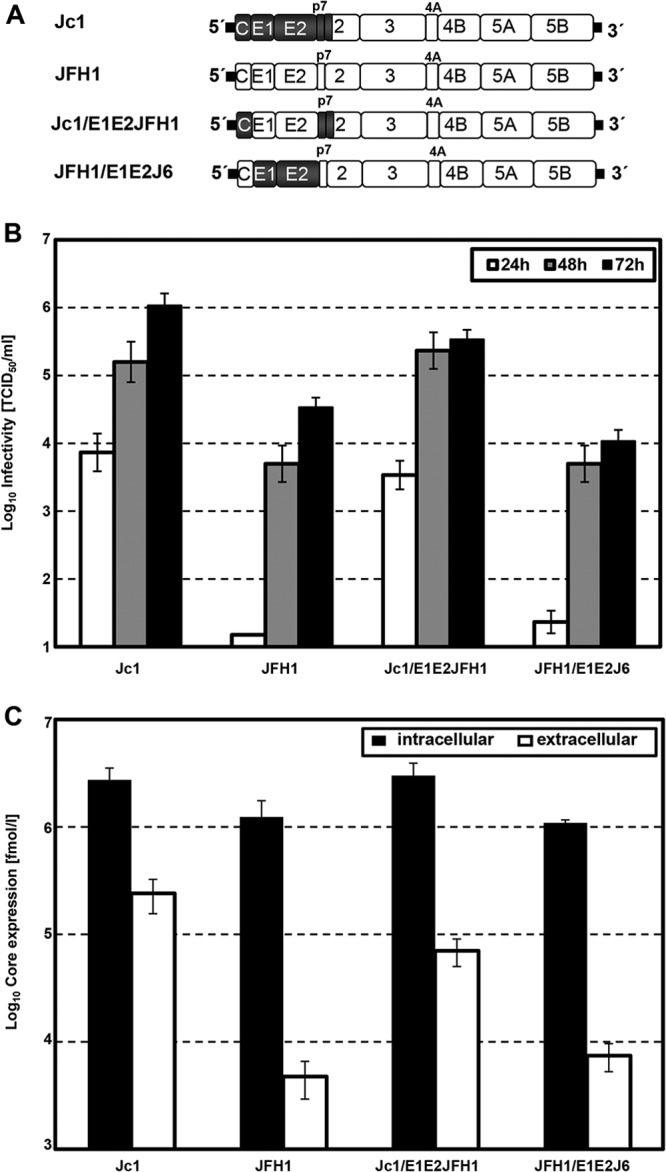
Influence of intragenotypic glycoprotein chimeras on the production of HCV infectious particles. (A) Schematic representation of the parental Jc1 and JFH1 genomes (top) and the intragenotypic glycoprotein chimeras (bottom). JFH1 and J6 sequences are depicted as white and gray boxes, respectively. JFH1-derived 5′ and 3′ nontranslated regions are drawn as thick black lines. Note that the junction site of J6 and JFH1 in the Jc1 chimera resides in the N-terminal membrane domain of NS2. (B) Huh7.5 cells transfected with RNA of full-length chimeras. After 24 h, 48 h, and 72 h, cell supernatants were harvested, and extracellular infectivity was determined by TCID50 titration on Huh7.5 cells. (C) Intra- and extracellular core levels from cells transfected with the HCV genomes specified in panel A at 48 posttransfection. Data from a representative experiment conducted with six replicates (TCID50 assay) and two replicates (core ELISA), representative of three independent experiments in total, are shown.
