

Abstract
We sought proof of principle that tumor-targeting ligands can be displayed on the surface of vesicular stomatitis virus (VSV) by engineering its glycoprotein. Here, we successfully rescued VSVs displaying tumor vasculature-targeting ligands. By using a rational approach, we investigated various feasible insertion sites on the G protein of VSV (VSV-G) for display of tumor vasculature-targeting ligands, cyclic RGD (cRGD) and echistatin. We found seven sites on VSV-G that tolerated insertion of the 9-residue cRGD peptide, two of which could tolerate insertion of the 49-amino acid echistatin domain. All of the ligand-displaying viruses replicated as well as the parental virus. In vitro studies demonstrated that the VSV-echistatin viruses specifically bound to targeted integrins. Since the low-density lipoprotein receptor (LDLR) was recently identified as a major receptor for VSV, we investigated the entry of ligand-displaying viruses after masking LDLR. The experiment showed that the modified viruses can enter the cell independently of LDLR, whereas entry of unmodified virus is significantly blocked by a specific monoclonal antibody against LDLR. Both parental and ligand-displaying viruses displayed equal oncolytic efficacies in a syngeneic mouse myeloma model. We further demonstrated that single-chain antibody fragments against tumor-specific antigens can be inserted at the N terminus of the G protein and that corresponding replication-competent VSVs can be rescued efficiently. Overall, we demonstrated that functional tumor-targeting ligands can be displayed on replication-competent VSVs without perturbing viral growth and oncolytic efficacy. This study provides a rational foundation for the future development of fully retargeted oncolytic VSVs.
INTRODUCTION
Vesicular stomatitis virus (VSV) is an enveloped, negative-strand RNA virus that belongs to the Vesiculovirus genus of the Rhabdoviridae family. VSV has the ability to infect and kill cancer cells while sparing normal cells (1–4). Exploitation of this oncolytic property provides a promising alternative approach for the treatment of cancer. For disseminated cancer, virotherapy should ideally be administered systemically (2, 5–7), but this route of delivery brings its own set of problems. The major concerns for VSV virotherapy are neurotoxicity, antibody neutralization, and sequestration in off-target organs, especially the liver and spleen. Many attempts have been made to address these drawbacks. To reduce the neurotoxicity, the matrix protein of VSV was mutated (8, 9), and microRNA targets (10) or picornaviral internal ribosome entry sites (11) were engineered into the VSV genome. Serum neutralization has been avoided by PEGylating the virus (12, 13) or loading onto antigen-specific T cells (14), which ultimately improved virotherapy outcomes.
To circumvent all of the above hurdles in a single step, pseudotyping VSV with other viral envelope glycoproteins was considered a potentially feasible approach. Recent studies demonstrated that VSV neurotoxicity can be circumvented by pseudotyping with the surface glycoproteins of lymphocytic choriomeningitis virus (15) or measles virus (16). However, the reported viruses were replication incompetent and had substantially reduced titers compared to unmodified VSVs. For oncolytic applications, an ideal VSV should have the following characteristics: (i) it lacks neurotoxicity; (ii) it evades serum neutralization; (iii) it extravasates efficiently into tumor tissue; (iv) it targets only tumor tissue. To be an ideal oncolytic agent, VSV therefore has to be fully retargeted. As a first step to reach that goal, we attempted to display tumor-targeting ligands on a replication-competent VSV. Our approach was to identify novel sites in the G protein of VSV (VSV-G) to insert and display foreign peptides without compromising viral replication kinetics or oncolytic efficacy.
Several previous attempts to insert foreign peptides into VSV-G were conducted purely in the interest of lentivirus targeting and purification (17–20). Additionally, one previous study identified a site that could tolerate insertion of a 16-residue peptide that was an antigenic HIV epitope (21). However, there are no previous reports of ligand display on the G protein of a replication-competent VSV. In the current study, we successfully rescued recombinant vesicular stomatitis viruses displaying functional tumor-targeting ligands incorporated into their engineered G proteins. We selected cyclic RGD (cRGD), a 9-amino-acid integrin-binding peptide, to evaluate potential insertion sites that were identified by analysis of the VSV-G crystal structure. Integrins are cell surface glycoproteins that bind to extracellular matrix components and cell surface and soluble ligands and are involved in transmembrane cell signaling (22). Integrins also play a major role in tumor initiation, progression, and metastasis (23), which makes them attractive targets for cancer therapy. RGD binds to five αV integrins (αvβ1, αVβ3, αvβ5 αvβ6, and αvβ8), two β1 integrins (α5 and α8), and αIIbβ3 (24). RGD in its disulfide-bonded cyclic form (CDCRGDCFC) binds more strongly to integrins than does its linear form (25). RGD has been extensively studied in targeting and killing tumor cells (26). Early studies demonstrated that displaying the RGD peptide on the surface of oncolytic parvoviruses, adenoviruses, or measles viruses enhanced their interaction with tumor vasculature and/or tumor cells (19, 27, 28). cRGD also has been used to label gold nanoparticles to target tumor cells for destruction or imaging (29, 30). Thus, this peptide was considered a good candidate to explore possible insertion sites on the VSV glycoprotein and to target VSV. We also used echistatin, a 49-amino-acid snake venom disintegrin peptide that has very high affinity toward αVβ3, α5β1, and αIIbβ3 integrins (31) to explore the possibilities of rescuing replication-competent VSV. Previously, echistatin was displayed on oncolytic measles viruses that were subsequently shown to target tumor vasculature (32).
Based on the results of the above studies, we further examined the feasibility of displaying very large polypeptide-binding domains on VSV by fusing them to the extreme N terminus of the G protein. We successfully inserted single-chain antibody variable fragments (scFv) against tumor-specific receptors at this site and rescued antibody-displaying viruses that were still capable of normal receptor binding, infection, and propagation. The demonstration that it is possible to display a variety of tumor-targeting ligands on the VSV G protein may have important implications for the development of a fully retargeted oncolytic VSV platform.
MATERIALS AND METHODS
Cell culture and reagents.
BHK and MPC-11 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in 5% CO2. M36 mutant cells (33) were a generous gift from Felix Randow, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom, and were maintained in RPMI with 10% FBS and 50 μM 2-mercaptoethanol. K562-αvβ3 and K562-α5β3 cells were a kind gift from S. Blystone (Upstate Medical University, Syracuse, NY). K562 cells were maintained in Iscove's modified Dulbecco's medium (IMDM; Life Technologies, NY) supplemented with 10% fetal bovine serum, 0.5 U/liter penicillin-streptomycin, and 2 mM l-glutamine. K562-αvβ3 and K562-α5β3 cells were maintained in the same medium containing 500 μg/ml of G418 (Gibco). Anti-human integrin αvβ3 (monoclonal antibody [MAb] 1976) was purchased from Millipore (Billerica, MA). Monoclonal antibodies against the low-density lipoprotein receptor (LDLR; 6E2) were a kind gift from Ross Milne, Diabetes and Atherosclerosis Laboratory, University of Ottawa Heart Institute, Ottawa, Canada.
