

Abstract
Herpes simplex virus 1 (HSV-1) establishes a lifelong latent infection in sensory neurons and can reactivate from latency under stress conditions. To promote lytic infection, the virus must interact with specific cellular factors to evade the host's antiviral defenses. The HSV-1 E3 ubiquitin ligase, infected cell protein 0 (ICP0), activates transcription of viral genes, in part, by mediating the degradation of certain cellular proteins that play a role in host antiviral mechanisms. One component of the cellular defenses that ICP0 disrupts is the suborganelle, nuclear domain 10 (ND10), by inducing the degradation and dissociation of the major organizer of ND10, a promyelocytic leukemia (PML) and ND10 constituent, Sp100. Because previously identified domains in ICP0 explain only partially how it directs the degradation and dissociation of PML and Sp100, we hypothesized that additional regions within ICP0 may contribute to these activities, which in turn facilitate efficient viral replication. To test this hypothesis, we used a series of ICP0 truncation mutants and examined PML protein levels and PML and Sp100 immunofluorescence staining in human embryonic lung cells. Our results demonstrate that two overlapping regions within the central N-terminal portion of ICP0 (residues 212 to 311) promoted the dissociation and degradation of PML and dissociation of Sp100 (residues 212 to 427). In conclusion, we have identified two additional regions in ICP0 involved in altering ND10 antiviral defenses in a cell culture model of HSV-1 infection.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is an alphaherpesvirus that establishes latency in humans. Infections of individuals with HSV-1 can cause cold sores, potentially blinding ocular infections, and life-threatening encephalitis. The virus has a characteristic temporal cascade of gene expression ordered into three classes: immediate early (IE), early (E), and late (L). ICP0 (infected cell protein 0) is one of the five IE proteins that facilitates viral gene expression and impairs the host's antiviral responses to infection. Although not an essential protein, ICP0 plays a key role in the establishment of lytic infection and reactivation from latency (1–3). ICP0 is a RING-finger-containing E3 ubiquitin ligase (4). E3 Ub ligases direct the attachment of ubiquitin molecules to target proteins, regulating their function or marking them for degradation by the proteasome. The ubiquitin-proteasome system has been characterized as a major component for cellular protein degradation, whose biological functions extend to the regulation of basic cellular processes (5–8). Through its E3 ligase function, ICP0 transactivates viral gene expression from all three kinetic classes and impairs cellular intrinsic (9, 10) and innate (11–13) antiviral responses, which include chromatinization of the viral genome (14–16), inactivation of the DNA damage response (10, 17, 18), and the induction and establishment of the type 1 interferon response (11–13).
As previously mentioned, ICP0 performs an important role during viral infection by counteracting host antiviral defenses. In addition to impairing the interferon response, ICP0 interferes with intrinsic defenses, which act to immediately suppress one or more steps in the HSV-1 life cycle (19–22). Key components of the host's intrinsic resistance to viruses are PML (promyelocytic leukemia) nuclear bodies, also known as nuclear domain 10 (ND10) (23–25). This dynamic subnuclear complex contains a variety of proteins that are recruited to these nuclear domains by PML, a major constituent of ND10 (20, 26). Other components of ND10 include the cellular proteins Sp100, hDaxx, and ATRX (20, 25, 27). ND10 is associated with cellular processes that include DNA damage, protein degradation, apoptosis, senescence, and interferon signaling (20, 22, 28, 29).
It has been hypothesized that ND10 and its constituent proteins repress transcription of the virus by enveloping viral genomes and limiting their interactions with host factors that stimulate viral transcription (30, 31). Consequently, the activation of viral gene expression by ICP0 is linked to its ability to disrupt ND10 by directing the dissociation and/or degradation of ND10 constituents such as PML and Sp100 and their SUMO-modified forms (27, 32). Further support for the idea that ND10 components play important roles in the function of ICP0 and the HSV-1 life cycle comes from the key observation that an ICP0 null mutant can be partially complemented by cells depleted of PML, Sp100, hDaxx, or ATRX (19, 21, 27, 31, 33), with a further enhancement in viral replication when cells are depleted of two or more of these proteins (21, 31).
To date, only three domains or motifs in ICP0 that mediate the disruption of ND10 via the dissociation and/or degradation of PML, Sp100, hDaxx, and/or ATRX, have been identified. Specifically, the RING-finger motif (Fig. 1), which directs the E3 ubiquitin ligase activity of ICP0, is required for the dissociation of ND10 components (34). In addition, a portion of the C terminus (amino acids 633 to 767; Fig. 1) of ICP0 contains an ND10 localization domain (35, 36); this domain and the nuclear localization signal (NLS; Fig. 1) of ICP0 facilitate its dissociation and degradation of PML and Sp100 and ND10-disrupting activities (36–38). Recently, Cuchet-Lourenço et al. reported interactions between an N-terminal region of ICP0 (amino acids 1 to 388) and a single PML isoform (I) in yeast two-hybrid assays (39), suggesting one potential mechanism by which ICP0 directs the degradation and/or dissociation of PML. Because all of these domains in ICP0 provide only a partial explanation as to how ICP0 alters PML and Sp100 levels, we hypothesized that additional regions within ICP0 likely contribute to PML and/or Sp100 dissociation/degradation, which in turn facilitates efficient viral replication. Consequently, we performed structure-function analyses using a series of ICP0 truncation mutants (Fig. 1) and examined PML levels by flow cytometry and Western blot analyses and the subcellular localization of PML, Sp100, and ICP0 by immunofluorescence. Overall, our results indicate that we have identified two minimal regions in ICP0 that affect PML and Sp100. Specifically, the first 311 N-terminal amino acids of ICP0 promote the dissociation and degradation of PML and the first 427 amino acids the dissociation of Sp100; residues from 212 to 311 and from 212 to 427 within this N-terminal half of ICP0 are required for both of these activities, respectively.
Fig 1.

Functional domains of full-length ICP0 and ICP0 truncation mutants. (A) Schematic of WT ICP0, expressed from WT HSV-1, and mutant forms of ICP0, expressed from the viruses n720, n680, n525, n428, and n212; each ICP0 mutant contains a nonsense linker insertion within the ICP0 gene (1). Selected domains and their amino acid residue boundaries are shown as follows: a RING-finger motif, a central transactivation domain, a nuclear localization signal (NLS), and a C-terminal transactivation domain (TRANS.), which includes an ND10 localization sequence (ND10-loc). Other domains include phosphorylated region I (224 to 232), SLS-4 (asterisk; 362 to 634), phosphorylated region II (365 to 371), a SIAH-1 binding site (401 to 410), and a USP7 binding site (618 to 638). (B) Schematic of additional mutant forms of ICP0 expressed from n389 and n312 viruses.
