

Abstract
The mode and timing of virally induced cell death hold the potential of regulating viral yield, viral transmission, and the severity of virally induced disease. Orbiviruses such as the epizootic hemorrhagic disease virus (EHDV) are nonenveloped and cytolytic. To date, the death of cells infected with EHDV, the signal transduction pathways involved in this process, and the consequence of their inhibition have yet to be characterized. Here, we report that the Ibaraki strain of EHDV2 (EHDV2-IBA) induces apoptosis, autophagy, a decrease in cellular protein synthesis, the activation of c-Jun N-terminal kinase (JNK), and the phosphorylation of the JNK substrate c-Jun. The production of infectious virions decreased upon inhibition of apoptosis with the pan-caspase inhibitor Q-VD-OPH (quinolyl-valyl-O-methylaspartyl-[-2,6-difluorophenoxy]-methyl ketone), upon inhibition of autophagy with 3-methyladenine or via the knockout of the autophagy regulator Atg5, or upon treatment of infected cells with the JNK inhibitor SP600125 or the cyclin-dependent kinase (CDK) inhibitor roscovitine, which also inhibited c-Jun phosphorylation. Moreover, Q-VD-OPH, SP600125, and roscovitine partially reduced EHDV2-IBA-induced cell death, and roscovitine diminished the induction of autophagy by EHDV2-IBA. Taken together, our results imply that EHDV induces and benefits from the activation of signaling pathways involved in cell stress and death.
INTRODUCTION
The epizootic hemorrhagic disease virus (EHDV) is an arbovirus (genus orbiviruses) of the Reoviridae family that is transmitted by biting midges and infects ruminants. In recent years, outbreaks of epizootic hemorrhagic disease in cattle have been reported in Israel and Turkey (1, 2), suggesting that EHDV is an emerging threat to the cattle industry in Europe (3). EHDV presents structural and sequence similarities to the better-studied bluetongue virus (BTV), sharing its repertoire of infection targets and symptoms of disease (3, 4). However, in spite of structural similarities between these viruses, a recent study suggests that preexisting immunity to BTV does not protect against EHDV infection (5). The EHDV genome is organized in 10 double-stranded RNA (dsRNA) segments encoding seven structural proteins (VP1 to VP7) and the nonstructural (NS) proteins NS1 to NS3. Recently, an additional nonstructural protein, NS4, has been identified in BTV (6, 7), raising the possibility that the same protein occurs in EHDV. The present study mainly employs the Ibaraki strain of EHDV2 (EHDV2-IBA), originally isolated from infected cattle in 1959, in Ibaraki, Japan (8). Selected experiments were also carried out with EHDV7-Israel (EHDV7-ISR), isolated from infected cattle in 2006 in Israel (1).
For different types of reoviruses, including BTV, apoptosis is integral to the cellular pathogenesis they induce (9–20). Yet the molecular mechanisms that govern reovirus-mediated induction of apoptosis are a contentious matter (10, 17, 19, 20). For orbiviruses in general and EHDV in particular, these mechanisms and the functional consequences of apoptosis on the virus replication cycle remain uncharacterized and merit further study. Similarly, while the induction of autophagy (a cellular process also associated with viral pathogenesis [21]) by mammalian reovirus (MRV), avian reovirus, and BTV has been recently identified (22–25), its occurrence in the realm of EHDV infection and its functional significance to the infection process remain unstudied.
Mitogen-activated protein kinases (MAPKs) in general (26, 27) and c-Jun N-terminal kinase 1 (JNK1) in particular (28, 29) regulate autophagy, while JNK activation also mediates apoptosis (reviewed in reference 30). Specifically, JNK activation mediates the apoptosis induced by BTV (31) and, depending on the strain, by MRV (32, 33). Similarly, different strains of MRV also differ in their potentials to induce and benefit from integrated cell stress responses (34). Taken together, these studies exemplify the interconnectivity of virally activated stress and death-related cellular programs, demonstrate differences in the potential of induction of these processes by different reovirus strains, and support the notion of usurpation of cell stress and autophagy machinery by some reoviruses.
Here, we report that EHDV2-IBA induces apoptosis, autophagy, the activation of JNK and c-Jun, and the inhibition of protein synthesis in the course of the infection of mammalian cells in culture. Moreover, through the use of specific inhibitors of these processes, we identify their contributions to the generation of infectious virions.
MATERIALS AND METHODS
Cell culture and viruses.
The following cells were employed in this study: spontaneously immortalized ovine kidney (OK) cells (Kimron Veterinary Institute, Beit Dagan, Israel), calf pulmonary aortal endothelial (CPAE) cells (a gift of Eyal Klement, Hebrew University of Jerusalem), and Vero cells (ATCC CCL-81). Cells were grown in either modified Eagle's medium (MEM; CPAE and OK) or Dulbecco's modified Eagle's medium (DMEM; Vero) supplemented with 10% fetal calf serum, 5 mM glutamine, and penicillin-streptomycin (all from Beit HaEmek Biological Industries, Israel) at 37°C and 5% CO2. Atg5−/− and Atg5+/+ mouse embryonic fibroblasts (MEFs) were cultured as described by Schmukler et al. (35), who also confirmed that these cells are defective in autophagy. OK cells stably expressing LC3-GFP (light chain 3 tagged with green fluorescent protein) were generated by transfection with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and selection with 1 mg/ml G418. EHDV2-IBA was a gift to H. Yadin from T. Tsuda (Kyushu Research Station, National Institute of Animal Health, Chuzan, Kagoshima, Japan). The virus was isolated in 1959, in Ibaraki, Japan, and passaged for 21 times prior to the present study. The 2006 Israeli isolate of EHDV7-ISR was contributed by H. Yadin.
Antibodies and reagents.
For the production of polyclonal rabbit and monoclonal mouse anti-NS3 antibodies, mice (8-week-old BALB/c female mice) and rabbits were immunized with the following peptide: CDEMSLVPYQENVRPPS (which includes amino acids 19 to 34 of EHDV2-IBA NS3 (GeneScript Corporation, NJ, USA). Mouse anti-p62/SQSTM1 was from MBL International (Woburn, MA, USA); rabbit anti-LC3B was from Sigma-Aldrich (St. Louis, MO, USA); anti-JNK, anti-phospho-JNK (Thr183/Tyr185), anti-caspase 3, anti-MEK1/MEK2 (MEK1/2), and anti-eukaryotic initiation factor 2α (eIF2α) antibodies, all anti-rabbit, were from Cell Signaling (Beverly, MA, USA). Rabbit anti-c-Jun and monoclonal mouse anti-phospho-c-Jun (Ser63) antibodies were from Santa-Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-phospho-eIF2α (Ser52) antibodies were from Invitrogen (Invitrogen, Carlsbad, CA, USA). Mouse anti-actin was from MP Biomedicals (Irvine, CA, USA). Alexa Fluor 555-conjugated secondary antibodies were from Molecular Probes (Eugene, OR, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). DAPI (4′,6′-diamidino-2-phenylindole) was from Sigma-Aldrich (St. Louis, MO, USA).
Drugs and treatments.
Reagents were employed at the following final concentrations: staurosporine (STS; A.G. Scientific Inc., San Diego, CA, USA), 1 μM; caspase inhibitor Q-VD-OPH (quinolyl-valyl-O-methylaspartyl-[-2,6-difluorophenoxy]-methyl ketone; Kamiya Biomedical, Seattle, WA, USA), 20 μM; 3-methyladenine (3MA; Sigma-Aldrich, St. Louis, MO, USA), 10 mM; JNK inhibitor SP600125 (Merck KGaA, Darmstadt, Germany), 20 μM; roscovitine (Sigma-Aldrich, St. Louis, MO, USA), 20, 40, or 80 μM.
Plasmids.
LC3-GFP was a gift from Z. Elazar (Weizmann Institute of Science, Israel).
Immunoblotting.
Cells were lysed in lysis buffer [50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EDTA, pH 8, 1 mM EGTA, pH 8, 1.5 mM MgCl2, 200 μM Na3VO4, 150 nM aprotinin, 1 μM leupeptin, and 500 μM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride; Sigma-Aldrich, St. Louis, Mo, USA]. Depending on the experiment, 30 to 100 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) through 10% to 12% polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked for 1 h in TBST buffer (0.05 M Tris-HCl, pH 7.5, 0.15 M NaCl, and 0.1% Tween 20) containing 5% milk and blotted with primary antibodies for 2 h. Secondary antibody linked to HRP was then added for 1 h. Immunoreactive bands were detected by chemiluminescence. Quantification of SDS-PAGE immunoblots was performed with TINA and ImageJ software programs.
Microscopy.
Images were acquired with a spinning-disk confocal microscope (CSU-22 confocal head [Yokogawa] and Axiovert 200 M instrument [Carl Zeiss MicroImaging]) under the control of the SlideBook program (Intelligent Imaging Innovations), using a 63× or 100× oil immersion objective (Plan Apochromat; numerical aperture [NA], 1.4) and an Evolve electron-multiplying charge-coupled-device (EMCCD) camera (Photometrics). Samples were illuminated with 473-nm or 561-nm 40-mW solid-state lasers. Typical exposure times were between 0.1 and 0.5 s.
Immunofluorescence.