Construction and generation of ligand-displaying recombinant VSVs.
The VSV full-length plasmid pVSV-MC11, described previously (11), was modified, and a VSV mutant plasmid, pVSV-Δ51-MC11 (with methionine deleted at position 51 of the M protein) was created as described earlier (8). For safety concerns, this plasmid, pVSV-Δ51-MC11, was used as a backbone to make all the constructs mentioned in this study. Echistatin was obtained by PCR amplification of previously described measles virus plasmids (32). Echistatin and cRGD (CDCRGDCFC) sequences were inserted into appropriate places in the full-length VSV genome by overlapping PCR. Recombinant VSVs (rVSVs) were generated as previously described (11). Briefly, BHK cells were plated at a density of 1 × 106 cells/well in 6-well plates. The cells were infected with vaccinia virus encoding T7 polymerase at a multiplicity of infection (MOI) of 10. After 1 hour, vaccinia virus was removed, and the cells were transfected with 1 μg pVSV, 0.5 μg pN, 0.4 μg pP, and 0.2 μg pL by using 6 μl of Lipofectamine LTX transfection reagent (Life Technologies, NY) according to the manufacturer's instructions. The cells were incubated for 6 h at 37°C, and then the medium was replaced with DMEM with 5% FBS. After 48 h, culture medium was harvested, filtered twice through a 0.2-μm filter, and overlaid onto new BHK cells in a 6-well plate. Forty-eight hours later, the culture medium was harvested, subjected to low-speed centrifugation, and titrated on fresh BHK cells. When necessary, the recombinant viruses were further passaged to amplify the viral titer. Viral titers were determined by plaque assay as described previously (34). All the recombinant viruses were verified by sequence analysis.
Growth curve analysis.
Growth curve analysis was carried out as described earlier (11). For multistep growth curves, BHK cells were incubated with rVSV at an MOI of 0.01 for 1 h at 37°C. Following this incubation, supernatant was removed, the monolayer was washed, and fresh growth medium was added. Supernatant was collected at predetermined time points (6, 12, 18, 24, 30, and 36 h), and the virus titer was determined in a standard plaque assay.
Western blotting, immunofluorescence, and FACS analysis.
Western blotting was done as described earlier (11). To detect viral protein expression levels, VSV-infected cells were harvested at the indicated time points and incubated with 100 μl of RIPA buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) at 4°C for 10 min. Cell lysate was centrifuged, and the supernatant was collected and stored at −20°C until used. For immunoblotting, the proteins were electrophoretically separated in a 12% sodium dodecyl sulfate-polyacrylamide gel. VSV proteins were detected by Western blotting using polyclonal antibody against wild-type VSV, and echistatin was detected using polyclonal serum against echistatin peptide (Millipore, MA). Immunofluorescence microscopy was used to analyze and image green fluorescent protein (GFP)-expressing cells. We used ImageJ software (http://rsbweb.nih.gov/ij/) to quantify GFP-positive cells. The surface expression level of integrin was detected by fluorescence-activated cell sorting (FACS) analysis. M36 cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde. Cells were blocked with 2% horse serum for 20 min at room temperature and then incubated with anti-α5β1 antibody (Millipore, MA) for 1 h followed by secondary antibody for 1 h at room temperature. After a final washing with PBS, the cells were analyzed by flow cytometry using a FACScan system and CellQuest software (Becton, Dickinson).
Virus binding and quantitative RT-PCR.
Binding assays, using wild-type and echistatin-displaying viruses, were carried out in suspensions as described earlier (35, 36). Briefly, K562 cells were pelleted and washed with PBS three times. The final cell pellet was dissolved in PBS, and cells were counted and aliquoted into different tubes. Cells and viruses (MOI, 0.1) were incubated for 2 h at 4°C or for 30 min at 37°C. Cells were washed three times in cold PBS to remove unbound viruses. Then, cell pellets were stored at −80°C for further processing. The cells were lysed in 400 μl RLT buffer (Qiagen, CA), and total RNA was then extracted using the Qiagen RNeasy kit following the manufacturer's instructions. Quantitative two-step reverse transcription-PCR (RT-PCR) was performed according to the manufacturer's instructions (LightCycler 480 real-time PCR system; Roche Applied Science, IN). For reverse transcription, a random hexamer was used, followed by quantitative real-time PCR carried out using primers directed against the N gene. A standard curve was made using serial dilutions of the known virus genome. The copy numbers for the samples were calculated by using LightCycler 480 software.
In vivo experiments.
All animal protocols were reviewed and approved by the Mayo Clinic Institutional Care and Use Committee. BALB/c mice, females, 4 to 6 weeks old, were purchased from Jackson Laboratories. Mice were implanted with 5 × 106 mouse plasmacytoma (MPC-11) cells in the right flank. When tumors reached an average size of 0.2 to 0.5 cm3, mice were treated with a single intravenous injection of recombinant VSV. Tumor volume was measured using a hand-held caliper. The mice were monitored daily until the end of the study or when they reached the euthanasia criteria. The euthanasia criteria were as follows: clinical signs of neurotoxicity, tumor ulceration, tumor volume greater than 10% of body weight, weight loss greater than 10%, or mice were unable to gain access to food or water.
Immunohistochemistry.
Tumors harvested were frozen in optimal cutting medium (OCT) for sectioning. Tumor sections were analyzed by immunofluorescence for VSV antigens by using polyclonal rabbit anti-VSV antibody and for endothelial cells by using anti-CD31 antibody (AbD Serotec, NC) followed by Alexa-labeled anti-rabbit IgG secondary antibody (Life Technologies, NY), and cellular nuclei were stained with Hoescht 33342 (Life Technologies).
Statistical analyses.
An unpaired two-tailed Student t test was carried out to compare the values. Survival curve analysis was done using the Prism 4.0 program (GraphPad Software, San Diego, CA). Survival curves were plotted according to the Kaplan-Meier method, and survival function across treatment groups was compared using log rank test analyses.
RESULTS
Construction and rescue of VSVs displaying tumor-targeting peptides.