MATERIALS AND METHODS
Cells and viruses.
HEL-299 (human embryonic lung) cells and Vero (African green monkey kidney) cells were maintained at 37°C in 5% CO2 and cultured in either alpha minimum essential medium (αMEM) for HEL-299 cells or Dulbecco's modified Eagle medium (DMEM) for Vero cells, supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM l-glutamine and 10% or 5% fetal bovine serum (FBS). A human embryonic lung-derived cell line, WI-38+PML-green fluorescent protein (GFP) cells, and L7 cells (Vero cells stably transformed with the ICP0 gene) were grown and maintained as previously described (40, 41). Wild-type (WT) HSV-1 (strain KOS), strain 7134 (an ICP0 null mutant), and ICP0 truncation mutants (n212, n428, n525, n680, and n720) were propagated as previously described (1, 3). Titers for the ICP0 mutant viruses were determined on L7 cells.
pAlter-1+ICP0 was used to create the n312 and n389 ICP0 truncation mutants by inserting a nonsense Spel linker while removing a specific restriction site (either NruI or NotI) with the pAlter site-mutagenesis kit following the manufacturer's instructions. Introduction of the mutations into the plasmid was confirmed by DNA sequencing. Mutant viruses were generated by cotransfection of the respective mutation-containing plasmid and 7134 genomic DNA at a ratio of 3 μg:1 μg into Vero cells plated in 60-mm-diameter dishes at 3 × 105 cells per plate. Selection of mutant viruses was performed using white/blue selection in the presence of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and Phenol red for at least three subsequent rounds and confirmed by Southern blotting.
Flow cytometry.
WI-38+PML-GFP cells were plated at 5 × 105 cells per well in 6-well plates. At 24 h later, the cells were infected at a multiplicity of infection (MOI) of 5, 10, or 20 (as shown in Fig. 2) with KOS or one of the ICP0 mutant viruses for 1 h at 37°C. WI-38+PML-GFP cells express the first 539 N-terminal amino acids of PML fused with GFP. Infected cells were washed 3 times with phosphate-buffered saline (PBS) to remove unabsorbed viruses and placed back into growth medium. For each time point, cells were washed three times with PBS, trypsinized, resuspended twice in 0.5% bovine serum albumin (BSA)–PBS, and fixed with 4% paraformaldehyde–0.5% BSA–PBS. Fluorescence levels were monitored in a Beckman flow cytometer (BD, Biosciences), collecting at least 20,000 events per sample. Results were analyzed using CellQuest software (BD Biosciences).
Fig 2.

Loss of PML-GFP fluorescence by strain KOS and truncation mutants quantified by flow cytometry. PML-GFP-expressing cells were mock infected or infected at an MOI of 5 with KOS or each ICP0 mutant virus. At 6 or 12 hpi, the cells were trypsinized, fixed, and analyzed by flow cytometry. (A) Representative histograms taken from cells at 6 hpi. A total of 20,000 events were analyzed for each histogram. (B) The MFI (mean fluorescence intensity) of each sample was used to calculate the relative fluorescence compared to that of the mock-infected cells (assigned the value of 100%). 6 hpi, n ≥ 13 independent experiments; 12 hpi, n ≥ 5 independent experiments. Data were analyzed using one-way analysis of variance (ANOVA) and Tukey's b multiple-comparison posttest. Asterisks indicate significant differences (P ≤ 0.05) relative to mock-infected cells. (C) PML-GFP-expressing cells were mock infected or infected at an MOI of 5, 10, or 20 with KOS, n212, or 7134. The MFI of each sample was used to calculate the relative fluorescence compared to that of the mock-infected cells (assigned the value of 100%). 12 hpi, n ≥ 2 independent experiments for each MOI. Data were analyzed using one-way ANOVA and Tukey's b multiple-comparison posttest. Asterisks indicate significant differences (P ≤ 0.05) relative to mock-infected cells.
Western blot analyses.
HEL-299 cells were plated at 5 × 105 in 6-well plates. At 24 h later, the cells were infected at an MOI of 5 with KOS or ICP0 mutant viruses. An MOI of 5 was used in these experiments and in our immunofluorescence assays (see the next section) to ensure that >95% of cells in culture were infected with HSV-1. At 6 or 9 h postinfection (hpi), the cells were washed 3 times with PBS prior to being scraped into boiling 1× Laemmli buffer containing protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride), vortexed, and heated for additional 5 min at 95°C. A 7.5% proportion of each sample was resolved using 4% to 12% Bis-Tris gradient gel (Invitrogen) and subsequently transferred to nitrocellulose membranes. Membranes were blocked for 1 h at room temperature with 2% nonfat dry milk in Tris-buffered saline with 0.05% Tween 20 (TBS-T). Blots were probed for ICP0 overnight at 4°C (H11060; Santa Cruz Biotechnology) in blocking buffer. After three washes with TBS-T, membranes were probed with goat anti-mouse IgG-horseradish peroxidase (HRP) (Jackson ImmunoResearch). After an additional three washes with TBS-T, the membranes were developed using ECL reagents (SuperSignal West Femto chemiluminescent substrate; ThermoFisher Scientific). The membranes were then washed with TBS-T prior being stripped for 30 min at 50°C in 100 mM 2-mercaptoethanol–62.5 mM Tris (pH 6.7)–2% SDS. Blots were again blocked as before prior to being probed overnight for PML (A301-167A; Bethyl Laboratories) and β-actin [(I-19)-R; Santa Cruz Biotechnology], both diluted in blocking buffer. After washing, the membrane was incubated with goat anti-rabbit IgG-HRP (Jackson ImmunoResearch) and developed as before. All images were assembled using Adobe Photoshop and Adobe Illustrator.
Immunofluorescence.