These findings raised the hope that non-GT2a-derived envelope protein genes could also be transferred to the highly efficient GT2a chimera, permitting efficient virus production of particles carrying these glycoproteins. To test this, we next generated a chimeric Jc1 genome encoding the glycoproteins of GT1a isolate H77 (Jc1/E1E2H77). In parallel, we inserted the J6CF-derived envelope proteins into the GT1a-GT2a chimera JFH1/H77/C3 (34), to create JFH1/H77/C3/E1E2J6 (Fig. 3A). As noted previously (34), Jc1 and JFH1/H77/C3 differ with respect to the extent and kinetics of accumulation of infectious virions, with 106 TCID50/ml for Jc1 and only 102 TCID50/ml for JFH1/H77/C3 (Fig. 3B). The replacement of J6CF glycoproteins by H77 envelope proteins suppressed release of infectious particles more than 1,000-fold. Remarkably, exchange of J6CF-derived glycoproteins into JFH1/H77/C3 enhanced production of infectious particles 50-fold (Fig. 3B). To rule out that these effects were due merely to isolate-specific differences in assembly competence of J6CF- and H77-derived glycoproteins, we performed similar experiments with intergenotypic glycoprotein chimeras between J6CF and Con1 (GT1b) and observed similar results (Fig. 3D and E). The replacement of J6CF glycoproteins by Con1 envelope proteins suppressed infectious particle production dramatically, and at 48 h, viral titers were 50-fold enhanced for JFH1/Con1/C3/E1E2J6 in comparison to those for JFH1/Con1/C3 (Fig. 3D). Thus, both GT1-derived E1-E2 genes strongly reduced virus production in the context of the GT2a chimeric genome, whereas J6CF-derived genes facilitated release of infectious progeny from the GT1a-JFH1 and the GT1b-JFH1 chimeric genomes (Fig. 3). Intracellular core amounts were similar for all tested genomes, demonstrating comparable replication efficiencies, and the levels of extracellular infectivity correlated with the extracellular core levels, indicating that virus assembly/release was affected by the glycoprotein exchanges (Fig. 3C and E).
Fig 3.

Influence of intergenotypic glycoprotein chimeras on HCV particle production. (A) Schematic representation of the parental Jc1, JFH1/H77/C3, and JFH1/Con1C3 genomes (top) and the intergenotypic glycoprotein chimeras (bottom). JFH1, J6, H77, and Con1 sequences are depicted as white, dark gray, light gray, and striped boxes, respectively. JFH1-derived 5′ and 3′ nontranslated regions are drawn as thick black lines. Note that the junction site of J6 and JFH1 in the Jc1 chimera resides in the N-terminal membrane domain of NS2. (B) Huh7.5 cells transfected with RNA of full-length chimeras. After 24 h, 48 h, and 72 h, cell supernatants were harvested, and extracellular infectivity was determined by TCID50 titration on Huh7.5 cells. (C) Intra- and extracellular core levels from cells transfected with the HCV genomes specified in panel A at 48 posttransfection. Data from a representative experiment conducted with six replicates (TCID50 assay) and two replicates (core ELISA), representative of three independent experiments in total, are shown. (D and E) Experiments conducted as described above for panels B and C but with JFH1/Con1 chimeras.
These data suggested that genotype 1a glycoproteins in the Jc1 backbone decrease the number of secreted infectious particles. This may be attributable to defective virion assembly or impaired release of virions. To distinguish between these two possibilities, we compared the quantities of intracellular infectious viruses prepared by repetitive freeze-and-thaw cycles and extracellular infectious virions produced upon transfection (Fig. 4). In the case of Jc1/E1E2H77, total infectivity was reduced by several orders of magnitude (Fig. 4A), and the parallel decrease of intra- and extracellular infectivity indicates a specific impairment of HCV assembly rather than release (Fig. 4B). The titer-enhancing effect of JFH1/H77/C3/E1E2J6 is also reflected in the amounts of intracellular infectious particles and core protein levels, consistent with more efficient assembly of this glycoprotein chimera than of the parental construct JFH1/H77/C3 (Fig. 4B and C). In conclusion, transfer of GT1a-derived glycoprotein into Jc1 abrogated efficient HCV assembly, indicating genetic incompatibilities between the glycoproteins and other viral factors. On the other hand, insertion of J6CF-derived E1-E2 genes into the JFH1/H77/C3 or JFH1/Con1/C3 chimeras increased virus assembly of these chimeras. This finding suggests that glycoprotein compatibility with viral factors other than core, p7, and the N-terminal portion of NS2, which are derived from JFH1 (GT2a) in these chimeras, could influence virus production.
Fig 4.

Efficiency of virus assembly and release of glycoprotein chimeras. Huh7.5 cells were transfected with RNA encoding full-length HCV chimeras. After 48 h, cell supernatants were harvested, and intracellular lysates were prepared by repetitive cycles of freezing and thawing. Intra- and extracellular infectivity was determined by TCID50 titration on Huh7.5 cells (A and B), and core levels were quantified by ELISA (C). Data from a representative experiment conducted with six replicates (TCID50 assay) and two replicates (core ELISA), representative of three independent experiments in total, are shown.