OK cells stably expressing LC3-GFP were seeded (8 × 104 cells/well) onto glass coverslips placed in a 24-well plate and either left uninfected or infected with EHDV2-IBA. At 72 h postinfection (hpi), cells were washed twice with cold phosphate-buffered saline (PBS; at 4°C), fixed (in 4% paraformaldehyde [PFA] for 30 min), blocked, permeabilized (for 30 min in PBS–1% bovine serum albumin–0.1% Triton X [PBS-BSA-T]), and stained with polyclonal anti-NS3 antibodies (1:300 dilution in PBS-BSA-T) and Alexa Fluor 555-conjugated goat anti-rabbit antibodies (1:200 dilution in PBS-BSA-T). Mounting was with Fluorescence Mounting Medium (Golden Bridge, Mukilteo, WA, USA).
Virus purification.
EHDV-infected OK cells (at 48 hpi) were collected and pelleted at 4°C. The pellet was resuspended in 6 ml of TNET buffer (50 mM Tris-HCl, pH 8.0, 0.2 M NaCl, 5 mM EDTA, 0.5% Triton X-100) and homogenized (10 strokes) using a glass homogenizer (7 ml). The homogenate was layered onto a sucrose cushion comprised of 66% and 40% sucrose, each prepared in 0.2 M Tris. Samples were centrifuged in a Beckman Ultracentrifuge using an SW41 rotor at 23,000 rpm and 4°C for 3 h. Purified virus was extracted from the interface of the sucrose cushions, and 10 mM dithiothreitol (DTT) was added to prevent virus aggregation.
UV inactivation of virus.
EHDV2-IBA (0.2 × 103 PFU/ml) was irradiated with 250,000 μJ/cm2 with a CL-1000 UV cross-linker (UVP). Virus inactivation was experimentally verified.
Plaque assay.
EHDV2-IBA was collected from infected OK cells or MEFs (untreated or under different inhibitory treatment conditions as specified in the text and figure legends). Virus was collected from the medium and released from attached and detached cells by sonication. Serial dilutions, in 500 μl, were prepared and employed in the infection of reporter cultures of Vero cells seeded in 12-well plates at ∼ 60 to 70% confluence. One well was left uninfected as a control. Plates were incubated with virus at 37°C for 1 h, after which cells were washed and overlaid with 0.6% tragacanth (Sigma-Aldrich, St. Louis, MO, USA) in MEM. Fixation (after 2 to 4 days) was with crystal violet (Sigma-Aldrich, St. Louis, MO, USA) and formaldehyde. Virus titer (PFU/ml) was calculated according to number of plaques/dilution factor.
Determination of MOI.
A total of 100,000 OK cells, plated on glass coverslips placed in 24-well plates, were infected with serial dilutions of purified EHDV2-IBA. At 12 hpi, cells were fixed, permeabilized, and stained against the NS3 protein (with monoclonal anti-NS3 antibodies) and with DAPI (0.1 to 0.3 μg/ml). Stained cells were imaged by microscopy, and the percentage of infected cells was calculated. The multiplicity of infection (MOI) was calculated according to the following formula: MOI = −ln (1 − portion of infected cells). Infections were carried out at MOIs of 0.9 to 1.2, termed ∼1 throughout the text.
Cell cycle analysis by flow cytometry.
Cells were harvested, washed with PBS, fixed with 4% PFA for 8 min, and resuspended in 0.5 ml of PBS supplemented with 0.1% Triton X-100 and 50 μg/ml propidium iodide. Samples were analyzed by fluorescence-activated cell sorter (FACS) flow cytometry (FACSort; BD Bioscience) using CellQuest Pro software.
35S incorporation measurements.
A total of 100,000 OK cells were seeded in 24-well plates. At 12 h postplating and at 12-h intervals, cells were infected with EHDV2-IBA (MOI of ∼1). Triplicate samples were prepared at each time point. After 48 h, cells were washed and starved in cysteine-methionine-free medium (30 min at 37°C) (Beit HaEmek Biological Industries, Israel). Cells were pulse-labeled at a concentration of 20 μCi/ml [35S]cysteine-methionine-containing medium. Proteins (30 μg) from the cell lysates were spotted on Whatman paper filters and allowed to dry. Filters were incubated with boiling 15% trichloroacetic acid (TCA; 5 min) and subsequently washed three times with 5% TCA and once with 70% ethanol. After the drying step, samples were suspended in scintillation fluid and counted with a Beckman beta particle counter for 1 min.
DNA ladder.
DNA from OK cells under the different experimental conditions was extracted using a GenElute mammalian genome DNA Miniprep Kit (Sigma-Aldrich, St. Louis, MO, USA) and separated on a 2% agarose gel containing ethidium bromide.
Trypan blue exclusion assay.
OK cells were seeded at a density of 7 × 104 cells/well of a 24-well plate and either left uninfected or infected under the different experimental conditions (for different time periods and under different treatment conditions). The entire cell population in each well (detached and attached) was collected, precipitated by centrifugation, resuspended in staining medium (MEM mixed with 0.4% trypan blue at a 4:1 ratio), and classified for trypan blue exclusion by light microscopy. Trypan blue (0.4%) was from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysis.
Data are expressed as means ± standard errors (SE). Significant differences in mean values were assessed by a one-tailed Student's t test. A P value of ≤0.05 was considered significant. All experiments were performed at least three times.
RESULTS
EHDV induces apoptosis.
Prior to studying the mechanisms by which EHDV induces the death of mammalian cells, we were interested in establishing the time frame in which this process occurs. Toward this end, we infected spontaneously immortalized ovine kidney (OK) cells, as sheep are a natural orbivirus host, with EHDV2-IBA at a multiplicity of infection (MOI) of ∼1 (MOI was calculated based on the percentage of cells staining for the nonstructural protein NS3 at 12 hpi) (see Materials and Methods). We quantified the percentage of cell death of infected and noninfected cell cultures at different time points, employing a trypan blue exclusion assay to detect the loss of plasma membrane impermeability. In short, we gathered cells from the cell medium and resuspended attached cells with trypsin, prior to their staining with trypan blue and classification by microscopy. As seen in Fig. 1A, at all the measured time points (24, 48, and 72 hpi), the percentage of dead cells in the infected cultures was significantly higher than that in noninfected cultures. Moreover, the percentage of dead cells increased with the progression of infection, peaking at 72 hpi. These data point to virus-mediated induction of cell death already at early infection stages (24 hpi) and the culmination of this process at 72 hpi, establishing thus a time frame for the study of EHDV-induced cell death. To obtain further insight into the dynamics of the infection process within this window, we assessed the levels of the nonstructural protein NS3 at different times postinfection. For this, lysates from cells (infected or noninfected) were separated by SDS-PAGE and subjected to immunoblotting against NS3 and actin (Fig. 1B). NS3 appeared as two major bands in addition to a smear of higher apparent molecular weight, representing mainly different forms of NS3 glycosylation (data not shown). A calculation of the ratios of the intensities of the NS3 and actin signals in multiple immunoblots of infected cell cultures presented peak values at 48 hpi (Fig. 1C). Death by EHDV2-IBA was found to be slightly faster in calf pulmonary aortal endothelial (CPAE) cells (maximal by 48 to 60 hpi), which also originate from a natural orbivirus host, and was slower (up to ∼96 hpi) in Vero African green monkey kidney cells (data not shown). Also, similar infection and cell death kinetics were observed with a different EHDV strain (EHDV7-ISR) (1; also data not shown).
Fig 1.