HEL cells were plated 24 h prior to infection at 50% confluence on collagen-coated coverslips. Cells were infected at an MOI of 5 with KOS or each mutant virus in αMEM as described above. At specific time points, the coverslips were washed with PBS and fixed and permeabilized as previously described (42) Coverslips were labeled with primary antibody by incubation for 1 h in a tissue culture incubator at 37°C, washed three times with PBS, labeled with secondary antibody in the same manner as the primary antibody, washed three times with PBS, and then mounted onto slides using ProLong antifade (Invitrogen). The primary antibodies used were ICP0 (H11060 or rabbit polyclonal IgG PAC 3678; Pacific Immunology), PML (A301-167A, Bethyl Laboratories, or sc-5621, Santa Cruz Biotechnology), and Sp100 (sc-16328; Santa Cruz Biotechnology); the secondary antibodies were goat anti-mouse IgG2b Dylight 488 (Jackson ImmunoResearch), goat anti-rabbit IgG Rhodamine Red-X (Jackson ImmunoResearch), donkey anti-rabbit IgG Dylight 594 (Jackson ImmunoResearch), and cow anti-goat IgG Dylight 488 (Jackson ImmunoResearch). All antibodies were diluted in 0.5% FBS–0.5% BSA–PBS. Slides were examined using a Nikon Eclipse TE-2000 U fluorescence microscope and photographed using with a CoolSNAP EZ digital camera (Photometrics) with NSI Elements Software (Nikon). Images were assembled in Adobe Photoshop and Adobe Illustrator. At least 100 cells that expressed ICP0 were examined for PML or Sp100 staining to quantify the extent to which the WT or each mutant form of ICP0 altered the staining of either cellular protein. Cells were categorized as staining either positive or negative for PML or Sp100. Criteria for negative staining included cells that lacked antigen expression, had half or less the intensity of staining, or had half or less the number of puncta seen with the negative controls (mock- and 7134-infected cells). In addition, we observed a minor degree of HSV Fc receptor staining and took this staining profile into account when we examined PML localization in HSV-1-infected cells.
RESULTS
ICP0 mutants mediate the dissociation and degradation of exogenous PML.
ICP0 has been shown to promote the degradation of the PML antiviral protein (38, 43, 44). To better understand how ICP0 induces the dissociation and degradation of PML from ND10, our approach was to identify a new region(s) within ICP0 other than its RING finger and its C terminus that plays a role in these functions. For these studies, we used a series of viruses in which nonsense linkers had been inserted into the ICP0 gene, giving rise to forms of ICP0 progressively truncated at its C terminus (Fig. 1A) (1). To initially monitor the dissociation of PML, we examined exogenous PML-GFP fluorescence in a human embryonic cell line by flow cytometry, a method successfully used by our laboratory to monitor ICP0's E3 ubiquitin ligase activity (40). To examine and quantify the loss of PML-GFP fluorescence induced by WT HSV-1 (strain KOS) and each ICP0 mutant virus, cells were mock infected or infected with KOS or each ICP0 mutant, and the fluorescence signal of each sample was examined at 6 and 12 hpi (Fig. 2A and B). In Fig. 2A, histograms for mock-infected cells and cells virally infected at 6 hpi are vertically aligned. The “y” vertical line intersects the peak for the PML-GFP mock-infected sample. The “x” vertical line intersects the KOS and n720 peaks at 6 hpi, which showed a reduction in fluorescence compared to the mock-infected cell results. n680, n525, and n428 had curves with PML-GFP levels that fell between those of the mock- and KOS-infected samples. n212- and 7134-infected samples had curves with fluorescence intensities nearly identical to that of the mock-infected cells.
When we quantified the levels of PML-GFP fluorescence in cells, we noted that there were three statistically distinguishable groups. KOS and n720 were very efficient at inducing the loss of PML-GFP, with 12% to 20% levels compared to mock-infected cells, which were given a value of 100%. The next group of mutant viruses that were significantly different from KOS- and mock-infected cells consisted of n680, n525, and n428. Reduced (45% to 62%) levels of PML-GFP were observed for n428, n525, and n680, although these reductions were not as large as those resulting from infection by n720 or KOS (Fig. 2B). The form of ICP0 encoded by n212 was the only truncation mutant unable to significantly decrease PML-GFP fluorescence (∼90% levels) for both time points, which were comparable to mock- and 7134-infected cell levels (Fig. 2B). Furthermore, even when PML-GFP-expressing cells were infected with n212 at higher MOIs (10 and 20) for up to 12 h, PML-GFP signals were still largely unaffected (Fig. 2C), suggesting that the defect was due to the inability of this mutant form of ICP0 to promote the efficient dissociation of PML.
Overall, these data show that the first 427 amino acids of ICP0 help facilitate the dissociation (and degradation) of PML-GFP from ND10 and that residues from 212 to 427 in this portion of ICP0 are required for these activities. Additionally, our results indicate that residues from 679 to 719 are necessary for ICP0 to efficiently promote the loss of PML-GFP. These data are in agreement with a previous study indicating a role for this region in the targeting of ICP0 to ND10s, facilitating the dissociation of PML from ND10 and PML's degradation (38).
A region of ICP0 spanning residues 212 to 427 is involved in the degradation of endogenous PML.
Because we had examined the effects that ICP0 truncation mutants have on exogenously expressed PML, we next wanted to determine whether these mutants affected endogenous PML in a comparable manner. Consequently, we analyzed endogenous PML protein levels at 6 hpi in HEL cells (Fig. 3). The results were generally consistent with those obtained by flow cytometry analysis of PML-GFP (Fig. 2A and B). Again, there was a link between the size of each mutant form of ICP0 and its ability to promote the degradation of PML. 7134- and n212-infected cells had PML levels similar to that of mock-infected cells. The n428 mutant form of ICP0 was capable of reducing PML protein levels, although not to same extent as the larger truncated forms of ICP0. This experiment indicated that the first 427 amino acids of ICP0 assist in the degradation of PML by 6 hpi, with the region spanning residues 212 to 427 being required for this function. Furthermore, the residues from 428 to 524 are required for the efficient degradation in these assays.
Fig 3.
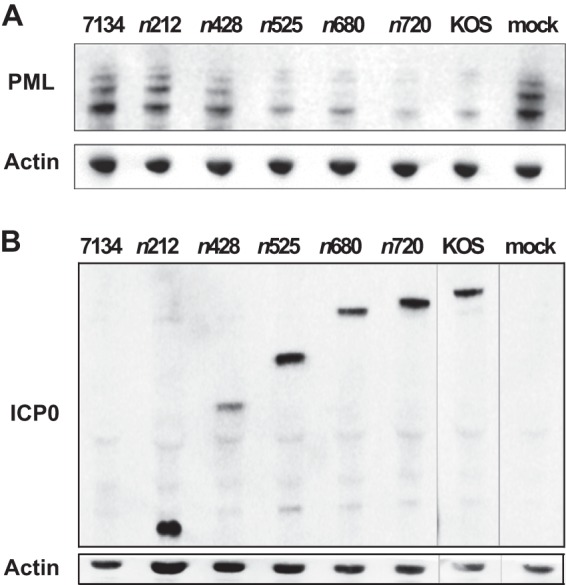
KOS and ICP0 truncation mutants induce the degradation of endogenous PML protein levels. Virally infected or mock-infected HEL cells were harvested and lysed at 6 hpi. PML (A) and ICP0 (B) protein levels were examined by Western blot analyses. β-Actin levels were included as loading controls. A representative set of images from 5 independent experiments is shown.
Subcellular localization of ICP0 truncation mutants.