Heterologous glycoproteins within intergenotypic chimeras do not cause gross changes in polyprotein processing, protein interactions, and subcellular localization of viral proteins.
To explore the molecular mechanism underlying the assembly defect apparent in intergenotypic glycoprotein chimeras, we first compared the subcellular localizations of core, E2, and NS2 between the different chimeric genomes. As shown in Fig. 5A, E2 was localized at the ER compartment, and no obvious differences between the parental constructs Jc1 and JFH1/H77/C3 and the glycoprotein chimeras Jc1/E1E2H77 and JFH1/H77/C3/E1E2J6 were observed (Fig. 5A). Comparable staining patterns of the different glycoprotein chimeras were also observed for the core protein and NS2, indicating no major influence of heterologous glycoprotein insertion on viral protein subcellular localization.
Fig 5.

Characterization of glycoprotein chimeras in subcellular localization of viral proteins and interaction partners. (A) The indicated genomes were transfected into Huh7.5 cells, fixed after 48 h, and subjected to immunofluorescence staining of E2 (AP33 antibody [green]) and core (C7-50 antibody [red]) or NS2 (C6H6 antibody [red]). Nuclei were counterstained with DAPI (blue). Representative pictures are shown. (B) Huh7.5 cells were transfected with the indicated genomes and after 24 h were pulse labeled with [35S]methionine-cysteine-containing medium for 6 h before lysis. Cells transfected with a subgenomic JFH1 replicon encoding HCV NS3-NS5B proteins only (SG-JFH1) served as a negative control. HCV proteins were immunoprecipitated by using an antibody monospecific for E2 and are identified by arrows on the left. Note that the H77-derived E1 displays a lower electrophoretic mobility than E1 from J6CF. (C) Transfected cells were harvested at 48 h posttransfection by cell lysis, and immunoprecipitation (IP) with antibody monospecific for E2 was performed. NS2 was detected in cell lysates (input) and in immunoprecipitates by Western blotting.
To assess whether the intergenotypic glycoprotein chimeras possessed a defect in glycoprotein processing, we analyzed the processing of E2, p7, and NS2 by pulse labeling of Huh7.5 cells transfected with the parental Jc1 and JFH1/H77/C3 genomes and the corresponding glycoprotein chimeras. Immunoprecipitations using an E2-specific antibody showed that E2, E1, and NS2 were precipitated in the case of Jc1, as previously described for JFH1 (Fig. 5B) (23). No major differences in precipitation patterns of the different mutants could be observed; larger amounts of viral proteins were detected only for Jc1/E1E2H77, which might be attributed to differences in the affinity of the antibody for E2 proteins of different genotypes (Fig. 5B). Furthermore, these results show that processing at the E2-p7 and p7-NS2 junctions occurred and yielded comparable quantities of E1-E2 complexes. To confirm these findings, we investigated the physical interaction by immunoprecipitation of E2 followed by Western blotting against NS2 (Fig. 5C). Huh7.5 cells were transfected with the indicated constructs, and E2-containing immunocomplexes were captured from lysates that were prepared at 48 h posttransfection. As observed by radioimmunoprecipitation, similar quantities of NS2 could be pulled down for the different mutants (Fig. 5C). As noted above, we were able to detect a truncated form of NS2, especially in the input. The nature and function of this product still remain elusive (43). In summary, swapping of genotype 1 glycoproteins into Jc1 had no major influence on subcellular localization, processing, and interactions of viral proteins.
HCV glycoproteins are important for core protein envelopment in a genotype-specific fashion.