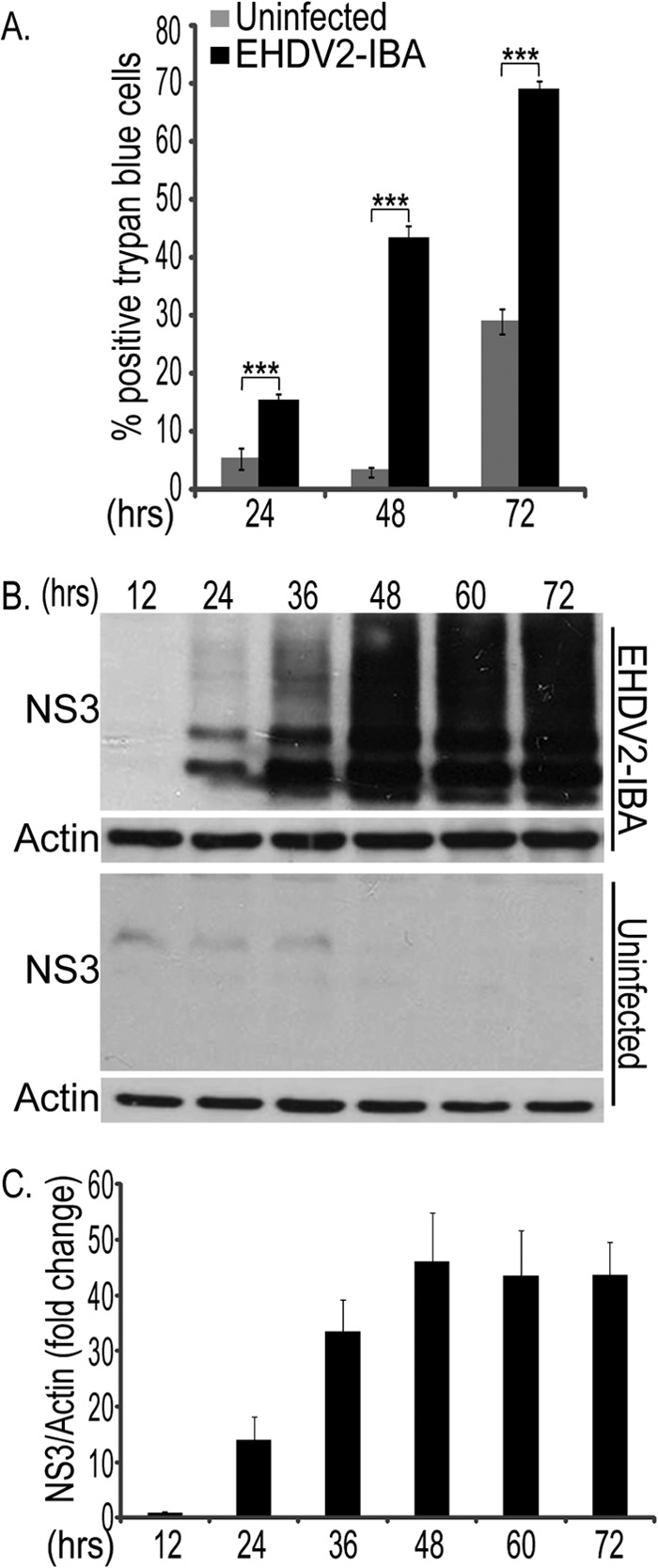
Time course of EHDV2-IBA-induced cell death. OK cells, at 24 h postplating, were either noninfected or infected with EHDV2-IBA (here and throughout this study, at an MOI of 1) (for the method of calculation, see Materials and Methods). At indicated time points, cells were suspended by trypsinization, stained with trypan blue, and scored for trypan blue exclusion by microscopy. (A) The graph depicts the percentage of cells stained with trypan blue under the different experimental conditions (noninfected or infected with EHDV2-IBA) at different time points. ***, P < 0.005. (B) Representative immunoblot of lysates of OK cells, either noninfected or infected with EHDV2-IBA for the indicated times, separated by SDS-PAGE, and probed with anti-NS3 and antiactin antibodies. (C) Graph depicts the average ± SE fold change in the NS3/actin ratio at different time points of infection relative to the NS3/actin ratio at 12 hpi (taken as 1; n = 3).
MRV, the avian reovirus, and BTV were shown to induce apoptosis in infected cells (10, 13, 19, 36). To assess if EHDV2-IBA induces apoptosis in OK cells, we initially measured the DNA content of infected and noninfected cell cultures by staining cells with propidium iodide and submitting them to fluorescence-activated cell sorting (FACS) analysis. As seen in Fig. 2A and B, a progressive increase in the percentage of infected cells presenting sub-G1 DNA content was observed. Maximal values for this percentage (34.2% ± 9.3%) were observed at 72 hpi. A similar percentage of cells presenting sub-G1 DNA content (28.1% ± 7.0%) was observed upon a 20-h treatment with staurosporine (STS), a broadly employed positive control for apoptosis induction. Next, we probed for caspase 3 activation by immunoblotting. Here, a reproducible activation of caspase 3 (detected by the appearance of its lower-molecular-weight cleaved form) was observed at 72 hpi (Fig. 2C). A well-established indicator of apoptosis is the laddering of genomic DNA. Thus, we examined if this phenomenon occurred in EHDV2-IBA-infected cells. As seen in Fig. 2D a clear laddering of DNA was observed in infected cells at 72 hpi and in cells treated with STS (20 h). From these data, we conclude that EHDV2-IBA induces the apoptosis of OK cells, a phenomenon that becomes clearly detectable at 72 hpi. Apoptosis induction by EHDV2-IBA was also observed in Vero and CPAE cells (data not shown), indicating that this phenomenon is not restricted to OK cells. The progressive increase in sub-G1 DNA content (Fig. 2A and B) suggested that the induction of apoptosis could be dependent on productive infection. To directly address this matter, we infected OK cells with UV-irradiated EHDV2-IBA. A comparison of the effects of nonirradiated- and UV-irradiated EHDV2-IBA on OK cells revealed marked decreases in the percentages of cells presenting sub-G1 DNA content (Fig. 2E) and in the virus-induced laddering of DNA (Fig. 2F) in cells incubated with UV-irradiated EHDV2-IBA. These data support the notion that under the experimental conditions employed here, productive infection with EHDV2-IBA plays an important role in the induction of apoptosis.
Fig 2.

EHDV2-IBA induces the apoptosis of OK cells. (A and B) Subconfluent cultures of OK cells, at 24 h postplating, were either noninfected or infected for the indicated times and left untreated or treated with staurosporine (STS; 1 μM for 20 h). Cells were collected from the medium or suspended by trypsinization and processed for FACS analysis. (A) The graphs depict the FACS measurements of DNA content of propidium iodide-stained OK cells: 1, sub-G1; 2, G0/G1; 3, S; 4,G2/M. (B) The graph shows the average ± SE of the percentage of cells presenting sub-G1 DNA content under the indicated experimental conditions (n = 3; *, P < 0.05; **, P < 0.01). (C) OK cells at 24 h postplating were noninfected or infected with EHDV2-IBA. At indicated times points, cells were collected and processed for 10% SDS-PAGE (see Materials and Methods). The panel depicts a typical immunoblot probed with the anti-NS3, anti-caspase 3, and anti-MEK (loading control) antibodies. tMEK, total MEK (MEK1/2). (D) Cells cultured and infected as described for panel C were processed for DNA laddering (see Materials and Methods) at the indicated time points. The panel depicts a representative DNA laddering experiment (n = 5) employing equal amounts of genomic DNA, extracted from cells under the indicated conditions, and separated on a 2% agarose gel. (E) OK cells, cultured as described for panel A and either noninfected or infected with UV-irradiated EHDV2-IBA for 72 h, were processed for FACS analysis of DNA content. Graphs depict typical FACS measurements of cells infected with EHDV2-IBA (left) or UV-irradiated EHDV2-IBA (right). (F) Representative agarose gel from a DNA laddering experiment of OK cells noninfected or infected with EHDV2-IBA or UV-irradiated EHDV2-IBA (60 hpi) and left untreated or treated with STS (1 μM for 20 h).
In order to probe for the significance of the induction of apoptosis to the infection process and to cell death induced by EHDV2-IBA, we treated infected or STS-treated cells with the pan-caspase inhibitor Q-VD-OPH. In a typical experiment, Q-VD-OPH (20 μM) markedly reduced the percentage of cells showing sub-G1 DNA content upon infection with EHDV2-IBA (72 hpi) or treatment with STS (1 μM; 20 h) (Fig. 3A). Analogously, the DNA laddering typical of apoptotic cells and induced by both the virus infection and STS treatment was also inhibited by Q-VD-OPH (Fig. 3B). Next, we assessed the effects of Q-VD-OPH on EHDV2-IBA-induced cell death by a trypan blue exclusion assay. As can be seen in Fig. 3C, Q-VD-OPH partially and significantly inhibited EHDV2-IBA-induced cell death during the entire course of infection (24, 48, and 72 hpi). Accordingly, Q-VD-OPH also inhibited cell death induced by STS. These data imply that apoptosis contributes to EHDV-IBA-induced cell death. However, that Q-VD-OPH is only partially protective in infected cells suggests that EHDV2-IBA-induced cell death also occurs by means other than apoptosis. Thus, for example, while Q-VD-OPH inhibited ∼80% of the cell death induced by STS, at 72 hpi only ∼27% of the EHDV-IBA-induced cell death was inhibited by Q-VD-OPH. To examine if apoptosis inhibition with a pan-caspase inhibitor affects the production of infectious virions, we infected cell cultures, which were either untreated or treated with Q-VD-OPH, collected virions (48 hpi; present in the attached cells or in the cell culture medium), and infected semiconfluent cultures of naive Vero cells (reporter cell culture for plaque assay). The titer of the inoculum was of 0.2 × 103 PFU per ml; the titer of the untreated culture at 48 hpi was (7.4 ± 2.1) ×103 PFU/ml, representing a ∼37-fold increase in PFU/ml. Q-VD-OPH treatment resulted in a slight, but significant, reduction in the fold increase in titer to ∼22-fold [up to (4 ± 0.8) ×103 PFU/ml; P < 0.05]. Taken together, our data show that EHDV2-IBA induces apoptosis in an infection-dependent manner, that part of the EHDV2-IBA-induced cell death can be attributed to apoptosis, and that caspase inhibition results in a slight (but significant) reduction in the production of infectious virions.
Fig 3.