To identify possible sites within the VSV-G ectodomain that might tolerate insertion of a foreign peptide while allowing rescue of viable virus, we analyzed and compared amino acid sequences of various vesiculovirus G proteins. The crystal structure of the prefusion form of VSV-G was also used to predict feasible insertion sites. Three types of sites were identified as being more likely to tolerate insertion: highly variable regions, regions rich in hydrophilic residues, and loop regions. Regions showing high variability between homologous rhabdovirus G proteins were predicted to be more likely dispensable for protein stability and function. Foreign peptides inserted between hydrophilic residues were expected to be better exposed on the protein surface. Loop regions are usually flexible, and hence they are more likely to allow insertion of epitopes without disturbing protein folding and structure. To display the cyclic RGD peptide on VSV-G, we fused the CDCRGDCFC sequence in-frame into the G protein in a full-length viral genome carrying enhanced green fluorescent protein (EGFP) as a marker gene (Fig. 1A). The amino acid numbering used in this study was taken from that for the VSV-G crystal structure (37). Figure 1B to D shows the tested RGD insertion sites on the VSV G protein. Seven of the 13 sites allowed recovery of viable viruses (Fig. 2A) that could be efficiently amplified in BHK cells. Since our goal was to insert larger peptides into those sites, we focused on the four RGD insertion sites that gave rise to viruses that replicated with wild-type virus kinetics. We then replaced cRGD with the 49-amino-acid-long echistatin peptide in these four constructs. Among the four constructs, two of them yielded viable viruses: one with echistatin inserted at the N terminus of G next to the signal peptide, and the other one with an insertion between amino acids 351T and 352T (Fig. 2A).
Fig 1.

Ligand insertion sites on VSV-G. (A) Linear map of the VSV genome with GFP insertion. (B) Amino acid sequence of VSV-G (crystal structure), showing cRGD insertion sites. (C) Insertion site of ligands (bold letters) at the N terminus of the G protein. Only the signal peptide (italics) and a few residues of G are shown (underlined). *, an additional lysine residue was included to maintain the proper G sequence. (D) Crystal structure of the VSV G protein, showing cyclic RGD insertion sites. The Pymol program was used to edit the G protein crystal structure (PDB 2J6J).
Fig 2.

Virus recovery and analysis of growth properties. (A) Various VSV constructs with ligand insertion sites and the virus recovery results. (B) Western immunoblotting results, showing G and N/P proteins of purified recombinant VSVs that were probed with anti-VSV polyclonal antibody (upper panel) or antiechistatin polyclonal antibody (lower panel). (C) Multistep growth curve analysis of the indicated viruses (MOI, 0.01).
Wild-type and ligand-displaying viruses were analyzed by immunoblotting for viral structural proteins (Fig. 2B). Very slight variations were noticed in the mobility of the G protein among the RGD-displaying viruses. In the case of echistatin display, the G protein of the VSV Echi1 virus had significantly faster mobility than that of VSV Echi9. Sequence analysis of the G genes of all the rescued viruses showed intact ligand sequences. We remain uncertain as to what caused this mobility difference between the G proteins of the two echistatin-displaying viruses. Since echistatin in the VSV Echi1 virus is immediately adjacent to the signal peptide, there may be cryptic signal peptidase cleavage signal in the echistatin peptide. Alternatively, there may be a difference in the glycosylation status of the two chimeric G proteins. When the membrane was probed with antiechistatin polyclonal antibody, both Echi1 and Echi9 virus glycoproteins were efficiently recognized (Fig. 2B). Multistep growth curve analysis showed that all of the recombinant viruses had replication kinetics similar to the parental virus (Fig. 2C), although some of them were slightly inferior. Since VSV Echi9 displays an intact echistatin peptide and grows better than VSV Echi1 virus, we used VSV Echi9 virus for most of our subsequent studies.
Virus neutralization assays.
Virus neutralization assays were performed to determine whether antibodies against echistatin are able to neutralize the infectivity of echistatin-displaying VSV. Recombinant VSVs (103 PFU) were incubated with serial dilutions of either antiechistatin or anti-VSV serum before plating onto BHK cells. VSV displaying echistatin was effectively neutralized by antiechistatin serum, which had no effect on wild-type virus infectivity (Fig. 3A). This suggested that the echistatin peptide is exposed on the surface of the virion. On the other hand, anti-VSV serum neutralized both the viruses (Fig. 3B).
Fig 3.
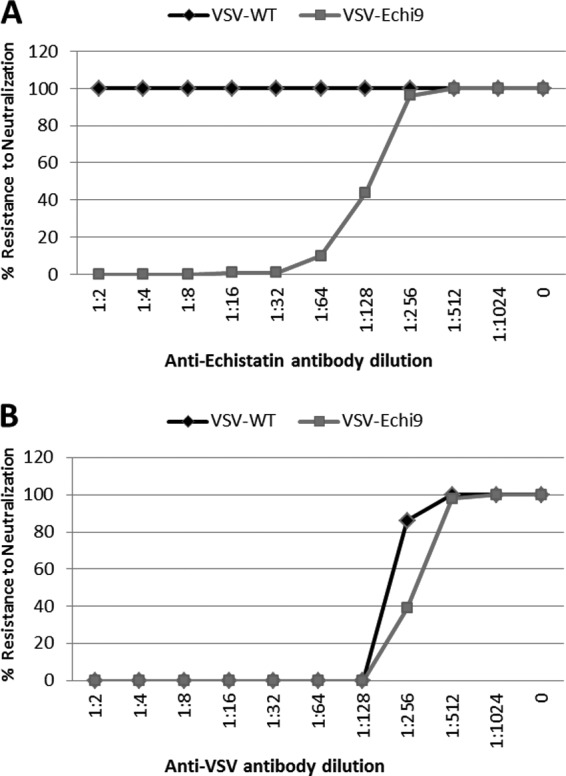
Neutralization assay results. A total of 1,000 PFU of VSV-WT or VSV-Echi9 were incubated with 2-fold serial dilutions of antibody against echistatin peptide (A) or wild-type VSV (B) for 1 h at 37°C before addition to BHK cells. After 30 min, the monolayer was washed twice with PBS and overlaid with methyl cellulose. Plaques were counted 48 h postinfection. Results are expressed as the percentage of plaques relative to that obtained with no antibody.
Solid-phase receptor-binding assay.
To determine whether echistatin displaying VSV can bind to its specific receptor, we coated microtiter wells with purified integrins. The viruses were allowed to bind to the immobilized integrins, and after 1 h of incubation unbound viruses were washed away with PBS. The bound viruses were rescued by addition of MPC-11 cells to the microtiter wells. The VSV-Echi9 virus bound significantly better to integrin αvβ3 than to the control integrin, α1β1 (Fig. 4). This study showed that a ligand-displaying VSV has the ability to bind to its targeted receptor thorough the displayed ligand and that the binding is highly specific.
Fig 4.