To further dissect the defect in morphogenesis caused by insertion of the GT1a-derived heterologous glycoproteins into Jc1, we assessed the resistance of intracellular core to proteolytic digestion by proteinase K. In these assays, HCV-transfected cells are ruptured by repetitive cycles of freezing and thawing. Subsequently, proteinase K is added, and the amount of core protein resistant to this treatment, presumably due to envelopment into a lipid membrane, is quantified by Western blotting and ELISA (44, 45). A fraction of these lysates is treated with Triton X-100, which dissolves all membranes and proves that the concentration of proteinase K used was sufficient to cleave core protein. As depicted in Fig. 6A, for the parental genomes Jc1 and JFH1/H77/C3, about 30% and 15% of intracellular core was resistant to protease digestion, respectively, and had already acquired an envelope (Fig. 6A). The transfer of genotype 1a-derived glycoproteins into Jc1 reduced the amount of protected core, indicating that the amount of core protein accessible to proteolytic digestion was increased. The insertion of E1/E2 from J6CF into JFH1/H77/C3 had no detectable influence on core protection, which may be due to the modest dynamic range of this assay (Fig. 6A). To further corroborate that insertion of H77-derived E1-E2 genes into Jc1 decreases core protein envelopment, we analyzed the sedimentation velocity of core protein structures derived from this chimera and the parental Jc1 genome. Previously, we had shown that deletion of glycoproteins or disruption of p7 function caused a defect in capsid envelopment, which resulted in an increase of core protein structures with a slow sedimentation behavior in these rate-zonal gradients (44). Similar to our previous observations (44), core proteins derived from the parental Jc1 genome sedimented rapidly, displaying a single peak in fraction 7 of our gradient (Fig. 6B). In contrast, the bulk of core protein structures originating after transfection of the glycoprotein chimera Jc1/E1E2H77 sedimented slowly, with a peak in fraction 3 representing nonenveloped capsids, as shown recently (Fig. 6B) (44). As observed for the protease digestion assay, no major differences were observed for the parental JFH1/H77/C3 genome and JFH1/H77/C3/E1E2J6 (Fig. 6B, bottom). Notably, however, both constructs yielded clearly lower levels of rapidly sedimenting core protein species than did Jc1, suggesting that large numbers of these complexes are a hallmark of viral genomes with highly efficient virus production. In conclusion, intergenotypic glycoprotein chimeras accumulated as nonenveloped capsid-like structures, revealing an important role for the glycoproteins in membrane envelopment.
Fig 6.

Proteinase K digestion and rate-zonal separation of HCV glycoprotein chimeras. (A) HCV genome-transfected detergent-free cell lysates were subjected to a proteolytic digestion protection assay as follows. Lysates were separated into three parts: (i) untreated, (ii) treated with 50 μg/ml proteinase K (PK) for 1 h on ice, or (iii) lysed in 5% Triton X-100 prior to proteinase K treatment. The amount of protease-resistant core was determined by a core-specific ELISA. Mean values and standard errors of data from 5 independent experiments are shown. (B) Postnuclear supernatants of cell lysates obtained by repetitive cycles of freezing and thawing at 48 h posttransfection were layered on top of a preformed continuous 0 to 30% sucrose density gradient and subjected to rate-zonal centrifugation for 1 h at 270,000 × g. Core content was measured along the gradient by ELISA and normalized to the total core amount in the lysate. Representative values of 3 independent experiments are plotted.
DISCUSSION
HCV envelope glycoproteins play important roles in different steps of the viral life cycle. During viral entry, they interact with host cell receptors and are involved in fusion of the viral and cellular membranes. In the late steps of the viral life cycle, E1 and E2 are crucial for viral assembly, as deletion of the glycoprotein abrogates viral particle production (11, 44). However, the precise role of the glycoproteins in HCV morphogenesis is poorly understood. To further dissect the role of the E1 and E2 glycoproteins in virion morphogenesis, we generated chimeric viral genomes, replacing the glycoprotein regions with corresponding regions from different isolates and genotypes. Intragenotypic chimeric HCV genomes revealed that core and p7 are crucial determinants of viral particle production, whereas glycoprotein chimeras between J6CF and JFH1 were fully functional and produced particles with comparable efficiencies. These findings raise the hope that at least within GT2a, different GT2a-derived glycoprotein genes could be shuffled into the Jc1 backbone in order to permit assessment of their functional properties. In contrast, HCV assembly of intergenotypic glycoprotein constructs was highly attenuated, although no major effect on subcellular localization and interaction of viral proteins was observed. However, swapping of GT1 glycoproteins into a GT2a genome led to the accumulation in nonenveloped capsid-like structures, revealing an important, genotype-dependent role of the glycoproteins in viral envelopment.