We then infected cells with KOS or each ICP0 truncation mutant for 3, 6, and 9 hpi. For WT ICP0 expressed from KOS, our results matched those of a previous report (45) in which ICP0 localization changes over time from primarily nuclear ICP0 during the initial stages of infection (e.g., 3 hpi) to a mix of nuclear and cytoplasmic ICP0 between 3 and 6 hpi and finally to predominantly cytoplasmic ICP0 at 6 hpi and later (Fig. 4). When we examined the subcellular localization of the ICP0 truncation mutants, the mutant forms of ICP0 expressed from n525, n680 and n720 were predominantly nuclear at 6 and 9 hpi (Fig. 4). These mutant forms possess a nuclear localization signal (NLS) (Fig. 1A), and they did not localize extensively to the cytoplasm later during viral infection as observed for WT ICP0. The n212 and n428 mutant forms of ICP0 were found in both the nucleus and cytoplasm at 6 and 9 hpi (Fig. 4). Interestingly, ICP0 expressed from n212 and n428 lacks WT ICP0's NLS, but both proteins were able to diffuse into the nucleus during infection; this localization is likely associated with their apparent molecular masses (i.e., ∼35 kDa and ∼58 kDa; see Fig. 7A), as proteins that are ≤60 kDa have been reported to be capable of diffusing through the nuclear pore (46).
Fig 4.
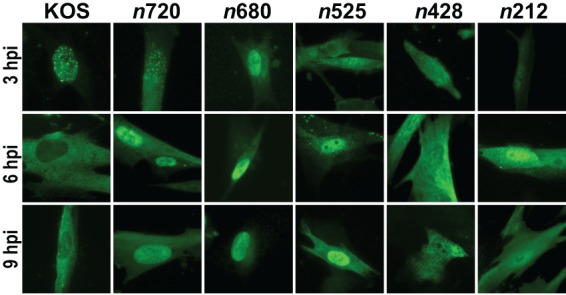
ICP0 localization in cells infected with KOS and ICP0 truncation mutants. HEL cells were infected (MOI = 5) with KOS or each ICP0 truncation mutant for 3, 6, and 9 h. Cells were fixed, permeabilized, and immunostained for ICP0 and visualized by fluorescence microscopy at ×400 magnification.
Fig 7.

Truncation mutants n312 and n389 induce the degradation of endogenous PML protein levels and alter the staining of ND10 constituents. (A) Infected or mock-infected HEL cells were harvested and lysed at 9 hpi. PML and ICP0 protein levels were examined by Western blot analyses. β-Actin levels were included as loading controls. A representative set of images from 4 independent experiments is shown. (B to E) HEL cells were infected at an MOI of 5 with KOS, ICP0 truncation mutant n212, n312, n389, or n428, or 7134 or were mock infected for 6 h. Cells were subsequently fixed, permeabilized, and immunostained with antibodies that recognize ICP0 and either PML (B) or Sp100 (D). Arrows indicate an infected cell. (C and E) Graphs representing the percentages of ICP0-expressing cells that stained positive (+) and negative (−) for PML (C) or Sp100 (E) by immunofluorescence were determined for each mutant (except 7134) by examining at least 100 ICP0-expressing cells using ImageJ software.
Two N-terminal regions of ICP0 are necessary for ICP0 to dissociate PML and Sp100 from ND10.
To determine how these truncated mutant forms of ICP0 affected PML localization over time, we performed immunofluorescence assays in HEL cells. Initially, cells were infected with KOS or each ICP0 truncation mutant for 3, 6, and 9 hpi. We then examined PML and ICP0 staining in virally infected cells at 6 hpi because this time point resulted in >90% loss of PML staining with WT ICP0. The n720 and n680 ICP0 mutants appeared to be similar to KOS in directing the dissociation of PML (Fig. 5A), as ≤2% of cells that expressed WT ICP0 or the n720 mutant form or the n680 mutant form of ICP0 stained positive for PML (Fig. 5B). Limited loss of PML staining was detected in n525- and n428-infected cells, and 8% to 32% of cells that expressed ICP0 were positive for PML (Fig. 5). In the case of n212 at 6 hpi, the majority (61%) of cells that expressed this truncated form of ICP0 stained positive for PML (Fig. 5). Notably, this mutant form of ICP0 partially colocalized with PML by 9 hpi (M. Perusina Lanfranca and D. J. Davido, unpublished data). PML staining of 7134-infected cells was comparable to that of mock-infected cells (Fig. 5A). To exclude the possibility that the n212 mutant protein was capable of dissociating PML in a delayed manner, infections with n212 were allowed to continue for 12 to 24 hpi; even at these later time points, the n212 truncation was unable to promote the further loss of PML staining. Thus, all truncation mutants were capable of promoting different degrees of PML dispersal from ND10 structures over time. Comparable results for PML staining for all of the truncation mutants were observed with two other PML antibodies (M. Perusina Lanfranca and D. J. Davido, unpublished data), demonstrating the relative specificities and reproducibilities of PML staining by the WT and mutant forms of ICP0 in this assay.
Fig 5.