Solid-phase receptor-binding assay. (A) Binding of unmodified (VSV-WT) or echistatin-displaying VSV (VSV-Echi9) to PBS (A) or α1β1 (B) or αvβ3 (C) integrins. Microtiter wells were coated (0.2 μg/ml) with PBS, α1β1, or αvβ3 and incubated with either VSV-WT or VSV-Echi9 (103 PFU). After 1 h of incubation, wells were washed three times with PBS, and MPC-11 cells were added to rescue bound viruses. (D) GFP-positive cells were counted using the ImageJ program. Results are expressed as means ± standard errors of triplicate wells. Significantly higher binding was noted in αvβ3-coated wells with VSV-Echi9 (*, P = 0.0001) than to WT VSV.
Infection of GP96 mutant (VSV receptor-negative) cells by ligand-displaying viruses.
In a recent study, Bloor and coworkers (33) demonstrated that cells without the endoplasmic reticulum chaperone GP96 or with a catalytically inactive gp96 do not bind VSV-G. When the GP96 mutant M36 cells were infected with unmodified and recombinant VSVs, the ligand-displaying viruses (especially VSV-Echi9 and VSV-RGD9) showed significantly higher infection than the unmodified virus (Fig. 5). Since M36 cells do not express αvβ3 and αIIβ3 integrins (38, 39), we postulated that the ligand-displaying viruses might enter through α5β1. We therefore analyzed M36 cells by flow cytometry, and we confirmed low-level expression of α5β1 (Fig. 5C), consistent with the hypothesis that VSV-Echi9 and VSV-RGD9 viruses bind the cells through this receptor. However, infectivity of the ligand-displaying viruses with different insertion sites was not significantly higher than with unmodified VSV. This observation warrants further detailed study to determine whether the precise positioning of the displayed ligand plays a role in receptor affinity.
Fig 5.

Infectivity of ligand-displaying VSVs in GP96 mutant cells. The experiment was performed in triplicate with M36 cells at an MOI of 1 or 10 and analyzed at 16 h postinfection. (A) GFP expression of infected cells at an MOI of 10.0. (B) GFP-positive cells were counted by using the ImageJ program. (C) Flow cytometry analysis of M36 cells for α5β1expression. Significantly higher infection rates were found for VSV-Echi9 and VSV-RGD9 (*, P < 0.01) than for WT VSV.
Specific binding of echistatin displaying VSV to cells overexpressing targeted integrins.
We next investigated whether the virally displayed echistatin domain could mediate specific viral binding to cells overexpressing the targeted integrin, αvβ3. For this, we incubated K562 (human erythroleukemia) cells with VSV wild type (WT) or VSV-Echi9 for 30 min at 37°C and also at 4°C for 2 h. After washing, the bound virus particles were subjected to RNA extraction and quantitative RT-PCR. Analysis showed that the affinities of unmodified and echistatin virus toward K562 wild-type cells were almost the same (Fig. 6A). On K562-αvβ5 cells, the affinity of VSV-Echi9 was slightly higher than that of unmodified VSV (Fig. 6B). At the same time, the affinity of VSV-Echi9 to the K562-αvβ3 cell surface ligand increased significantly compared to unmodified virus (Fig. 6C). These results showed that VSV displaying echistatin has the ability to bind to is specific receptor, in this case αvβ3, with high affinity.
Fig 6.
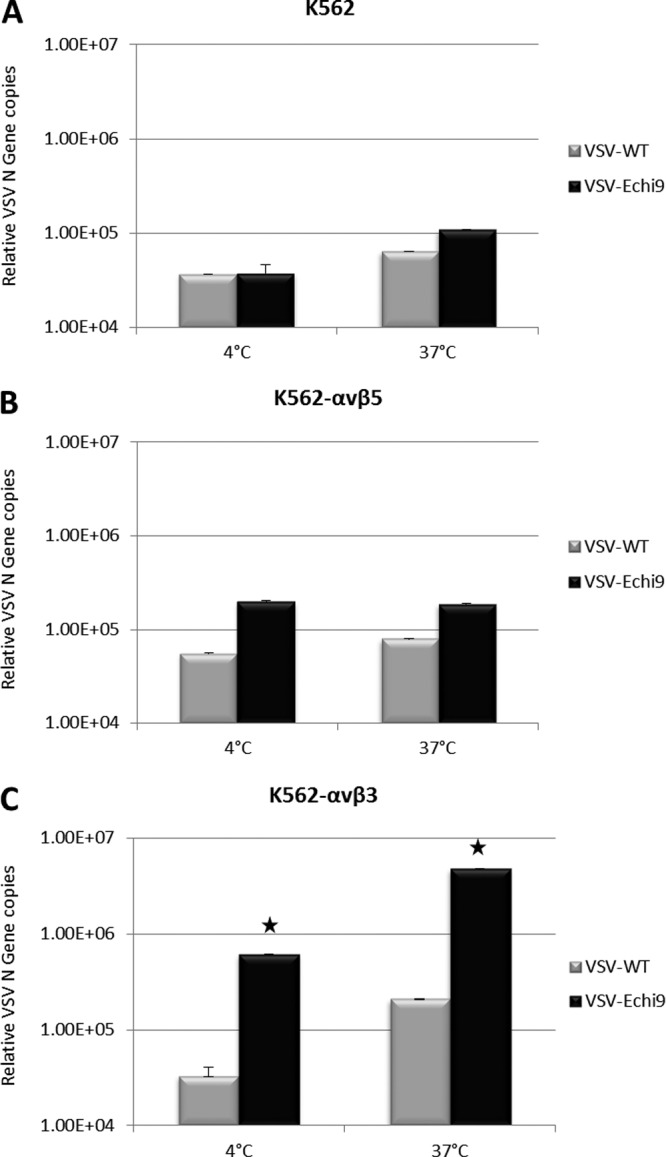
Affinity of echistatin-displaying VSV toward an integrin overexpressing cell line. Wild-type (A), αvβ3-overexpressing (B), or αvβ5-overexpressing (C) K562 cells were incubated with WT VSV or VSV-Echi9 viruses at an MOI of 0.1. After 2 h (4°C) or 30 min (37°C) of incubation, cells were washed twice with PBS, and total RNA was extracted as described in Materials and Methods. Quantitative analysis of N gene copies was carried out using quantitative RT-PCR. Error bars represent the standard errors of the means. Where error bars are not visible, the standard error was negligible. Significantly higher affinity was found with VSV-Echi9 (*, P < 0.01) compared to WT VSV.
Infection via the targeted receptors in vitro.