Chimeric HCV genotype genomes like Jc1, consisting of J6 and JFH1 segments, produce much higher virus titers than the parental JFH1 or genotype 1 viral chimeras (34). To identify determinants in the core to NS2 proteins that modulate the efficiency of virus production, we generated chimeric viruses between J6CF and JFH1. Combination of both J6CF-derived proteins core and p7 enhanced viral titers 100-fold, which are comparable to those obtained with Jc1 and indicate a minor role of the envelope protein in virus production efficiency (Fig. 1 and 2). These results are in agreement with results from previous studies focusing on either core or p7 but not in combination. Shavinskaya et al. showed previously that J6CF core contains determinants that enhance virus production, which reside in domain 2 of core, and that the binding strength of domain 2 for lipid droplets is crucial for the efficiency of viral assembly (37). With regard to the role of p7 in HCV morphogenesis, we showed that p7 variants differed in their abilities to promote virus production, and J6CF-derived p7 boosted virus production (23). Recently, we also described that p7 is part of a membrane-bound “recipient complex” that provides a scaffold to initiate unloading of core protein from lipid droplets for capsid assembly and membrane envelopment (44).
Transfer of genotype 1a glycoproteins into the Jc1 backbone caused a strong reduction in intra- and extracellular infectivity, implying a defect in HCV assembly (Fig. 3). The genetic compatibility between the envelope proteins themselves has been addressed in several studies that identified structural regions in the glycoproteins important for viral entry and assembly. A combination of E1 from genotype 2a and E2 from genotype 1a was nonfunctional in the HCV pseudoparticle (HCVpp) system and revealed the stem region of E2 as an important segment for entry and the intergenotypic variable region (IgVR) hypervariable region 2 (HVR2) as well as another segment in domain II for assembly in the cell culture-derived HCV (HCVcc) system (46). Carlsen et al. also characterized HCV recombinants by using genotype 1a/2a JFH1-based recombinants with exchanges of E2 by sequences of another genotype, 1a, 1b, or 2a (47). Compensatory mutations were identified at residues 706, 707, and 710 in the E2 stem region, which increased infectivity 100-fold by enhancing viral entry efficiency. Furthermore, cross talk between E1 and E2 using E1-E2 chimeras of H77 and Con1 was described (48). Maurin et al. discovered that the N terminus and transmembrane domain of E1 are crucial determinants of optimal HCV entry (48). In the present study, E1 and E2 were swapped together into the backbone of another genotype, and all isolates used have been shown to be entry competent (49). Interestingly, the transfer of genotype 1 glycoproteins into Jc1 had no major influence on subcellular localization of the viral proteins E2, core, and NS2, at least as far as they are detectable by the resolution of our assay (Fig. 5A). In addition, polyprotein processing at the E2-p7 and p7-NS2 junctions was not influenced by the heterologous glycoproteins (Fig. 5B). The HCV NS2 protein has been implicated as a key regulator of HCV assembly and interacts with several proteins, including E2, during HCV assembly (21, 22, 50–52). We therefore tested the interaction of E2 with NS2 by immunoprecipitation experiments and observed no defects in interaction profiles between the parental genomes and the glycoprotein chimeras. A genetic incompatibility with p7 might be possible, but we were recently unable to show a direct interaction between p7 and E2 (38). However, rate-zonal centrifugation of detergent-free cell lysates demonstrated an accumulation of slow-sedimenting, core complexes in the case of the genotype 1a glycoprotein chimera (Fig. 6). These core protein structures resemble the ones accumulating upon transfection of Jc1 with deleted glycoproteins or inactive p7, which lacked a lipid envelope (44). In line with this, core protein derived after transfection of the glycoprotein chimera Jc1/E1E2H77 was less resistant to protease K digestion. These findings corroborate the notion that insertion of the heterologous GT1a-derived envelope proteins had decreased membrane envelopment of core protein, which in turn limits assembly and release of infectious progeny. Notably, JFH1/H77/C3 and JFH1/H77/C3/E1E2J6 produced modest infectious titers and intermediate levels of rapidly sedimenting core protein structures compared with the highly efficient Jc1 (Fig. 6B). Thus, efficient membrane envelopment of core, which is estimated by the proteinase K assay and the rate-zonal gradient, albeit with modest dynamic ranges of these assays, may be a hallmark of those viral genomes with highly efficient assembly growing to high infectious titers. Future work should address which viral determinants and host factors specifically determine this critical step in virus morphogenesis.