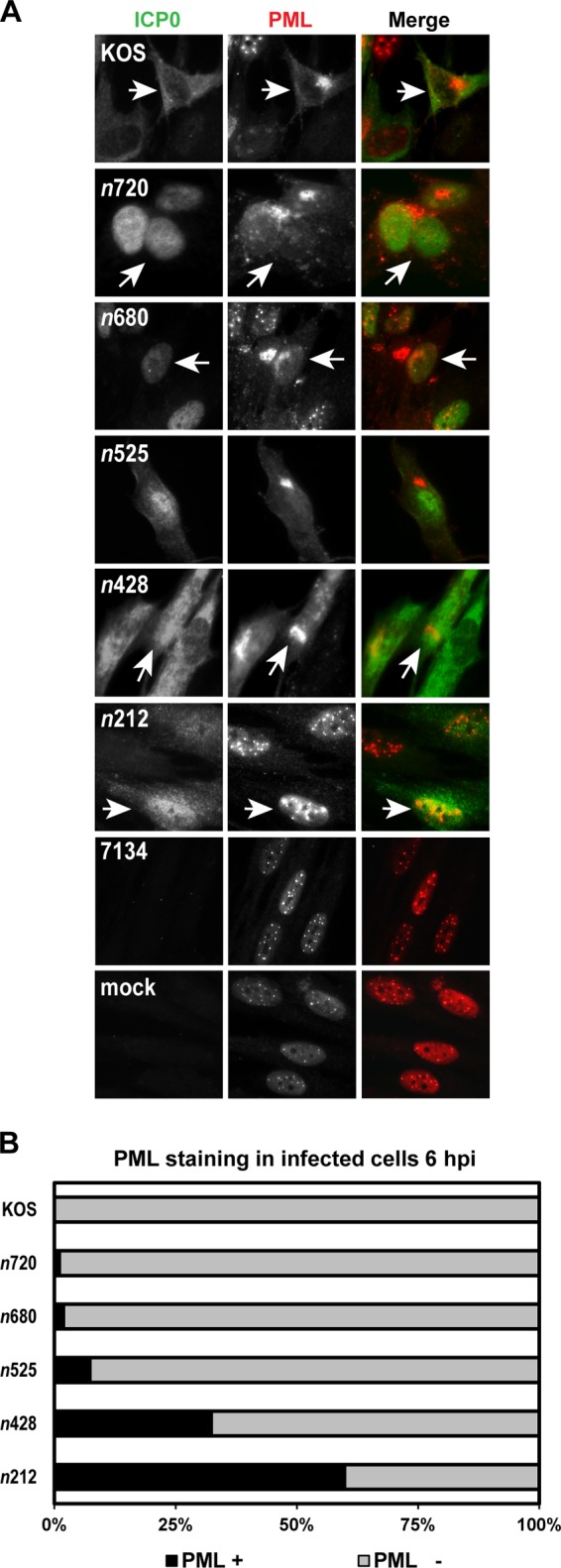
KOS and ICP0 truncation mutants mediate the loss of endogenous PML staining. (A) HEL cells were infected at an MOI of 5 with KOS, each ICP0 truncation mutant, or 7134 or were mock infected. At 6 hpi, cells were fixed, permeabilized, and immunostained with antibodies that recognize ICP0 and PML. ICP0 is labeled in green, and PML is labeled in red in the Merge images. Arrows indicate an infected cell. (B) Graph representing the percentages of ICP0-expressing cells that stained positive (+) and negative (−) for PML by immunofluorescence. For each mutant (except 7134), at least 100 ICP0-expressing cells were analyzed with ImageJ software.
Sp100 has been shown to stabilize ND10 by protecting PML from ICP0-mediated degradation while promoting the repression of HSV-1 IE expression (47). Because the ICP0 mutants showed a spectrum of PML dissociation phenotypes, we then asked how these mutant forms of ICP0 affected the staining of Sp100. Infections were carried out as described for the PML immunofluorescence experiments, and ICP0 and Sp100 staining was monitored at 6 hpi (Fig. 6A). As previously reported, WT ICP0 expressed from KOS efficiently dispersed Sp100 from ND10 by 6 hpi (Fig. 6) (32, 48). The staining profiles of Sp100 for the ICP0 truncation mutant viruses were similar to the results obtained with PML. The n720 and n680 mutant forms of ICP0 were ≤12% positive for Sp100 staining, a range comparable to that seen with WT ICP0 expressed by KOS (Fig. 6). There was a gradual increase in Sp100 staining for n525 and n428, where 23% and 29% of the n525- and n428-expressing cells, respectively, were Sp100 positive (Fig. 6). Most (77%) cells that expressed the n212 mutant form of ICP0 contained Sp100 at 6 hpi (Fig. 6). 7134- and mock-infected cells maintained punctate Sp100 staining (Fig. 6A). To confirm our results obtained with Sp100 staining, we tried to examine Sp100 protein levels by Western blot analysis using the same primary antibody as was used in our Sp100 immunofluorescence experiments. Unfortunately, this antibody was not functional in Western blot assays.
Fig 6.
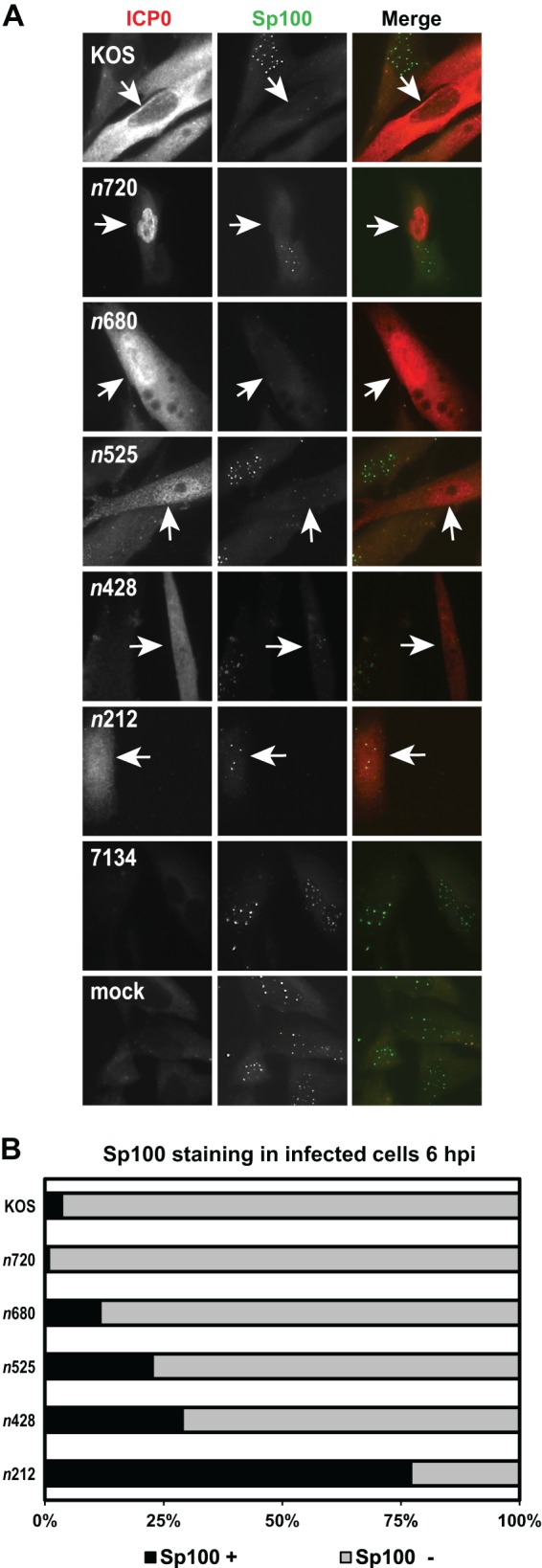
KOS and ICP0 truncation mutants direct the loss of endogenous Sp100 staining. (A) HEL cells were infected at an MOI of 5 with KOS, each ICP0 truncation mutant, or 7134 or were mock infected for 6 h. Cells were subsequently fixed, permeabilized, and immunostained with antibodies that recognize ICP0 and Sp100. ICP0 is labeled in red, and Sp100 is labeled in green in the Merge images. Arrows indicate an infected cell. (B) Graph representing the percentages of ICP0-expressing cells that stained positive (+) and negative (−) for Sp100 by immunofluorescence. For each mutant (except 7134), at least 100 ICP0-expressing cells were analyzed with ImageJ software.