We next investigated the entry of ligand-displaying VSVs into human cell lines expressing integrins after masking one of the major VSV receptors (40). To this end, we blocked the LDLR with a specific monoclonal antibody against human LDLR (40, 41). K562-avβ3 cells were incubated with equal amounts of anti-LDLR monoclonal antibody for 1 hour at 37°C. Then, VSV (103 PFU) was added, and the mixture was incubated for 30 min at 37°C, cells were washed twice with PBS, growth medium was added, and 16 h later infected cells were analyzed for GFP expression to calculate the virus titer (Fig. 7A to C). Since the wild type has a minor growth advantage over the ligand-displaying viruses, it produces a slightly higher titer in antibody-untreated cells than other viruses (Fig. 7B). Conversely, in anti-LDLR antibody-treated cells, the infectivity of wild-type VSV was significantly blocked, whereas the ligand-displaying viruses (especially VSV-Echi9 and VSV-RGD3) retained their infectivities.
Fig 7.

Receptor specificity of targeted viruses. K562-αvβ3 cells were treated with anti-LDLR and/or anti-αvβ3 monoclonal antibody for 1 h before addition of VSV. After 1 hour, cells were washed and incubated for 16 h before measuring virus titers and imaging. (A) Infectivity in untreated (left panels) and in anti-LDLR-treated K562-αvβ3 cells (right panels). (B and C) Titers of viruses from untreated cells (B) or anti-LDLR-treated K562-αvβ3 cells (C). Significantly higher infection rates were found with VSV-Echi9 and VSV-RGD3 (*, P < 0.01 compared to WT VSV). (D) Infectivity of WT VSV and VSV-Echi9 viruses in K562-αvβ3 cells treated with anti-LDLR and anti-αvβ3 antibodies. (E) Titers of viruses from panel D (*, P = 0.0105, compared to the titer of VSV-Echi9 from K562-αvβ3 cells treated only with anti-LDLR).
To further confirm that the ligand-displaying VSVs infect via an integrin receptor, we treated the K562-avβ3 cells with a functional blocking anti-avβ3 monoclonal antibody in addition to the anti-LDLR antibody. Individually, neither antibody significantly inhibited the infectivity of the VSV-Echi9 virus, but in combination the antibodies could inhibit infectivity. Conversely, the infectivity of the wild-type virus was efficiently blocked by the LDLR antibody but not by the anti-avβ3 antibody (Fig. 7D and E). This experiment strongly suggests that echistatin-displaying VSV can enter cells via the targeted integrin receptor, even when the LDL receptor is effectively masked.
Oncolytic activity of targeted viruses in vivo after intravenous administration.
We next evaluated the oncolytic activities of the recombinant VSVs in mice with established syngeneic plasmacytomas (MPC-11). Compared to unmodified VSV, the RGD- and echistatin-displaying viruses showed similar therapeutic potencies. No significant difference in oncolytic efficacy was observed (Fig. 8). Interestingly, the Echi-1, RGD-3, and RGD-7 viruses cured all treated mice, whereas a single relapse was seen in groups of five mice treated with WT, Echi9, or RGD-9 viruses, and two relapses were found in the group treated with RGD-1 virus. One tumor spontaneously subsided in the untreated control group. In a separate study, tumors were harvested 24 h post-intravenous administration of viruses for immunohistochemical analysis (Fig. 9A). Since tumor blood vessels express high levels of integrins, the RGD and echistatin viruses were expected to infect the blood vessels. Dual staining with anti-mouse CD31 and anti-VSV polyclonal antibody was therefore performed to identify endothelial cells of tumor blood vessels and VSV infection, respectively. The staining results showed no difference between unmodified and ligand-displaying VSVs. Figure 9B shows uninfected tumor blood vessels surrounded by VSV-infected cells.
Fig 8.

Oncolytic efficacies of ligand-displaying viruses. (A) Mice (BALB/c, 4-weeks old, n = 5) bearing subcutaneous MPC-11 tumors were treated with a single intravenous dose (5 × 106 PFU) of saline or WT VSV or RGD- or echistatin-displaying VSV. Tumor size was measured by serial caliper measurements. (B) Kaplan-Meier survival curves for the mice for which data are shown in panel A. No significant differences were noted between WT VSV and the other ligand-displaying viruses.
Fig 9.

Analysis of virus spread in myeloma tumors in vivo. MPC-11 tumor-bearing mice were injected with a single intravenous dose (5 × 106 PFU) of the indicated virus. Tumors were harvested and sectioned at 24 h posttreatment and analyzed for VSV antigen (red) and cell nuclei (Hoescht; blue). Magnification, ×4. (B) Tumor sections were stained for VSV antigen (green) and endothelial cell marker CD31 (red). Tumor blood vessels appeared to be uninfected.
Rescue of recombinant VSVs displaying single-chain antibodies.
To determine whether larger polypeptide ligands could be displayed on VSV as G protein fusions, we fused single-chain fragment variables (scFv) against epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) at the N terminus of the glycoprotein (Fig. 10A). Even though initial virus recovery was slow, multiple passages (∼5) yielded high titers of viruses. Sequencing results showed that the nucleotide sequence of the displayed scFv domains was fully intact in the passaged viruses, with a few nucleotide changes in the underlying G protein, which may have been selected to allow better accommodation of the larger peptides (data not shown). VSVs displaying scFvs achieved lower maximum titers (by 1 to 2 orders of magnitude) than unmodified VSV on BHK cells (Fig. 10B). Western blotting showed that the EGFR-scFv fusion was partially cleaved from the G protein, whereas the Her2-scFv-G protein fusion remained fully intact with no cleavage (Fig. 10C). Since VSV can adapt during replication in cell culture, additional passages may further increase its replication potential; also, codon optimization of the insert may be necessary to obtain high-replicating viruses.
Fig 10.

Rescue of scFv-displaying VSVs. (A) Schematic of a VSV genomic construct with an N-terminal scFv. (B) Multistep growth curve of scFv-displaying VSVs. (C) Western blot analysis of purified VSV-scFv proteins probed with anti-VSV polyclonal antibody.
DISCUSSION
Vesicular stomatitis virus is a rhabdovirus with inherent oncolytic properties that effectively kill cancer cells while sparing normal cells (42). A phase I clinical trial was recently initiated at Mayo Clinic in which VSV is being given as an experimental intratumoral therapy to patients with hepatocellular carcinoma. However, for future trials of intravenous VSV therapy, it will be important to engineer the virus for greater safety and efficacy. To achieve this goal, it will be important to develop oncolytic VSVs that are highly tumor specific, free of neurotoxicity, and that can clear all the hurdles (neutralization, sequestration, and lack of extravasation) of systemic administration (7). Toward this goal, and with a view to retargeting the virus, we sought to display tumor-targeting ligands on the surface of the virion. Our data demonstrated that it is feasible to insert peptides as large as single-chain antibody fragments into the G protein and rescue replication-competent VSVs that display the inserted domains on their surface.