Notably, we observed that insertion of J6CF-derived E1-E2 into JFH1/H77/C3 and JFH1/Con1/C3 chimeras increased virus production by these chimeras. Presently, we cannot strictly rule out that this is due to a higher permissiveness of J6CF-derived E1-E2 for HCV assembly than H77- or Con1-derived E1-E2. However, our observation that J6CF-derived E1-E2 did not increase virus production in the context of JFH1 makes this explanation less likely. Alternatively, it is possible that insertion of the J6CF-derived E1-E2 proteins increases virus production of the intergenotypic GT1-GT2a chimeras JFH1/H77/C3 and JFH1/Con1/C3 by facilitating the interplay of the envelope proteins with other viral proteins derived from the GT2a portion of these chimeras. Thus, these findings suggest that the HCV envelope proteins may cooperate with viral determinants resident downstream of the N-terminal domain of NS2 in a genotype-specific fashion. In turn, this interplay may be important to coordinate core protein envelopment during assembly. Clearly, more work is needed to fully understand the network of envelope protein interactions during HCV assembly.
In summary, we characterized intra- and intergenotypic glycoprotein chimeras in the HCV assembly process. First, we demonstrate that the concerted action of core and p7 facilitates efficient virion production in the context of GT2a chimeric genomes. While exchange of glycoproteins from the same genotype was fully functional, genotype 1-derived transfer of E1-E2 drastically reduced virus particle production. Capsid protection and envelopment assays uncovered a novel role of the HCV glycoproteins in viral membrane envelopment.
ACKNOWLEDGMENTS
We are grateful to Takaji Wakita for the gift of the JFH1 isolate, Jens Bukh for the J6CF strain, Charles Rice for Huh7.5 cells and the 9E10 monoclonal antibody, Darius Moradpour for the core-specific C7-50 antibody, and Genentech and Arvind Patel for providing AP33. We also thank all members of the Institute of Experimental Virology for helpful suggestions.
E.S. was supported by the DFG (STE 1954/1-1) and an intramural young investigator award of the Helmholtz Centre for Infection Research. T.P. was supported by grants from the DFG (PI 734/2-1) and the Helmholtz Association (SO-024).
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567 [DOI] [PubMed] [Google Scholar]
- 2.Hoofnagle JH. 1997. Hepatitis C: the clinical spectrum of disease. Hepatology 26:15S–20S [DOI] [PubMed] [Google Scholar]
- 3.Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 85:3173–3188 [DOI] [PubMed] [Google Scholar]
- 4.Negro F. 2010. Hepatitis C virus-induced steatosis: an overview. Dig. Dis. 28:294–299 [DOI] [PubMed] [Google Scholar]
- 5.Pearlman BL. 2012. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect. Dis. 12:717–728 [DOI] [PubMed] [Google Scholar]
- 6.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 [DOI] [PubMed] [Google Scholar]
- 7.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113 [DOI] [PubMed] [Google Scholar]
- 8.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 11.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinmann E, Pietschmann T. 2013. Cell culture systems for hepatitis C virus. Curr. Top. Microbiol. Immunol. 369:17–48 [DOI] [PubMed] [Google Scholar]
- 14.Chang KS, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81:13783–13793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Jr, Ye J. 2007. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 104:5848–5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035. 10.1371/journal.ppat.1000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J. Virol. 86:8987–8997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 81:8374–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 83:2163–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y, Yates J, Liang Y, Lemon SM, Yi M. 2008. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 82:7624–7639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phan T, Beran RK, Peters C, Lorenz IC, Lindenbach BD. 2009. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83:8379–8395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. 2007. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 3:e103. 10.1371/journal.ppat.0030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yi M, Ma Y, Yates J, Lemon SM. 2007. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81:629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choukhi A, Ung S, Wychowski C, Dubuisson J. 1998. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J. Virol. 72:3851–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubuisson J, Rice CM. 1996. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 70:778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merola M, Brazzoli M, Cocchiarella F, Heile JM, Helenius A, Weiner AJ, Houghton M, Abrignani S. 2001. Folding of hepatitis C virus E1 glycoprotein in a cell-free system. J. Virol. 75:11205–11217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cocquerel L, Duvet S, Meunier JC, Pillez A, Cacan R, Wychowski C, Dubuisson J. 1999. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 73:2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cocquerel L, Meunier JC, Pillez A, Wychowski C, Dubuisson J. 1998. A retention signal necessary and sufficient for endoplasmic reticulum localization maps to the transmembrane domain of hepatitis C virus glycoprotein E2. J. Virol. 72:2183–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Op De Beeck A, Voisset C, Bartosch B, Ciczora Y, Cocquerel L, Keck Z, Foung S, Cosset FL, Dubuisson J. 2004. Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 78:2994–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michalak JP, Wychowski C, Choukhi A, Meunier JC, Ung S, Rice CM, Dubuisson J. 1997. Characterization of truncated forms of hepatitis C virus glycoproteins. J. Gen. Virol. 78(Part 9):2299–2306 [DOI] [PubMed] [Google Scholar]
- 32.Goffard A, Dubuisson J. 2003. Glycosylation of hepatitis C virus envelope proteins. Biochimie 85:295–301 [DOI] [PubMed] [Google Scholar]
- 33.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 84:11905–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570–574 [DOI] [PubMed] [Google Scholar]
- 36.Yanagi M, Purcell RH, Emerson SU, Bukh J. 1999. Hepatitis C virus: an infectious molecular clone of a second major genotype (2a) and lack of viability of intertypic 1a and 2a chimeras. Virology 262:250–263 [DOI] [PubMed] [Google Scholar]
- 37.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. 2007. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282:37158–37169 [DOI] [PubMed] [Google Scholar]
- 38.Vieyres G, Brohm C, Friesland M, Gentzsch J, Wolk B, Roingeard P, Steinmann E, Pietschmann T. 2013. Subcellular localization and function of an epitope-tagged p7 viroporin in hepatitis C virus-producing cells. J. Virol. 87:1664–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Owsianka A, Tarr AW, Juttla VS, Lavillette D, Bartosch B, Cosset FL, Ball JK, Patel AH. 2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol. 79:11095–11104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dentzer TG, Lorenz IC, Evans MJ, Rice CM. 2009. Determinants of the hepatitis C virus nonstructural protein 2 protease domain required for production of infectious virus. J. Virol. 83:12702–12713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vieyres G, Pietschmann T. 2013. Entry and replication of recombinant hepatitis C viruses in cell culture. Methods 59:233–248 [DOI] [PubMed] [Google Scholar]
- 42.Clayton RF, Owsianka A, Aitken J, Graham S, Bhella D, Patel AH. 2002. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol. 76:7672–7682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jirasko V, Montserret R, Appel N, Janvier A, Eustachi L, Brohm C, Steinmann E, Pietschmann T, Penin F, Bartenschlager R. 2008. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J. Biol. Chem. 283:28546–28562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gentzsch J, Brohm C, Steinmann E, Friesland M, Menzel N, Vieyres G, Perin PM, Frentzen A, Kaderali L, Pietschmann T. 2013. Hepatitis C virus p7 is critical for capsid assembly and envelopment. PLoS Pathog. 9:e1003355. 10.1371/journal.ppat.1003355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menzel N, Fischl W, Hueging K, Bankwitz D, Frentzen A, Haid S, Gentzsch J, Kaderali L, Bartenschlager R, Pietschmann T. 2012. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog. 8:e1002829. 10.1371/journal.ppat.1002829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, Descamps V, Duverlie G, Rey F, Penin F, Dubuisson J. 2011. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J. Virol. 85:1777–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlsen TH, Scheel TK, Ramirez S, Foung SK, Bukh J. 2013. Characterization of hepatitis C virus recombinants with chimeric E1/E2 envelope proteins and identification of single amino acids in the E2 stem region important for entry. J. Virol. 87:1385–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maurin G, Fresquet J, Granio O, Wychowski C, Cosset FL, Lavillette D. 2011. Identification of interactions in the E1E2 heterodimer of hepatitis C virus important for cell entry. J. Biol. Chem. 286:23865–23876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, Manns MP, Ott M, Wedemeyer H, Meuleman P, Pietschmann T, Steinmann E. 2011. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 54:1947–1955 [DOI] [PubMed] [Google Scholar]
- 50.Jirasko V, Montserret R, Lee JY, Gouttenoire J, Moradpour D, Penin F, Bartenschlager R. 2010. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6:e1001233. 10.1371/journal.ppat.1001233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popescu CI, Callens N, Trinel D, Roingeard P, Moradpour D, Descamps V, Duverlie G, Penin F, Heliot L, Rouille Y, Dubuisson J. 2011. NS2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 7:e1001278. 10.1371/journal.ppat.1001278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stapleford KA, Lindenbach BD. 2011. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 85:1706–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]