Within the N terminus of ICP0, a 100-amino-acid region is crucial for the degradation and dissociation of PML from ND10.
To define the subregion from amino acids 212 to 427 of ICP0 that is necessary for directing the dissociation and degradation of PML, we generated two new truncation mutants, n312 and n389, which express the first 311 and 388 amino acids of ICP0, respectively (Fig. 1B and 7A). Using these and other ICP0 mutants (e.g., n428, n212, and 7134), we examined endogenous PML levels in HSV-1-infected or mock-infected HEL cells at 9 hpi by Western blot analysis. We chose to examine PML levels at 9 hpi to determine whether these mutant forms of ICP0 would be capable of directing the degradation of PML if given additional time. As shown in Fig. 7A, all of the ICP0 linker insertion mutants we tested were capable of mediating the loss of PML, with the exception of n212 by 9 hpi. Notably, n312, n389, and n428 significantly directed degradation of PML. In the case of n428, there appeared to be a greater degree of PML degradation at 9 hpi than at 6 hpi (Fig. 4A). Thus, we have defined the region from amino acids 212 to 311 of ICP0 involved in the degradation of PML by 9 hpi.
Using these ICP0 mutants, we examined the dissociation of PML and Sp100 from ND10 in HSV-1- or mock-infected HEL cells at 6 hpi. Only 0% to 26% of the cells that expressed the n428, n389, and n312 mutant forms of ICP0 stained positive for PML, whereas >65% of the n212-expressing cells were positive for PML staining (Fig. 7B and C). These data largely mirrored our results from the PML Western blots shown in Fig. 7A. When Sp100 immunofluorescence was examined, there was a clear boundary between n428 and the other truncation mutants examined (Fig. 7D and E). Specifically, 22% of the cells that expressed the n428 mutant form of ICP0 stained positive for Sp100, whereas 55% to 77% of n389-, n312-, and n212-expressing cells were positive for Sp100 (Fig. 7E). Furthermore, our immunofluorescence experiments with n389 and n312 indicated that these viral mutants are not as efficient in dissociating Sp100 from ND10 as PML.
Summarizing our results for PML and Sp100 from Fig. 3 to 7 in Table 1, the first 311 amino acids assist in mediating the dissociation and degradation of PML, whereas the first 427 amino acids of ICP0 promote the dissociation of Sp100 from ND10. Thus, residues from 212 to 311 contribute to ICP0's PML dispersal and degradation activities, given that the truncated n212 form of ICP0 was unable to promote the degradation of PML and efficiently alter the staining of PML. Amino acids 212 to 427 are involved in the dispersal of Sp100 from ND10. Lastly, the C-terminal half of ICP0 (amino acids 428 to 720) facilitates the efficient dissociation of PML and Sp100.
Table 1.
PML and Sp100 phenotypes of WT HSV-1 and ICP0 mutants
| Virus | PML |
Sp100 |
Plating efficiencyb | ||
|---|---|---|---|---|---|
| Dissociation/loss | Degradation |
Dissociation/loss | |||
| 6 h | 6 h | 9 h | 6 h | ||
| KOS | Nearly complete | Complete | Complete | Nearly complete | 1.2 |
| n720 | Nearly complete | Nearly complete | Completea | Nearly complete | 13 |
| n680 | Nearly complete | Nearly complete | Completea | Nearly complete | 6.5 |
| n525 | Nearly complete | Nearly complete | Complete | Partial | 4.5 |
| n428 | Partial | Partial | Nearly complete | Partial | 12 |
| n389 | Complete | Partiala | Nearly complete | Limited | 48 |
| n312 | Partial | Partiala | Nearly complete | Very limited | 79 |
| n212 | Very limited | Not evident | Not evident | Very limited | 114 |
| 7134 | Not evident | Not evident | Not evident | Not evident | 310 |
Unpublished data.
The efficiency of plating for each virus is the ratio of viral titers on complementing L7 versus Vero cells. Values are derived from 2 to 4 independent experiments.
DISCUSSION
ICP0 is a multifunctional protein, and many of its functions have been linked to its E3 ubiquitin ligase activity, which requires its RING-finger domain (49). The E3 ubiquitin ligase activity of ICP0 has been associated with the proteasome-dependent degradation of several cellular proteins, including PML (30, 32, 38, 39, 48, 50–52), Sp100 (32, 38, 50, 53), CENP-A, -B, and -C (54–57), DNA-PKcs (18, 53, 58), RNF-8 and RNF-168 (18), CD83 (59), USP7 (60, 61), IκBα (62), p53 (63), IFI16 (64, 65), and E2FBP1 (66), as well as certain SUMOylated targets (67). Degradation of these targets allows HSV-1 to evade the host's intrinsic and innate responses, thereby facilitating viral replication.
In the current study, we sought to understand how ICP0 mediates the dissociation of PML and Sp100, degradation of PML, and disruption of ND10, as PML, Sp100, and ND10 play important roles in intrinsic and innate immunity (9, 10). While the RING-finger motif, NLS, and particular regions of the C terminus of ICP0 have been shown to facilitate the dissociation and degradation of PML and Sp100 and the degradation of PML (38, 43), we hypothesized that other regions or domains in ICP0 are required to impair PML and Sp100 function. This hypothesis was tested in structure-function analyses using a series of progressively shorter ICP0 truncation mutants (Fig. 1). We observed that there was a gradation in the ability of the mutants to mediate the dissociation of exogenous PML-GFP, the degradation of endogenous PML, and the dissociation of endogenous PML and Sp100 from ND10. Specifically, our studies showed that the smallest truncation mutant, n212, had no effect or limited effects on PML-GFP, PML, and Sp100 staining and did not affect PML levels (Fig. 2 to 7) by 6 or 9 hpi. This mutant form of ICP0 has been reported to possess E3 ubiquitin ligase activity in vitro (42). Thus, our data indicate that the first 211 amino acids of ICP0 are partially capable of dispersing PML but do not mediate its degradation, suggesting that these two activities of ICP0 are not entirely coupled. In contrast, n428 moderately affected PML-GFP, PML, and Sp100 staining and PML levels. The remaining viral mutants, n525, n680, and n720, showed efficiency that was similar to or greater than that of n428, which does not contain the NLS of ICP0, with respect to these activities of ICP0 (Fig. 1A). Thus, it is possible that the predominantly nuclear localization of the n525 and n720 mutant forms of ICP0 facilitates its ND10-disrupting activities (Fig. 4). Notably, the PML and Sp100 staining profiles and PML levels of these ICP0 mutants largely correlate with their respective transactivation, complementation, and replication phenotypes (Table 1) (1, 3). With the addition of ICP0 mutants n312 and n389, we were able to better define the boundaries of ICP0 that result in the loss of PML protein levels and PML and Sp100 staining. Overall, our data indicate that there is a minimal region of ICP0 (amino acids 1 to 311) that directs the dispersal and degradation of PML and which lies within the minimal region (amino acids 1 to 427) of ICP0 that dissociates Sp100 from ND10; the residues from 212 to 311 and 212 to 427 are required for their respective functions.