Previously, it was shown that peptides up to 16 amino acids in length could be inserted into a permissive site on the VSV G protein of a replication-competent VSV (21). To our knowledge, no other successful attempt to display a ligand on a replication-competent VSV has been reported. Since our laboratory has had some success targeting other virus types for the purpose of oncolytic virotherapy (32, 43, 44), we decided to explore the feasibility of targeting VSV by displaying tumor-targeting ligands on its G protein.
We initially selected the integrin-binding cRGD peptide to explore feasible insertion sites in the VSV G protein, because of its smaller size and ability to target tumor blood vessels. The RGD motif has been displayed previously on the surfaces of many viruses (measles virus [45], parvovirus [27], adenovirus [46–48], and adeno-associated virus [49]) with the goal of enhancing their oncolytic specificities and/or transduction efficiencies. In selecting potential insertion sites for the VSV G protein, we relied heavily on the crystal structure of the protein and sequence comparisons with the G proteins of various related vesiculoviruses. Based on that information, we identified 13 potential sites in VSV-G, 7 of which could tolerate the 9-amino-acid-long cRGD insertion, which can be displayed on the virion surface. Among these, two of the sites yielded viable viruses when the sequence encoding the 49-amino-acid-long echistatin polypeptide domain was inserted (Fig. 2A). The recovery and efficient replication of the Echi9 virus was really very interesting. The Echi9 site between amino acids 351 and 352 is located in the middle of the lateral domain of VSV-G. The fact that G could tolerate insertion of a functional domain within this domain, which is not only a structural but also a functional unit, was quite surprising. VSV-G is a multidomain protein comprising at least four major domains. Most multidomain proteins are formed by a linear sequence of independently folding domains that are secured by end-to-end linkages (50). VSV-G is an exception to this pattern, as it exhibits discontinuity in its domain arrangement that allows insertion of a fully functional domain within a domain without affecting its protein folding and structure. This study suggested that one can insert any reasonable size of functional domain into this site in VSV-G, and probably into a corresponding site in any rhabdovirus G protein, as long as the insert does not interfere with G protein folding and maturation.
Display of the echistatin peptide on the virion surface was confirmed in a neutralization assay (Fig. 3). At the same time, specific binding of the echistatin-displaying virus to αvβ3 integrin in a solid-phase capture assay demonstrated that VSV can bind to a targeted receptor thorough a polypeptide ligand displayed on its surface (Fig. 4). When the recombinant viruses were incubated with wild-type and integrin-overexpressing K562 cells, VSV-Echi9 bound with considerably higher affinity to K562-αvβ3 cells than wild-type virus (Fig. 6). These data further confirmed the higher affinity of echistatin-displaying VSV to its targeted receptor.
A recent publication proposed and demonstrated that the LDLR and its family members serve as natural receptors for vesicular stomatitis virus (40). Therefore, to determine whether VSV can enter cells via a targeted receptor recognized by a displayed ligand, we used an anti-LDLR monoclonal antibody to mask the LDLR on K562 cells that overexpress αvβ3 integrin (K562-αvβ3) (41). Figure 7 shows that wild-type virus infection was efficiently blocked by the LDLR monoclonal antibody, whereas the ligand-displaying viruses, especially VSV-Echi9, showed significantly higher infection levels. Surprisingly, VSV-RGD3 also showed significant infection. This study suggested that VSV can enter cells through its displayed ligand. This conclusion was further confirmed by our demonstration that infection by the echistatin-displaying VSV was significantly blocked when the cells were treated with monoclonal antibodies against both the LDLR and αvβ3 integrin.
The oncolytic potencies of the ligand-displaying viruses were tested in a mouse myeloma model. Immunocompetent BALB/c mice bearing syngeneic subcutaneous mouse plasmactyomas were treated with a single intravenous dose of VSV. There was no significant difference in tumor response or survival between wild-type and recombinant viruses (Fig. 8). Since cRGD and echistatin are theoretically capable of targeting the tumor vasculature, we were curious to see whether tumor blood vessels were infected with VSV. However, dual staining (anti-CD31 and anti-VSV) of tumor sections did not yield any conclusive results (Fig. 9B). Recently, it was shown in a different tumor model that VSV can infect tumor vasculature directly (51), but this was not evident in our model. We did observe VSV infection immediately adjacent to the intact tumor blood vessels, but not in the lumen, as was observed in a previous study from our laboratory that used a different syngeneic myeloma model (2). Whether the displayed RGD or echistatin peptides can target VSV attachment to endothelial cells is not clear. One possible scenario is that the displayed peptides may mediate initial binding of VSV to tumor endothelial cells, which express high levels of αvβ3 integrin and thereby facilitate extravasation of the virus into the tumor parenchyma without directly infecting the vascular endothelial cells. The 100% cure of MPC-11 tumors by some but not all of the targeted viruses (Echi1, RGD3, and RGD7) may give some support to this idea, but more detailed studies are needed to confirm this impression.
We further tested the ability of the VSV G protein to tolerate large polypeptide insertions by fusing single-chain antibody variable fragments (scFv) to its N terminus and rescuing replication-competent VSV. Previous studies from our laboratory demonstrated that measles virus displaying single-chain antibodies against various cancer antigens was efficiently targeted to tumors expressing the respective cognate receptors (44). We therefore fused single-chain antibodies against EGFR and HER2 to the N terminus of the G protein (Fig. 10) and successfully rescued viruses encoding the chimeric proteins. Western blot analysis showed that there was partial cleavage of the EGFR scFv from the chimeric G protein (Fig. 10), which may have been due to an improperly folding chimera, which was not apparent in the case of the HER2 antibody-displaying virus. While scFv-displaying VSVs grow to a maximum titer that is 1 or 2 logs lower than that with the unmodified virus, insertion of a large domain at this site is very appealing. Further optimization of the system may well yield viruses with high replication potentials. However, rescue of replication-competent VSVs displaying intact scFvs marks a necessary milestone in vesiculovirus targeting.
Vesicular stomatitis virus has recently made the transition into the clinic from the laboratory. Although safety is of prime importance for its use in clinical therapy, efficacy is also a key factor for successful curative therapy and should not be compromised. In the MPC-11 myeloma model, VSV has the ability to infect and kill most of the cancer cells before the virus gets eliminated by the immune system. Many replication-competent viruses have been attenuated to the point of compromising their ability to fight innate immunity. Since even attenuated VSVs retain some neurotoxic potential when administered as systemic therapy, there is a strong rationale to modify their tropism to allow effective binding and entry via tumor-specific receptors without compromising the oncolytic potency. To this end, we have successfully displayed tumor-targeting ligands on the surface of the G protein and have demonstrated that the modified viruses have oncolytic potencies equivalent to those of unmodified virus. This is a proof-of-concept study that has demonstrated the feasibility of display of tumor-targeting ligands on replication-competent VSVs, and our results suggest that full retargeting will indeed be possible if the natural receptor tropism can be ablated. Ligand-displaying VSV G proteins might also be of interest for retargeting of lentiviral vectors.