Treatment with the pan-caspase inhibitor Q-VD-OPH reveals roles for apoptosis in EHDV2-IBA-induced cell death and in the production of infectious virions. (A and B) OK cells, at 24 h postplating, were left untreated or treated with Q-VD-OPH (20 μM for 20 or 72 h) or STS (1 μM for 20 h) and either left noninfected or infected with EHDV2-IBA. (A) FACS analysis of DNA content of OK cells under the indicated infection and treatment conditions. The percentage of cells presenting sub-G1 DNA content in this representative experiment is indicated in each FACS graph. (B) Representative agarose gel (2%) from a DNA laddering experiment. Treatment and infection conditions were as indicated. (C) OK cells, cultured, infected, and treated as described for panels A and B, were resuspended by trypsinization at the indicated time points and assessed by trypan blue exclusion assay. Graph depicts the average ± SE of the percentage of trypan blue-positive cells under the indicated experimental conditions. *, P < 0.05; **, P < 0.01; ***, P < 0.005. (D) Plaque assay analysis of the titer of infectious virions generated upon the EHDV2-IBA infection (48 h) of cultures of OK cells, untreated or treated with Q-VD-OPH (20 μm for 48 h). At left, representative wells show plaques formed upon the infection of Vero cells (reporter cell culture). At right, the graph depicts the titer (average ± SE; PFU/ml) in the culture of infected OK cells. The titer of the inoculum was 0.2 × 103 PFU/ml and is represented by the dotted line. *, P < 0.05.
EHDV induces autophagy.
Based on the partial inhibitory effects of Q-VD-OPH, we set out to probe for the induction of additional cell stress-/cell death-related processes by EHDV2-IBA. One such process is autophagy (37). To examine if EHDV2-IBA induces autophagy, we separated the lysates of infected and uninfected cells by SDS-PAGE and probed for the alteration in molecular weight of the autophagic factor light chain 3 (LC3) and for the reduction in levels of the adapter protein p62, two well-accepted markers of autophagy (38). As shown in Fig. 4A, EHDV2-IBA infection induced a progressive increase in the ratio of the lower-molecular-weight form of LC3 (LC3-II, the lipidated form) to the higher-molecular-weight form of LC3 (LC3-I). Such an increase in the ratio became significant at 48 hpi and peaked at 60 hpi (quantification is shown in Fig. 4B). Accordingly, a progressive reduction in the cellular content of p62 was also observed in the EHDV2-IBA-infected cell cultures (Fig. 4A). A calculation of the p62/actin ratio in multiple immunoblots showed a significant reduction already at 36 hpi, a reduction which peaked at 48 hpi (Fig. 4C). To gain further insight into the induction of autophagy by EHDV2-IBA, we generated a cell line of OK cells stably expressing LC3-GFP to monitor LC3 aggregation, which is typical of autophagy induction, using real-time imaging. Initially, we probed for the ability of EHDV2-IBA to induce an increase in the ratio of LC3-II–GFP to LC3-I–GFP by immunoblotting. As shown in Fig. 4D, at 48 hpi, EHDV2-IBA induced both a reduction in the amount of LC3-I–GFP and an increase in the amount of LC3-II–GFP, indicating that the LC3-GFP-expressing OK cells undergo autophagy upon EHDV2-IBA infection. Next, we employed three-dimensional (3D) confocal imaging to analyze the intracellular distribution of LC3-GFP cells in infected and noninfected cells. To this end, LC3-GFP-expressing OK cells were either noninfected or infected with EHDV2-IBA, fixed, permeabilized, stained for NS3, and imaged by confocal microscopy. As see in Fig. 4D, in uninfected cells, which did not show specific staining for NS3, LC3-GFP presented a mainly diffuse distribution throughout the cell interior. In contrast, at 72 hpi, a clear NS3 signal is observed, and the intracellular distribution of LC3-GFP is altered and becomes prominently punctate. To follow the induction of autophagy in real time, we imaged the culture of infected LC3-GFP-expressing OK cells from 36 to 60 hpi (conditions in which at least 90% of cells stained positive for NS3) with spinning-disk confocal microscopy. In this time window, and in contrast to the mostly diffuse distribution of LC3-GFP at the beginning of the time-lapse sequence, the aggregation of LC3-GFP into discrete puncta was observed at later time points (Fig. 4D; see Movie S1 in the supplemental material). The alteration in the intracellular distribution of LC3-GFP upon infection is consistent with its recruitment to autophagosomes. In addition to the alteration in LC3-GFP distribution, infected cells were also characterized by extensive cell rounding (Fig. 4D; see also Movie S1). Notably, EHDV2-IBA infection also induced autophagy in Vero and CPAE cells (data not shown), indicating that this phenomenon is not restricted to OK cells. Moreover, EHDV7-ISR also induced autophagy in OK cells (data not shown). Next, to probe if the induction of autophagy by EHDV2-IBA is dependent on productive infection, we incubated OK cell cultures with UV-irradiated virus, separated cell lysates by SDS-PAGE, and probed for NS3, LC3, and p62. In these experiments, no NS3 was detected. In sharp contrast to the phenomena observed with nonirradiated EHDV2-IBA, no virus-induced increase in the LC3-II/LC3-I ratio or decrease in the p62 levels was observed at any time point postinfection (Fig. 4E). Together, these data indicate that autophagy is induced only upon productive EHDV2-IBA infection.
Fig 4.

EHDV2-IBA induces autophagy. (A to C) OK cells at 24 h postplating were either noninfected or infected with EHDV2-IBA. At indicated time points, cells were collected and processed for 12% SDS-PAGE (see Materials and Methods). Panel A depicts a typical immunoblot probed with anti-LC3, anti-p62, and antiactin (loading control) antibodies. In panel B the graph depicts the average ± SE of the fold changes in the LC3-II/LC3-I ratios under the indicated conditions relative to the LC3-II/LC3-I ratio of uninfected cells at 12 hpi (taken as 1; n = 3). *, P < 0.05. The graph in panel C depicts the average ± SE of the fold changes in the p62/actin ratio under the indicated conditions relative to the p62/actin ratio of uninfected cells at 12 hpi (taken as 1; n = 3). **, P < 0.01. (D) A stable cell line of OK cells expressing LC3-GFP (generated as described in Materials and Methods) was infected with EHDV2-IBA (MOI of 1; 48 h). Infected and noninfected cells were lysed, and lysates were separated by 10% SDS-PAGE. The top panel is a representative immunoblot probed with anti-GFP and antiactin antibodies. For the experiments shown in the middle panels, LC3-GFP-expressing cells were plated on glass-bottomed 35-mm dishes, and at 24 h after plating cells were either left uninfected or infected with EHDV2-IBA. At 72 h after infection, cells were fixed, permeabilized, stained with anti-NS3 and Alexa Fluor-555 goat anti-mouse antibodies, and imaged by spinning-disk confocal microscopy. Panels depict a 3D rendition of confocal z-stacks (0.3 μm between steps) of infected (lower panel) or noninfected (upper panel) cells. Grid, 10 μm. For the experiments show in the bottom panels, LC3-GFP-expressing OK cells were cultured and infected as described above. Beginning at 36 hpi, cells were imaged by spinning-disk confocal time-lapse microscopy. Staining of cells cultured and infected in this manner with anti-NS3 antibodies yielded at least 90% of infected cells (data not shown). Scale bar, 10 μm. (E) OK cells cultured and plated as described for panel A were either untreated or incubated with UV-irradiated (UV-irr) EHDV2-IBA (for the indicated time periods). The panel shows a representative immunoblot of lysates of such cells, separated by 12% SDS-PAGE and probed with antibodies against LC3, p62, NS3, and actin.