In addition to the ICP0 structure-function analyses highlighting the importance of other motifs/regions in ICP0 (i.e., RING-finger, NLS, and C terminus) in facilitating the dissociation and degradation of ND10 components, a recent study by Cuchet-Lourenço et al. indicated that ICP0 can interact with a particular isoform of PML (i.e., PML I) in coimmunoprecipitation assays (39). Those same investigators established by yeast two-hybrid assays that the N-terminal 388 amino acids of ICP0 interact with PML and that the residues from 242 to 388 are necessary for this interaction. The domain required for PML dissociation and degradation (amino acids 212 to 311) maps to this region of ICP0, suggesting that residues from 241 to 311 of ICP0's N terminus participate in the binding of at least one isoform of PML to mediate the ubiquitination, degradation, and dissociation of PML from ND10. Also, our ICP0-Sp100 data indicate that an additional region in ICP0 (residues 212 to 427) is required for the dissociation of Sp100, implying that ICP0 regulates the dispersal of Sp100 by mechanisms other than or in addition to those required for PML.
As previously mentioned, our mapping studies identified amino acids 212 to 311 as playing an important role in the dissociation and degradation of PML, whereas residues from 212 to 427 facilitate the dissociation of Sp100. In addition to the aforementioned PML interaction domain, functional motifs within the larger (residues 212 to 427) of the two domains of HSV-1 ICP0 (see Fig. 1A) include the two phosphorylated regions (region I [residues 224 to 232] and region II [residues 365 to 371]) (68, 69), a SUMO-interacting motif (SIM) (SLS-4; residues 362 to 364) (30), and a binding site for the E3 ubiquitin ligase, SIAH-1 (residues 401 to 410) (70). Of these motifs, only phosphorylated region I lies within the required PML dissociation/degradation domain of ICP0 (residues 212 to 311). Mutation of phosphorylated regions I and II of ICP0 in the absence of other viral factors diminished the E3 ubiquitin ligase and/or altered the ND10-disrupting activities of ICP0 (69). Furthermore, viral mutants containing these mutations (i.e., Phos 1 and Phos 2) were impaired for viral replication in cell culture and/or mice (69, 71). Mutation of the ICP0 SIM, SLS-4, diminished the complementation and reactivation capabilities of ICP0, especially when this mutation was expressed in truncated forms of ICP0 (residues 1 to 396 and 1 to 594) (30). In the case of SIAH-1, its depletion increased the stability of ICP0, and an ICP0 SIAH-1 binding site mutant in HSV-2 had a modest decrease in viral replication (70). For the PML- and Sp100-associated activities of ICP0, we speculate that these motifs likely serve collective or redundant roles and thus that their individual contributions are apparent only when expressed in the N-terminal fragment of ICP0. Future experiments will address this possibility.
Furthermore, while we have discovered that there are minimal regions of ICP0 that regulate these activities, our results also suggest that other regions/domains from amino acids 428 to 775 facilitate PML and Sp100 dispersal, PML degradation, and ND10 disruption. We propose that these additional domains in the C terminus, besides the NLS, stabilize the binding of ICP0 to its target proteins, including PML, for ubiquitination. It is known that ICP0 can preferentially target SUMO-conjugated proteins such as PML and Sp100 for proteasomal degradation (30). Furthermore, ICP0 contains several known SIMs, which appear to be redundant in function (30). Thus, SUMO-SIM interactions are linked to ICP0 functions, as PML and Sp100 are modified by SUMO, and PML, Sp100, and other ND10 constituents contain SIMs that are necessary for their recruitment to sites of incoming HSV-1 genomes (30, 31, 39, 72). Ultimately, all of these interactions contribute to ICP0's multifaceted role in impairing the host's intrinsic antiviral response.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grant RO1AI72357 from the National Institute of Allergy and Infectious Diseases.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
We thank Steve Benedict and members of the Davido laboratory for comments related to the manuscript and Priscilla Schaffer for providing many of the ICP0 mutants used in this study.
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Cai WZ, Schaffer PA. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4579–4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RA, Everett RD, Zhu XX, Silverstein S, Preston CM. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 63:3513–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai W, Astor TL, Liptak LM, Cho C, Coen DM, Schaffer PA. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67:7501–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everett RD, Barlow P, Milner A, Luisi B, Orr A, Hope G, Lyon D. 1993. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif of an alpha herpes virus protein family. J. Mol. Biol. 234:1038–1047 [DOI] [PubMed] [Google Scholar]
- 5.Schwartz AL, Ciechanover A. 1992. Ubiquitin-mediated protein modification and degradation. Am. J. Respir. Cell Mol. Biol. 7:463–468 [DOI] [PubMed] [Google Scholar]
- 6.Ciechanover A. 1994. The ubiquitin-proteasome proteolytic pathway. Cell 79:13–21 [DOI] [PubMed] [Google Scholar]
- 7.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu. Rev. Biochem. 67:425–479 [DOI] [PubMed] [Google Scholar]
- 8.Baumeister W, Walz J, Zuhl F, Seemuller E. 1998. The proteasome: paradigm of a self-compartmentalizing protease. Cell 92:367–380 [DOI] [PubMed] [Google Scholar]
- 9.Bieniasz PD. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 5:1109–1115 [DOI] [PubMed] [Google Scholar]
- 10.Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. 2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 7:e1002084. 10.1371/journal.ppat.1002084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mossman KL, Saffran HA, Smiley JR. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mossman KL, Macgregor PF, Rozmus JJ, Goryachev AB, Edwards AM, Smiley JR. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mossman KL, Smiley JR. 2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 76:1995–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coleman HM, Connor V, Cheng ZSC, Grey F, Preston CM, Efstathiou S. 2008. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following ICP0-mediated de-repression. J. Gen. Virol. 89:68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82:12030–12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferenczy MW, DeLuca NA. 2009. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J. Virol. 83:8514–8524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett RD. 2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol. 8:365–374 [DOI] [PubMed] [Google Scholar]
- 18.Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. 2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 29:943–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830 [DOI] [PubMed] [Google Scholar]
- 21.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82:2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 1783:2207–2221 [DOI] [PubMed] [Google Scholar]
- 23.Sternsdorf T, Grotzinger T, Jensen K, Will H. 1997. Nuclear dots: actors on many stages. Immunobiology 198:307–331 [DOI] [PubMed] [Google Scholar]
- 24.Maul GG. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20:660–667 [DOI] [PubMed] [Google Scholar]
- 25.Maul GG, Negorev D, Bell P, Ishov AM. 2000. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J. Struct. Biol. 129:278–287 [DOI] [PubMed] [Google Scholar]