A major receptor for VSV (the LDLR family) was proposed recently, but there are many questions yet to be answered. How many receptors really exist for VSV? Does VSV also use carbohydrates or lipids for attachment? How does VSV interact with its individual receptors when more than one is available? What is the role of receptor density and of steric hindrance for access to competing receptors? It is also unclear whether VSV uses the same receptor(s) on all cell types or uses a different receptor(s) on each cell line. Is there any interspecies variation in the nature or binding affinity of the receptor? Does the absence of one receptor alter the binding affinity of VSV toward another receptor? There are still a great many unanswered questions about the VSV receptor, and this is an important area of study. We anticipate that ligand display on the VSV G protein may open the door for finding answers to some of these questions. In addition, besides contributing to the understanding of VSV entry, this study may lead to the development of fully targeted oncolytic vesicular stomatitis virus constructs in the near future.
ACKNOWLEDGMENTS
This work was supported by funds from the NIH (grant R01 CA 129966-01-A1-05) and a gift from Alvin E. and Mary A. McQuinn.
We thank Rebecca Nace, Mayo Clinic, for mouse work, S. Blystone, Upstate Medical University, Syracuse, NY, for providing K562-αvβ3 and K562-αvβ5 cells, Felix Randow, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom, for providing M36 cells, and Ross Milne, Diabetes and Atherosclerosis Laboratory, University of Ottawa Heart Institute, Ottawa, Canada, for providing the anti-LDLR monoclonal antibody.
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Alajez NM, Mocanu JD, Shi W, Chia MC, Breitbach CJ, Hui AB, Knowles S, Bell JC, Busson P, Takada K, Lo KW, O'Sullivan B, Gullane P, Liu FF. 2008. Efficacy of systemically administered mutant vesicular stomatitis virus (VSVΔ51) combined with radiation for nasopharyngeal carcinoma. Clin. Cancer Res. 14:4891–4897 [DOI] [PubMed] [Google Scholar]
- 2.Naik S, Nace R, Federspiel MJ, Barber GN, Peng KW, Russell SJ. 2012. Curative one-shot systemic virotherapy in murine myeloma. Leukemia 26:1870–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shinozaki K, Ebert O, Woo SL. 2005. Treatment of multi-focal colorectal carcinoma metastatic to the liver of immune-competent and syngeneic rats by hepatic artery infusion of oncolytic vesicular stomatitis virus. Int. J. Cancer 114:659–664 [DOI] [PubMed] [Google Scholar]
- 4.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. 2000. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 6:821–825 [DOI] [PubMed] [Google Scholar]
- 5.Au GG, Lindberg AM, Barry RD, Shafren DR. 2005. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int. J. Oncol. 26:1471–1476 [DOI] [PubMed] [Google Scholar]
- 6.Peng KW, Facteau S, Wegman T, O'Kane D, Russell SJ. 2002. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat. Med. 8:527–531 [DOI] [PubMed] [Google Scholar]
- 7.Russell SJ, Peng KW, Bell JC. 2012. Oncolytic virotherapy. Nat. Biotechnol. 30:658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Publicover J, Ramsburg E, Robek M, Rose JK. 2006. Rapid pathogenesis induced by a vesicular stomatitis virus matrix protein mutant: viral pathogenesis is linked to induction of tumor necrosis factor alpha. J. Virol. 80:7028–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275 [DOI] [PubMed] [Google Scholar]
- 10.Kelly EJ, Nace R, Barber GN, Russell SJ. 2010. Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J. Virol. 84:1550–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ammayappan A, Nace R, Peng KW, Russell SJ. 2013. Neuroattenuation of vesicular stomatitis virus through picornaviral internal ribosome entry sites. J. Virol. 87:3217–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Croyle MA, Callahan SM, Auricchio A, Schumer G, Linse KD, Wilson JM, Brunner LJ, Kobinger GP. 2004. PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J. Virol. 78:912–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tesfay MZ, Kirk AC, Hadac EM, Griesmann GE, Federspiel MJ, Barber GN, Henry SM, Peng KW, Russell SJ. 2013. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J. Virol. 87:3752–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiao J, Wang H, Kottke T, Diaz RM, Willmon C, Hudacek A, Thompson J, Parato K, Bell J, Naik J, Chester J, Selby P, Harrington K, Melcher A, Vile RG. 2008. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 15:604–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muik A, Kneiske I, Werbizki M, Wilflingseder D, Giroglou T, Ebert O, Kraft A, Dietrich U, Zimmer G, Momma S, von Laer D. 2011. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J. Virol. 85:5679–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayala-Breton C, Barber GN, Russell SJ, Peng KW. 2012. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum. Gene Ther. 23:484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dreja H, Piechaczyk M. 2006. The effects of N-terminal insertion into VSV-G of an scFv peptide. Virol. J. 3:69. 10.1186/1754-422X-3-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guibinga GH, Hall FL, Gordon EM, Ruoslahti E, Friedmann T. 2004. Ligand-modified vesicular stomatitis virus glycoprotein displays a temperature-sensitive intracellular trafficking and virus assembly phenotype. Mol. Ther. 9:76–84 [DOI] [PubMed] [Google Scholar]
- 19.Padmashali RM, Andreadis ST. 2011. Engineering fibrinogen-binding VSV-G envelope for spatially- and cell-controlled lentivirus delivery through fibrin hydrogels. Biomaterials 32:3330–3339 [DOI] [PubMed] [Google Scholar]
- 20.Yu JH, Schaffer DV. 2006. Selection of novel vesicular stomatitis virus glycoprotein variants from a peptide insertion library for enhanced purification of retroviral and lentiviral vectors. J. Virol. 80:3285–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlehuber LD, Rose JK. 2004. Prediction and identification of a permissive epitope insertion site in the vesicular stomatitis virus glycoprotein. J. Virol. 78:5079–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takada Y, Ye X, Simon S. 2007. The integrins. Genome Biol. 8:215. 10.1186/gb-2007-8-5-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desgrosellier JS, Cheresh DA. 2010. Integrins in cancer: biological implications and therapeutic opportunities. Nat. Rev. Cancer 10:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humphries JD, Byron A, Humphries MJ. 2006. Integrin ligands at a glance. J. Cell Sci. 119:3901–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assa-Munt N, Jia X, Laakkonen P, Ruoslahti E. 2001. Solution structures and integrin binding activities of an RGD peptide with two isomers. Biochemistry 40:2373–2378 [DOI] [PubMed] [Google Scholar]