In order to probe for the significance of the induction of autophagy to the infection process and to cell death induced by EHDV2-IBA, we searched for conditions under which autophagy was inhibited by 3-methyladenine (3MA), a phosphatidyl-inositol-3-kinase inhibitor (39). To induce autophagy, we starved cells in Earle's balanced salt solution (EBSS) medium (3 h; devoid of amino acids or serum). Under these conditions, an increase in the LC3-II/LC3-I ratio is observed in the starved, untreated cultures (Fig. 5A). In contrast, no change in this ratio was observed in the starved, 3MA-treated culture (10 mM 3MA) (Fig. 5A). Next, we probed for the effect of 3MA on EHDV2-IBA-induced autophagy. This assay was carried out at 36 hpi because prolonged incubations with 3MA proved toxic to cells. Under these conditions, 3MA markedly attenuated the increase in the LC3-II/LC3-I ratio induced by EHDV2-IBA while also attenuating the decrease in the levels of p62 in infected cells (Fig. 5B) (for discussion on the remaining decrease in p62 that was observed, see below). We also probed for the effect of 3MA on the titer of infectious virions. Here, at 36 hpi, 3MA treatment resulted in a 62% reduction in the fold increase in titer [from (2.7 ± 0.5) ×103 PFU/ml to (1.1 ± 0.6) × 103 PFU/ml, with an initial inoculum of 0.2 × 103 PFU/ml] (Fig. 5C). We then examined the effects of autophagy inhibition with 3MA on EHDV2-IBA-induced cell death. Separately, at 32 to 36 hpi, each treatment (EHDV2-IBA infection and 3MA treatment) induced similar levels of cell death (31.2% ± 5.1% and 31.4% ± 4.1%, respectively) (data not shown). The combined infection-3MA treatment procedure induced more extensive killing of cells (51% ± 1.6% dead cells) (data not shown). Based on these data one cannot reject the assumption that EHDV2-IBA and 3MA work independently of each other, supporting the notion that autophagy does not play a major role in EHDV2-IBA-induced cell death. Next, we examined EHDV2-IBA infection in mouse embryonic fibroblasts (MEFs) that either express or are deficient in Atg5 (Atg5+/+ or Atg5−/−, respectively), a regulator of autophagosome formation (40). EHDV2-IBA infection of Atg5+/+ MEFs resulted in the induction of autophagy, as seen by the increase in the LC3-II/LC3-I ratio at 48 hpi (Fig. 5D). In Atg5−/− MEFs, LC3-II could not be identified under any condition, in accord with the inability of these cells to undergo autophagy (35). Notably, and similarly to OK cells infected with EHDV2-IBA and treated with 3MA, the levels of both LC3 and p62 were decreased in both types of MEFs at 48 hpi. Since this reduction also occurs in the absence of autophagy (in Atg5−/− MEFs and in 3MA-treated OK cells), we hypothesized that it could stem from a virally induced block in protein synthesis. Indeed, EHDV2-IBA infection of OK cells induced both a progressive increase in the phosphorylation of the regulator of translation eIF2α that was significant at 36 hpi (Fig. 6A and B) and a reduction in protein synthesis (measured by 35S labeling) (Fig. 6C). Thus, it is likely that the EHDV2-IBA-induced reduction in protein synthesis contributes to the observed decrease in p62 levels in infected cells. A similar mechanism may also contribute to the reduction in levels of other cellular factors, such as LC3, in infected cells. In this context, we observed a variation in the severity of the reduction in the levels of p62, LC3, and actin (consistent reduction of p62, variable reduction of LC3, and no significant reduction of actin). Such variation may stem from differences in the turnover rate of these proteins and will be the object of future study. Next, we measured the production of infectious virions in Atg5+/+ and Atg5−/− MEFs. Both cell types were inoculated at a titer of 0.2 × 103 PFU/ml. At 48 hpi, the titer of infectious virions in the Atg5+/+ cell culture was (7.7 ± 0.35) × 103 PFU/ml while the titer in the Atg5−/− cell culture was (4.7 ± 0.96) × 103 PFU/ml, indicating a ∼40% reduction in the production of infectious particles in the absence of autophagy (P < 0.05). Notably, we did not observe significant differences in the quantification of the percentages of trypan blue-stained Atg5+/+ and Atg5−/− MEFs at 48 hpi (36.1% ± 4.6% for Atg5+/+ versus 48.9% ± 6.5; P = 0.19) (data not shown). Together, these data suggest that EHDV2-IBA-induced autophagy contributes to the production of infectious virions but does not add to EHDV2-IBA-induced cell death.
Fig 5.
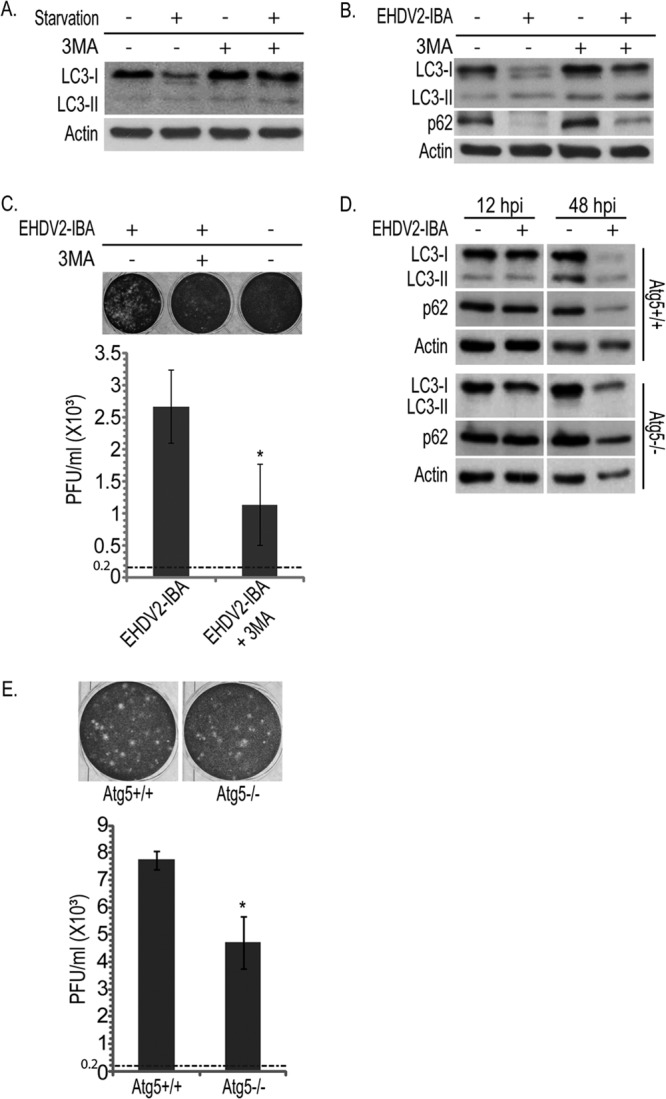
Inhibition of autophagy attenuates the production of infectious particles. OK cells at 24 h postplating were starved or given EBSS (3 h) and treated with 3MA (10 mM for 3 h) or left untreated. Cells were lysed, separated by 12% SDS-PAGE, blotted, and probed with anti-LC3 or antiactin antibodies. (B) Representative immunoblot of lysates of OK cells infected or not with EHDV2-IBA (36 hpi) and treated or not with 3MA (10 mM for 36 h), separated by 12% SDS-PAGE, and probed with the indicated antibodies. (C) Plaque assay analysis of the titer of infectious virions under untreated or 3MA treatment conditions. The upper panels show representative wells displaying plaques formed upon infection of Vero cells with different dilutions of the lysates of OK cells (uninfected or infected with EHDV2-IBA; 36 hpi), untreated or treated with 3MA (10 mM; 36 h). The graph in the lower panel depicts the titer (PFU/ml) produced in infected OK cells (untreated or treated with 3MA) at 36 hpi. The titer of the inoculum was 0.2 × 103 PFU/ml and is represented by the dotted line. *, P < 0.05. (D) Atg5+/+ or Atg5−/− MEFs at 24 h postplating were left uninfected or infected with EHDV2-IBA for 12 or 48 h. Cells were lysed, separated by 12% SDS-PAGE, blotted, and probed with antibodies against LC3, p62, or actin. (E) Plaque assay analysis of the titer of infectious virions generated in infected Atg5+/+ or Atg5−/− MEFs. Sample processing and data presentation are as described for panel C.
Fig 6.
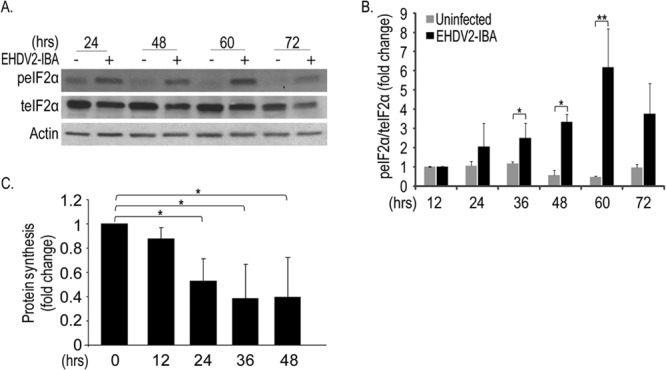
EHDV2-IBA induces phosphorylation of eIF2α and a decrease in protein synthesis. OK cells were cultured, plated, infected, and processed for immunoblotting as described in the legend of Fig. 4. (A) Representative immunoblot of lysates of OK cells, either uninfected or infected with EHDV2-IBA for the indicated periods and probed with anti-phosphorylated eIF2α (peIF2α), anti-total eIF2α (teIF2α), and antiactin antibodies. (B) The graph shows the fold change (average ± SE) in the ratio of phosphorylated eIF2α to total eIF2α relative to the ratio in uninfected cells at 12 hpi. (C) OK cells were seeded in 24-well plates. At 12 h postplating and at 12-h intervals, cells were infected with EHDV2-IBA. After 48 h, cells were pulse-labeled with [35S]cysteine-methionine-containing medium. 35S incorporation in TCA-precipitated samples was measured with a Beckman beta particle counter. The graph depicts the fold change (inhibition) in the ratio of calibrated 35S counts in infected cells to calibrated 35S counts in noninfected cells at each time point. *, P < 0.05.
EHDV induces the activation of JNK and the phosphorylation of c-Jun.