- 26.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF, III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147:221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 84:4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regad T, Chelbi-Alix MK. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274–7286 [DOI] [PubMed] [Google Scholar]
- 29.Bernardi R, Papa A, Pandolfi PP. 2008. Regulation of apoptosis by PML and the PML-NBs. Oncogene 27:6299–6312 [DOI] [PubMed] [Google Scholar]
- 30.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 7:e1002245. 10.1371/journal.ppat.1002245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 87:2174–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chelbi-Alix MK, de Thé H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941 [DOI] [PubMed] [Google Scholar]
- 33.Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, Heeren J, Bartelt A, Everett RD. 2011. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 124:280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Everett R, O'Hare P, O'Rourke D, Barlow P, Orr A. 1995. Point mutations in the herpes simplex virus type 1 Vmw110 RING finger helix affect activation of gene expression, viral growth, and interaction with PML-containing nuclear structures. J. Virol. 69:7339–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Everett RD. 1987. A detailed mutational analysis of Vmw110, a trans-acting transcriptional activator encoded by herpes simplex virus type 1. EMBO J. 6:2069–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Everett RD. 1988. Analysis of the functional domains of herpes simplex virus type 1 immediate-early polypeptide Vmw110. J. Mol. Biol. 202:87–96 [DOI] [PubMed] [Google Scholar]
- 37.Everett RD, Parsy ML, Orr A. 2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol. 83:4963–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuchet-Lourenço D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 86:11209–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilliard JG, Cooper AL, Slusser JG, Davido DJ. 2009. A flow cytometric assay for the study of E3 ubiquitin ligase activity. Cytometry A 75:634–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samaniego LA, Wu N, DeLuca NA. 1997. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 71:4614–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Everett RD, Maul GG. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 75(Pt 6):1223–1233 [DOI] [PubMed] [Google Scholar]
- 45.Paladino P, Collins SE, Mossman KL. 2010. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS One 5:e10428. 10.1371/journal.pone.0010428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding Q, Zhao L, Guo H, Zheng AC. 2010. The nucleocytoplasmic transport of viral proteins. Virol. Sin. 25:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Negorev DG, Vladimirova OV, Maul GG. 2009. Differential functions of interferon-upregulated Sp100 isoforms: herpes simplex virus type 1 promoter-based immediate-early gene suppression and PML protection from ICP0-mediated degradation. J. Virol. 83:5168–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu H, Roizman B. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. U. S. A. 100:8963–8968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Everett RD. 1988. Promoter sequence and cell type can dramatically affect the efficiency of transcriptional activation induced by herpes simplex virus type 1 and its immediate-early gene products Vmw175 and Vmw110. J. Mol. Biol. 203:739–751 [DOI] [PubMed] [Google Scholar]
- 50.Müller S, Dejean A. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 73:5137–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parkinson J, Everett RD. 2000. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 74:10006–10017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J. Virol. 84:3476–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parkinson J, Lees-Miller SP, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 73:650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Everett RD, Earnshaw WC, Findlay J, Lomonte P. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18:1526–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lomonte P, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J. Virol. 73:9456–9467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lomonte P, Morency E. 2007. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 581:658–662 [DOI] [PubMed] [Google Scholar]
- 57.Gross S, Catez F, Masumoto H, Lomonte P. 2012. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS One 7:e44227. 10.1371/journal.pone.0044227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. 1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70:7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kummer M, Turza NM, Muhl-Zurbes P, Lechmann M, Boutell C, Coffin RS, Everett RD, Steinkasserer A, Prechtel AT. 2007. Herpes simplex virus type 1 induces CD83 degradation in mature dendritic cells with immediate-early kinetics via the cellular proteasome. J. Virol. 81:6326–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Canning M, Boutell C, Parkinson J, Everett RD. 2004. A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279:38160–38168 [DOI] [PubMed] [Google Scholar]
- 61.Antrobus R, Boutell C. 2008. Identification of a novel higher molecular weight isoform of USP7/HAUSP that interacts with the herpes simplex virus type-1 immediate early protein ICP0. Virus Res. 137:64–71 [DOI] [PubMed] [Google Scholar]
- 62.Diao L, Zhang B, Fan J, Gao X, Sun S, Yang K, Xin D, Jin N, Geng Y, Wang C. 2005. Herpes virus proteins ICP0 and BICP0 can activate NF-[kappa]B by catalyzing I[kappa]B[alpha] ubiquitination. Cell. Signal. 17:217–229 [DOI] [PubMed] [Google Scholar]
- 63.Boutell C, Everett RD. 2003. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and ubiquitinates p53. J. Biol. Chem. 278:36596–36602 [DOI] [PubMed] [Google Scholar]
- 64.Johnson KE, Chikoti L, Chandran B. 20 February 2013. HSV-1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 10.1128/JVI.00082-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U. S. A. 109:E3008–E3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fukuyo Y, Horikoshi N, Ishov AM, Silverstein SJ, Nakajima T. 2011. The herpes simplex virus immediate-early ubiquitin ligase ICP0 induces degradation of the ICP0 repressor protein E2FBP1. J. Virol. 85:3356–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 94:465–481 [DOI] [PubMed] [Google Scholar]
- 68.Davido DJ, von Zagorski WF, Lane WS, Schaffer PA. 2005. Phosphorylation site mutations affect herpes simplex virus type 1 ICP0 function. J. Virol. 79:1232–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mostafa HH, Thompson TW, Kushnir AS, Haenchen SD, Bayless AM, Hilliard JG, Link MA, Pitcher LA, Loveday E, Schaffer PA, Davido DJ. 2011. Herpes simplex virus 1 ICP0 phosphorylation site mutants are attenuated for viral replication and impaired for explant-induced reactivation. J. Virol. 85:12631–12637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagel CH, Albrecht N, Milovic-Holm K, Mariyanna L, Keyser B, Abel B, Weseloh B, Hofmann TG, Eibl MM, Hauber J. 2011. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. J. Virol. 85:7644–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mostafa HH, Thompson TW, Davido DJ. 2013. N-terminal phosphorylation sites of herpes simplex virus 1 ICP0 differentially regulate its activities and enhance viral replication. J. Virol. 87:2109–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuchet-Lourenço D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 7:e1002123. 10.1371/journal.ppat.1002123 [DOI] [PMC free article] [PubMed] [Google Scholar]