- 26.Danhier F, Le Breton A, Preat V. 2012. RGD-based strategies to target αvβ3 integrin in cancer therapy and diagnosis. Mol. Pharm. 9:2961–2973 [DOI] [PubMed] [Google Scholar]
- 27.Allaume X, El-Andaloussi N, Leuchs B, Bonifati S, Kulkarni A, Marttila T, Kaufmann JK, Nettelbeck DM, Kleinschmidt J, Rommelaere J, Marchini A. 2012. Retargeting of rat parvovirus H-1PV to cancer cells through genetic engineering of the viral capsid. J. Virol. 86:3452–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Smith T, Idamakanti N, Mulgrew K, Kaloss M, Kylefjord H, Ryan PC, Kaleko M, Stevenson SC. 2002. Targeting adenoviral vectors by using the extracellular domain of the coxsackie-adenovirus receptor: improved potency via trimerization. J. Virol. 76:1892–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arosio D, Manzoni L, Araldi EMV, Scolastico C. 2011. Cyclic RGD functionalized gold nanoparticles for tumor targeting. Bioconjug. Chem. 22:664–672 [DOI] [PubMed] [Google Scholar]
- 30.Kim YH, Jeon J, Hong SH, Rhim WK, Lee YS, Youn H, Chung JK, Lee MC, Lee DS, Kang KW, Nam JM. 2011. Tumor targeting and imaging using cyclic RGD-PEGylated gold nanoparticle probes with directly conjugated iodine-125. Small 7:2052–2060 [DOI] [PubMed] [Google Scholar]
- 31.Kumar CC, Nie HM, Rogers CP, Malkowski M, Maxwell E, Catino JJ, Armstrong L. 1997. Biochemical characterization of the binding of echistatin to integrin αvβ3 receptor. J. Pharmacol. Exp. Ther. 283:843–853 [PubMed] [Google Scholar]
- 32.Hallak LK, Merchan JR, Storgard CM, Loftus JC, Russell SJ. 2005. Targeted measles virus vector displaying echistatin infects endothelial cells via alpha v beta 3 and leads to tumor regression. Cancer Res. 65:5292–5300 [DOI] [PubMed] [Google Scholar]
- 33.Bloor S, Maelfait J, Krumbach R, Beyaert R, Randow F. 2010. Endoplasmic reticulum chaperone gp96 is essential for infection with vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 107:6970–6975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts A, Kretzschmar E, Perkins AS, Forman J, Price R, Buonocore L, Kawaoka Y, Rose JK. 1998. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J. Virol. 72:4704–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnberg N, Pring-Akerblom P, Wadell G. 2002. Adenovirus type 37 uses sialic acid as a cellular receptor on Chang C cells. J. Virol. 76:8834–8841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jonsson N, Gullberg M, Israelsson S, Lindberg AM. 2009. A rapid and efficient method for studies of virus interaction at the host cell surface using enteroviruses and real-time PCR. Virol. J. 6:217. 10.1186/1743-422X-6-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848 [DOI] [PubMed] [Google Scholar]
- 38.Staron M, Yang Y, Liu B, Li J, Shen Y, Zuniga-Pflucker JC, Aguila HL, Goldschneider I, Li Z. 2010. gp96, an endoplasmic reticulum master chaperone for integrins and Toll-like receptors, selectively regulates early T and B lymphopoiesis. Blood 115:2380–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weekes MP, Antrobus R, Talbot S, Hor S, Simecek N, Smith DL, Bloor S, Randow F, Lehner PJ. 2012. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J. Proteome Res. 11:1475–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. 2013. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 110:7306–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen AT, Hirama T, Chauhan V, Mackenzie R, Milne R. 2006. Binding characteristics of a panel of monoclonal antibodies against the ligand binding domain of the human LDLr. J. Lipid Res. 47:1399–1405 [DOI] [PubMed] [Google Scholar]
- 42.Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H, Bell JC, McCart JA, Mahoney D, Stojdl DF. 2010. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol. Ther. 18:1440–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly EJ, Hadac EM, Greiner S, Russell SJ. 2008. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 14:1278–1283 [DOI] [PubMed] [Google Scholar]
- 44.Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, Russell SJ. 2005. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 23:209–214 [DOI] [PubMed] [Google Scholar]
- 45.Ong HT, Trejo TR, Pham LD, Oberg AL, Russell SJ, Peng KW. 2009. Intravascularly administered RGD-displaying measles viruses bind to and infect neovessel endothelial cells in vivo. Mol. Ther. 17:1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murugesan SR, Akiyama M, Einfeld DA, Wickham TJ, King CR. 2007. Experimental treatment of ovarian cancers by adenovirus vectors combining receptor targeting and selective expression of tumor necrosis factor. Int. J. Oncol. 31:813–822 [PubMed] [Google Scholar]
- 47.Pesonen S, Diaconu I, Cerullo V, Escutenaire S, Raki M, Kangasniemi L, Nokisalmi P, Dotti G, Guse K, Laasonen L, Partanen K, Karli E, Haavisto E, Oksanen M, Karioja-Kallio A, Hannuksela P, Holm SL, Kauppinen S, Joensuu T, Kanerva A, Hemminki A. 2012. Integrin targeted oncolytic adenoviruses Ad5-D24-RGD and Ad5-RGD-D24-GMCSF for treatment of patients with advanced chemotherapy refractory solid tumors. Int. J. Cancer 130:1937–1947 [DOI] [PubMed] [Google Scholar]
- 48.Wesseling JG, Bosma PJ, Krasnykh V, Kashentseva EA, Blackwell JL, Reynolds PN, Li H, Parameshwar M, Vickers SM, Jaffee EM, Huibregtse K, Curiel DT, Dmitriev I. 2001. Improved gene transfer efficiency to primary and established human pancreatic carcinoma target cells via epidermal growth factor receptor and integrin-targeted adenoviral vectors. Gene Ther. 8:969–976 [DOI] [PubMed] [Google Scholar]
- 49.Shi XQ, Fang GG, Shi WF, Bartlett JS. 2006. Insertional mutagenesis at positions 520 and 584 of adeno-associated virus type 2 (AAV2) capsid gene and generation of AAV2 vectors with eliminated heparin-binding ability and introduced novel tropism. Hum. Gene Ther. 17:353–361 [DOI] [PubMed] [Google Scholar]
- 50.Aroul-Selvam R, Hubbard T, Sasidharan R. 2004. Domain insertions in protein structures. J. Mol. Biol. 338:633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, Sun YY, Roy DG, Rintoul JL, Daneshmand M, Parato K, Stanford MM, Lichty BD, Fenster A, Kirn D, Atkins H, Bell JC. 2011. Targeting tumor vasculature with an oncolytic virus. Mol. Ther. 19:886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]