JNK is termed a stress-activated protein kinase (SAPK) and was shown to be activated in reovirus-infected cells (31, 33). To probe if EHDV2-IBA infection involves JNK activation, lysates of OK cells either noninfected or infected with EHDV2-IBA were separated by SDS-PAGE at different time points and subjected to immunoblotting with anti-total-JNK (tJNK) and anti-phospho-JNK (pJNK) antibodies (Fig. 7A). Quantification of the pJNK/tJNK ratio revealed significant phosphorylation of JNK already at 24 hpi (Fig. 7B), suggesting that the activation of this pathway is an early phenomenon in EHDV2-IBA infection. To examine if the EHDV2-IBA-induced phosphorylation of JNK was dependent on productive infection, we incubated OK cells with UV-irradiated EHDV2-IBA and probed for pJNK and tJNK by immunoblotting. As can be seen in Fig. 7C, no virus-dependent increase in the pJNK/tJNK ratio was observed with the UV-irradiated EHDV2-IBA. Next, we assessed the phosphorylation of c-Jun, a main downstream target of JNK. As seen in Fig. 8A, OK cells presented a low basal level of phosphorylated c-Jun (pc-Jun) in the absence of infection. Upon infection with EHDV2-IBA (48 hpi), a marked increase in pc-Jun was observed. Treatment of OK cells, either noninfected or infected with EHDV2-IBA, with the specific JNK inhibitor SP600125 (20 μM; 48 h) resulted in the abrogation of the basal levels of pc-Jun and in variable and partial attenuation (P = 0.066) in the infection-induced increase in the pc-Jun/total c-Jun (tc-Jun) ratio (Fig. 8A, immunoblot, and B, quantification). These data suggest a role for JNK in c-Jun phosphorylation in EHDV2-IBA-infected cells and the existence of additional mediators of c-Jun phosphorylation in infection. In addition to JNK, cyclin dependent kinases (CDKs) have also been shown to regulate c-Jun phosphorylation (41, 42). In a previous study, we have shown that 80 μM roscovitine impeded efficient entry into mitosis and the phosphorylation of Dab2, a mitotic phosphoprotein (43), thus suggesting that this concentration of inhibitor leads to an efficient inhibition of CDK activity. We next probed for the effects of the specific CDK inhibitor roscovitine on c-Jun phosphorylation in EHDV2-IBA-infected or noninfected OK cells. Roscovitine (80 μM; 48 hpi) markedly and significantly reduced the pc-Jun/tc-Jun ratio in EHDV2-IBA-infected cells (P < 0.05) (Fig. 8A and B). We then assessed the effects of SP600125 on EHDV2-IBA-induced cell death and on the production of infectious virions. SP600125 (20 μM; 48 h) significantly (P < 0.005) reduced the percentage of cells stained with trypan blue in the trypan blue exclusion assay (Fig. 8C), indicating the involvement of JNK in EHDV2-IBA-induced cell death. Also, SP600125 (20 μM; 48 h) reduced the increase in titer upon EHDV2-IBA infection (48 hpi) relative to that in untreated EHDV2-IBA-infected OK cells (∼74% reduction; P < 0.005) (Fig. 8D), suggesting a role for JNK in the production of infectious virions. Based on the dramatic reduction in pc-Jun levels upon the treatment of EHDV2-IBA-infected OK cells with roscovitine (Fig. 8A and B), we probed for the effects of roscovitine on EHDV2-IBA-induced cell death. Due to the induction of a considerable level of cell death by 80 μM roscovitine in the absence of infection (36.9% ± 2.8% trypan blue-positive cells) (data not shown), we tested for the effects of lower roscovitine concentrations (20 μM and 40 μM) on EHDV2-IBA-induced cell death. Here, in the absence of infection, a concentration-dependent level of roscovitine-induced cell death was observed (5.1% ± 2.5% and 19.2% ± 2.8% trypan blue-positive cells, with 20 μM and 40 μM roscovitine, respectively) (Fig. 8E). Notably, while both 20 μM and 40 μM roscovitine reduced EHDV2-IBA-induced cell death, this effect was significant only with 40 μM (55.5% ± 2.9% versus 38.2% ± 0.5% trypan blue-positive cells for 40 μM; P < 0.01) (Fig. 8E). Next, we examined the effects of roscovitine (20 μM, 40 μM, or 80 μM for 48 h) on the production of infectious virions. Here, all three concentrations induced marked, significant (P values of <0.05 and <0.005), and concentration-dependent reductions in the production of infectious virions (∼ 60%, 80%, or 90% attenuation in the fold increase in titer with 20 μM, 40 μM, or 80 μM, respectively) (Fig. 8F). These results support the notion of a role for CDKs in EHDV2-IBA infection.
Fig 7.
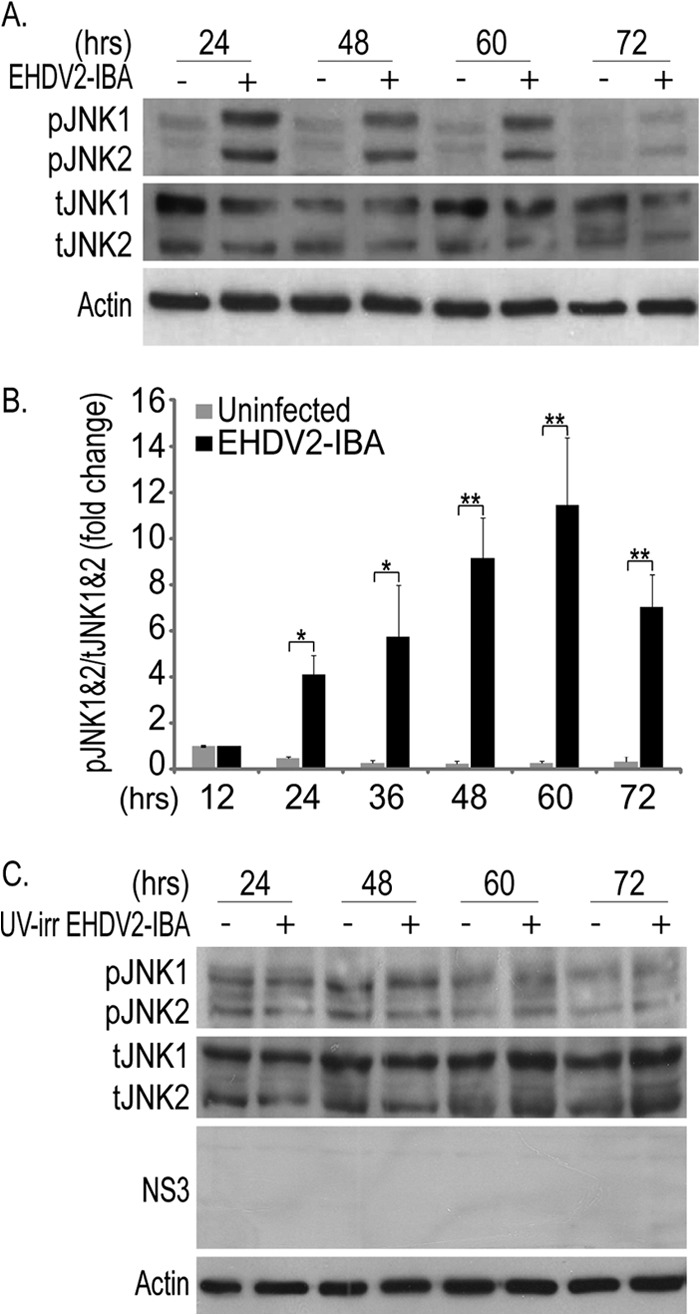
EHDV2-IBA induces JNK activation. OK cells were cultured, plated, infected, and processed for immunoblotting as described in the legend of Fig. 4. (A) A representative immunoblot probed with anti-pJNK, anti-tJNK, and antiactin antibodies. (B) The graph depicts the fold change (average ± SE) in the pJNK/tJNK ratio relative to that of uninfected cells at 12 hpi (n = 3; *, P < 0.05; **, P < 0.01). (C) Representative immunoblot of lysates of OK cells either untreated or incubated with UV-irradiated EHDV2-IBA for the indicated time periods, separated by 10% SDS-PAGE, and probed with anti-pJNK, anti-tJNK, and antiactin antibodies.
Fig 8.

SP600125 and roscovitine decrease EHDV2-IBA-mediated cell death and autophagy and the production of infectious virions. (A) OK cells at 24 h postplating were either noninfected or infected with EHDV2-IBA (for 48 h) and left untreated or treated with SP600125 (20 μM; 48 h) or roscovitine (80 μM; 48 h). Cell lysates were separated by 12% SDS-PAGE, blotted, and probed with antibodies against LC3, pJNK, tJNK, pc-Jun, tc-Jun, NS3, or actin. (B) Graph depicts the fold increase (average ± SE) in the pc-Jun/tc-Jun ratio (48 hpi) under the indicated treatment/infection conditions, relative to the uninfected untreated sample (taken as 1). *, P < 0.05. (C and E) OK cells were infected (for 48 h) and either left untreated or treated with SP600125 (20 μM; 48 h) (C) or roscovitine (20 μM, 40 μM or 80 μM; 48 h) (E). Graphs depict the average ± SE of the percentage of cells positive for trypan blue staining upon EHDV2-IBA infection and/or treatment. *, P < 0.05; ***, P < 0.005. (D and F) Plaque assay analyses of the effects of SP600125 (D) or roscovitine (F) on the increase in EHDV2-IBA titer at 48 hpi. Graphs depict the average ± SE of the titer (PFU/ml) of infectious virions under the indicated experimental conditions. The titer of the inoculum (0.2 × 103 PFU/ml) is indicated as a horizontal dotted line. Panels show typical wells of the plaque assays employed for the quantification. *, P < 0.05; ***, P < 0.005. (G) The graph shows the fold change (average ± SE) in the LC3-II/LC3-I ratio (from immunoblots as described for panel A), relative to that of uninfected untreated samples (taken as 1). *, P < 0.05.
Lastly, we examined the effects of SP600125 and roscovitine on the induction of autophagy by EHDV2-IBA, as measured by the LC3-II/LC3-I ratio. As shown in a representative immunoblot (Fig. 8A) and in the quantification of multiple experiments (Fig. 8G), SP600125 induced a minor reduction in the increase in the LC3-II/LC3-I ratio observed upon EHDV2-IBA infection. In contrast, roscovitine significantly attenuated the increase in LC3-II/LC3-I, suggesting the regulation of autophagy induction by cell stress-related pathways in infected cells.
DISCUSSION
A main finding of the present study is that EHDV2-IBA induces apoptosis in the course of its infection of mammalian cells in culture. This finding is supported by the evidence that EHDV2-IBA infection induces the progressive increase in the percentage of cells presenting sub-G1 DNA content (Fig. 2A and B), the activation of caspase 3 (Fig. 2C), and the laddering of genomic DNA (Fig. 2D). The notion of a determining role for productive infection in the levels of induction of apoptosis is supported by the detection of such indicators of apoptosis mainly at late stages of infection (48 to 72 hpi) and by the marked reduction in these phenomena upon the UV irradiation of EHDV2-IBA prior to its incubation with cells (Fig. 2E and F). However, the protective effect of caspase inhibition on the low levels of virus-induced cell death at 24 hpi (Fig. 3C) raises the possibility of induction of apoptosis early in infection, a stage where viral replication might play a lesser role. BTV, which presents extensive structural similarity to EHDV, was also shown to induce the apoptosis of mammalian cells in culture (10, 19, 44). While the BTV structural proteins VP2 and VP5, which are present on the incoming virus, were shown to be sufficient for the induction of apoptosis (10), a different study from the same group identified a central role for the BTV nonstructural protein NS1 (generated only upon productive infection) in cellular pathogenesis as impairment of its function with a single-chain antibody fragment inhibited cell rounding and death (45). Similarly, with MRV, both an outer capsid protein (μ1) and a nonstructural protein (σ1s) were implicated in the induction of apoptosis (14, 46). Interestingly, the requirement of productive infection for the induction of apoptosis by MRV may be cell type dependent as the antiviral drug ribavirin, which inhibits viral replication, was shown to inhibit MRV-induced apoptosis in HEK 293 cells but not in HeLa cells (47). Taken together, these results suggest a possible role for reovirus/orbivirus structural proteins in the induction of apoptosis early in infection in a replication-independent manner and the efficient induction of apoptosis by replication-dependent mechanisms later in infection.
What role does apoptosis play in EHDV2-IBA infection? The protective effect of the pan-caspase inhibitor Q-VD-OPH (Fig. 3) implies that EHDV2-IBA induces cell death by apoptosis. However, the partial nature of this protective effect points to caspase-independent mechanisms of induction of cell death by EHDV2-IBA. Such mechanisms may be similar to the caspase-independent death pathway activated by MRV (48). Interestingly, in addition to reducing cell death, caspase inhibition with Q-VD-OPH also induced a reduction in the titer of infectious virions (Fig. 3D). Notably, a similar correlation between apoptosis inhibition and a decrease in viral titer was observed upon expression of the Aedes albopictus inhibitor of apoptosis (Aaiap1) in cells infected with BTV. Aaiap1 expression inhibited BTV-induced apoptosis of mammalian BSR cells and also induced a 10-fold reduction in BTV titer (44).
An additional principal finding of this study is that EHDV2-IBA induces autophagy in the context of the infection of mammalian cells in culture. This finding is supported by the progressive increase in the ratio of the lipidated form of LC3 (LC3-II) to its unlipidated form (LC3-I) (Fig. 4, 5, and 8), the aggregation of LC3-GFP (Fig. 4), and the decrease in the levels of the adaptor protein p62 (Fig. 4). Of note, an infection-induced reduction in host cell protein synthesis (Fig. 6) may also contribute to the observed decrease in p62 levels. The appearance of all of the autophagy markers strictly depended on productive infection as they were not observed upon incubation of cells with UV-irradiated virus (Fig. 4E). What may induce autophagy in EHDV2-IBA-infected cells? The context of the infected cell provides multiple potential sources of signals for autophagy induction. For example, protein kinase R (PKR) and RNase L, which are activated by dsRNA, a pathogen-associated molecular pattern characteristic of viruses such as EHDV, were implicated in the induction of autophagy (23, 49, 50). Indeed, the infection-dependent phosphorylation of the translation regulator eIF2α (Fig. 6A and B), the reduction in protein synthesis levels (Fig. 6C), and the phosphorylation of PKR (data not shown) are all in line with PKR activation in EHDV2-IBA-infected cells. Moreover, EHDV2-IBA-infected cells are also characterized by JNK activation (Fig. 7 and 8), and JNK has been proposed as a regulator of the induction of autophagy, for example, via the phosphorylation of Bcl-2 (51). The attenuating effect of the specific JNK inhibitor SP600125 on the infection-induced increase in the LC3-II/LC3-I ratio (Fig. 8A and G) supports the notion of the importance of this pathway in EHDV2-IBA-induced autophagy. Viral factories (viroplasms), which are characteristic of reovirus infections in general and of the EHDV2-IBA infection in particular (52) and which present structural similarities to aggresomes (53), may serve as potential inducers of selective autophagy. Indeed, the sharp reduction in the levels of the adapter protein p62, a mediator of selective autophagy, upon EHDV2-IBA infection (Fig. 4A) is in line with such a scenario. However, the reduction in p62 levels observed in the Atg5−/− MEFs, which cannot undergo autophagy (35), suggests that additional processes, such as the virus-induced block in host protein synthesis (Fig. 6C), may also contribute to this phenomenon. Importantly and similarly to apoptosis, the induction of autophagy by EHDV2-IBA apparently contributes to the production of infectious virions as blocking autophagy via either genetic or chemical manipulations reduced the titer of infectious virions (Fig. 5).
The cyclin-dependent kinase inhibitor roscovitine has been reported to negatively regulate the infection of human cytomegalovirus (54), JC virus (55), varicella-zoster virus (56), and human immunodeficiency virus type I (57). Here, we expand on the notion of a role for CDKs in viral infection and show that roscovitine also inhibited the production of infectious EHDV2-IBA virions (Fig. 8F), EHDV2-IBA-induced autophagy (Fig. 8A and G), EHDV2-IBA-induced cell death (Fig. 8E), and the phosphorylation of c-Jun (Fig. 8A and B).
In summary, the observation of apoptosis, autophagy, and phosphorylation of JNK and c-Jun in EHDV2-IBA-infected cells and the attenuating effect of the inhibition of these cellular processes on the production of infectious viruses support a scenario whereby, in addition to the activation of cell stress and death pathways, EHDV usurps these cellular pathways to optimize the conditions for viral replication.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funding from the Binational Agricultural Research and Development Fund (IS-4192-09), Israeli Ministry of Agriculture (891-0230-09 and 891-0254-11/EMIDA-ERA-NET), the Binational Science Foundation (2005283), and the Middle East Research Collaboration fund (M29-060).
Footnotes
Published ahead of print 2 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02116-13.
REFERENCES
- 1.Yadin H, Brenner J, Bumbrov V, Oved Z, Stram Y, Klement E, Perl S, Anthony S, Maan S, Batten C, Mertens PP. 2008. Epizootic haemorrhagic disease virus type 7 infection in cattle in Israel. Vet. Rec. 162:53–56 [DOI] [PubMed] [Google Scholar]
- 2.Temizel EM, Yesilbag K, Batten C, Senturk S, Maan NS, Clement-Mertens PP, Batmaz H. 2009. Epizootic hemorrhagic disease in cattle, Western Turkey. Emerg. Infect. Dis. 15:317–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savini G, Afonso A, Mellor P, Aradaib I, Yadin H, Sanaa M, Wilson W, Monaco F, Domingo M. 2011. Epizootic haemorrhagic disease. Res. Vet. Sci. 91:1–17 [DOI] [PubMed] [Google Scholar]
- 4.Maclachlan NJ. 2011. Bluetongue: history, global epidemiology, and pathogenesis. Prev. Vet. Med. 102:107–111 [DOI] [PubMed] [Google Scholar]
- 5.Eschbaumer M, Wernike K, Batten CA, Savini G, Edwards L, Di Gennaro A, Teodori L, Oura CA, Beer M, Hoffmann B. 2012. Epizootic hemorrhagic disease virus serotype 7 in European cattle and sheep: diagnostic considerations and effect of previous BTV exposure. Vet. Microbiol. 159:298–306 [DOI] [PubMed] [Google Scholar]
- 6.Belhouchet M, Mohd Jaafar F, Firth AE, Grimes JM, Mertens PP, Attoui H. 2011. Detection of a fourth orbivirus non-structural protein. PLoS One 6:e25697. 10.1371/journal.pone.0025697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ratinier M, Caporale M, Golder M, Franzoni G, Allan K, Nunes SF, Armezzani A, Bayoumy A, Rixon F, Shaw A, Palmarini M. 2011. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 7:e1002477. 10.1371/journal.ppat.1002477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omori T, Inaba Y, Morimoto T, Tanaka Y, Kono M. 1969. Ibaraki virus, an agent of epizootic disease of cattle resembling bluetongue. II. Isolation of the virus in bovine cell culture. Jpn. J. Microbiol. 13:159–168 [DOI] [PubMed] [Google Scholar]
- 9.Lim SI, Kweon CH, Yang DK, Tark DS, Kweon JH. 2005. Apoptosis in Vero cells infected with Akabane, Aino and Chuzan virus. J. Vet. Sci. 6:251–254 [PubMed] [Google Scholar]
- 10.Mortola E, Noad R, Roy P. 2004. Bluetongue virus outer capsid proteins are sufficient to trigger apoptosis in mammalian cells. J. Virol. 78:2875–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeMaula CD, Jutila MA, Wilson DW, MacLachlan NJ. 2001. Infection kinetics, prostacyclin release and cytokine-mediated modulation of the mechanism of cell death during bluetongue virus infection of cultured ovine and bovine pulmonary artery and lung microvascular endothelial cells. J. Gen. Virol. 82:787–794 [DOI] [PubMed] [Google Scholar]
- 12.Stassen L, Huismans H, Theron J. 2012. African horse sickness virus induces apoptosis in cultured mammalian cells. Virus Res. 163:385–389 [DOI] [PubMed] [Google Scholar]
- 13.Clarke P, Debiasi RL, Goody R, Hoyt CC, Richardson-Burns S, Tyler KL. 2005. Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation. Viral Immunol. 18:89–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coffey CM, Sheh A, Kim IS, Chandran K, Nibert ML, Parker JS. 2006. Reovirus outer capsid protein μ1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J. Virol. 80:8422–8438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J. Virol. 82:161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. 2007. IκB kinase subunits α and γ are required for activation of NF-κB and induction of apoptosis by mammalian reovirus. J. Virol. 81:1360–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Dong CY, Li JK, Chen DE, Liang K, Liu J. 2008. Selective in vitro cytotoxic effect of human cancer cells by bluetongue virus-10. Acta Oncol. 47:124–134 [DOI] [PubMed] [Google Scholar]
- 18.Marcato P, Shmulevitz M, Pan D, Stoltz D, Lee PW. 2007. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 15:1522–1530 [DOI] [PubMed] [Google Scholar]
- 19.Nagaleekar VK, Tiwari AK, Kataria RS, Bais MV, Ravindra PV, Kumar S. 2007. Bluetongue virus induces apoptosis in cultured mammalian cells by both caspase-dependent extrinsic and intrinsic apoptotic pathways. Arch. Virol. 152:1751–1756 [DOI] [PubMed] [Google Scholar]
- 20.Stewart ME, Roy P. 2010. Role of cellular caspases, nuclear factor-kappa B and interferon regulatory factors in Bluetongue virus infection and cell fate. Virol. J. 7:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreux M, Chisari FV. 2010. Viruses and the autophagy machinery. Cell Cycle 9:1295–1307 [DOI] [PubMed] [Google Scholar]
- 22.Gu L, Musiienko V, Bai Z, Qin A, Schneller SW, Li Q. 2012. Novel virostatic agents against bluetongue virus. PLoS One 7:e43341. 10.1371/journal.pone.0043341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi PI, Huang WR, Lai IH, Cheng CY, Liu HJ. 2013. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/eIF2α signaling pathways. J. Biol. Chem. 288:3571–3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng S, Jiang K, Zhang X, Zhang M, Zhou Z, Hu M, Yang R, Sun C, Wu Y. 2012. Avian reovirus triggers autophagy in primary chicken fibroblast cells and Vero cells to promote virus production. Arch. Virol. 157:661–668 [DOI] [PubMed] [Google Scholar]
- 25.Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Gao H, Bahlis N, Neri P, Pho M, Stewart D, Mansoor A, Morris DG. 2012. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin. Cancer Res. 18:4962–4972 [DOI] [PubMed] [Google Scholar]
- 26.Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fenichel P, Hofman P, Poujeol P, Mograbi B. 2007. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 3:57–59 [DOI] [PubMed] [Google Scholar]
- 27.Roux PP, Blenis J. 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68:320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei Y, Sinha S, Levine B. 2008. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4:949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. 2009. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284:2719–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabapathy K. 2012. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 106:145–169 [DOI] [PubMed] [Google Scholar]
- 31.Mortola E, Larsen A. 2010. Bluetongue virus infection: activation of the MAP kinase-dependent pathway is required for apoptosis. Res. Vet. Sci. 89:460–464 [DOI] [PubMed] [Google Scholar]
- 32.Clarke P, Meintzer SM, Wang Y, Moffitt LA, Richardson-Burns SM, Johnson GL, Tyler KL. 2004. JNK regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 78:13132–13138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke P, Meintzer SM, Widmann C, Johnson GL, Tyler KL. 2001. Reovirus infection activates JNK and the JNK-dependent transcription factor c-Jun. J. Virol. 75:11275–11283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith JA, Schmechel SC, Raghavan A, Abelson M, Reilly C, Katze MG, Kaufman RJ, Bohjanen PR, Schiff LA. 2006. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 80:2019–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmukler E, Grinboim E, Schokoroy S, Amir A, Wolfson E, Kloog Y, Pinkas-Kramarski R. 2013. Ras inhibition enhances autophagy, which partially protects cells from death. Oncotarget 4:145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labrada L, Bodelon G, Vinuela J, Benavente J. 2002. Avian reoviruses cause apoptosis in cultured cells: viral uncoating, but not viral gene expression, is required for apoptosis induction. J. Virol. 76:7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labbe K, Saleh M. 2008. Cell death in the host response to infection. Cell Death Differ. 15:1339–1349 [DOI] [PubMed] [Google Scholar]
- 38.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8:445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. 1997. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243:240–246 [DOI] [PubMed] [Google Scholar]
- 40.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. 2001. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152:657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghahremani MH, Keramaris E, Shree T, Xia Z, Davis RJ, Flavell R, Slack RS, Park DS. 2002. Interaction of the c-Jun/JNK pathway and cyclin-dependent kinases in death of embryonic cortical neurons evoked by DNA damage. J. Biol. Chem. 277:35586–35596 [DOI] [PubMed] [Google Scholar]
- 42.Cho YY, Tang F, Yao K, Lu C, Zhu F, Zheng D, Pugliese A, Bode AM, Dong Z. 2009. Cyclin-dependent kinase-3-mediated c-Jun phosphorylation at Ser63 and Ser73 enhances cell transformation. Cancer Res. 69:272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chetrit D, Barzilay L, Horn G, Bielik T, Smorodinsky NI, Ehrlich M. 2011. Negative regulation of the endocytic adaptor disabled-2 (Dab2) in mitosis. J. Biol. Chem. 286:5392–5403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q, Li H, Blitvich BJ, Zhang J. 2007. The Aedes albopictus inhibitor of apoptosis 1 gene protects vertebrate cells from bluetongue virus-induced apoptosis. Insect Mol. Biol. 16:93–105 [DOI] [PubMed] [Google Scholar]
- 45.Owens RJ, Limn C, Roy P. 2004. Role of an arbovirus nonstructural protein in cellular pathogenesis and virus release. J. Virol. 78:6649–6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoyt CC, Richardson-Burns SM, Goody RJ, Robinson BA, Debiasi RL, Tyler KL. 2005. Nonstructural protein σ1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J. Virol. 79:2743–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clarke P, Meintzer SM, Moffitt LA, Tyler KL. 2003. Two distinct phases of virus-induced nuclear factor κB regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J. Biol. Chem. 278:18092–18100 [DOI] [PubMed] [Google Scholar]
- 48.Berger AK, Danthi P. 2013. Reovirus activates a caspase-independent cell death pathway. mBio 4(3):e00178–13. 10.1128/mBio.00178-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siddiqui MA, Malathi K. 2012. RNase L induces Autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 287:43651–43664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talloczy Z, Jiang W, Virgin HW, IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 99:190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30:678–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barhoom S, Kaur J, Cooperman BS, Smorodinsky NI, Smilansky Z, Ehrlich M, Elroy-Stein O. 2011. Quantitative single cell monitoring of protein synthesis at subcellular resolution using fluorescently labeled tRNA. Nucleic Acids Res. 39:e129. 10.1093/nar/gkr601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heath CM, Windsor M, Wileman T. 2001. Aggresomes resemble sites specialized for virus assembly. J. Cell Biol. 153:449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bresnahan WA, Boldogh I, Chi P, Thompson EA, Albrecht T. 1997. Inhibition of cellular Cdk2 activity blocks human cytomegalovirus replication. Virology 231:239–247 [DOI] [PubMed] [Google Scholar]
- 55.Orba Y, Sunden Y, Suzuki T, Nagashima K, Kimura T, Tanaka S, Sawa H. 2008. Pharmacological cdk inhibitor R-Roscovitine suppresses JC virus proliferation. Virology 370:173–183 [DOI] [PubMed] [Google Scholar]
- 56.Taylor SL, Kinchington PR, Brooks A, Moffat JF. 2004. Roscovitine, a cyclin-dependent kinase inhibitor, prevents replication of varicella-zoster virus. J. Virol. 78:2853–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang D, de la Fuente C, Deng L, Wang L, Zilberman I, Eadie C, Healey M, Stein D, Denny T, Harrison LE, Meijer L, Kashanchi F. 2001. Inhibition of human immunodeficiency virus type 1 transcription by chemical cyclin-dependent kinase inhibitors. J. Virol. 75:7266–7279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.