

Abstract
The reovirus outer capsid protein μ1 forms a lattice surrounding the viral core. In the native state, μ1 determines the environmental stability of the viral capsid. Additionally, during cell entry, μ1 undergoes structural rearrangements that facilitate delivery of the viral cores across the membrane. To determine how the capsid-stabilizing functions of μ1 impinge on the capacity of μ1 to undergo conformational changes required for cell entry, we characterized viruses with mutations engineered at charged residues within the μ1 loop formed by residues 72 to 96 (72-96 loop). This loop is proposed to stabilize the capsid by mediating interactions between neighboring μ1 trimers and between trimers and the core. We found that mutations at Glu89 (E89) within this loop produced viruses with compromised efficiency for completing their replication cycle. ISVPs of E89 mutants converted to ISVP*s more readily than those of wild-type viruses. The E89 mutants yielded revertants with second-site substitutions within regions that mediate interaction between μ1 trimers at a site distinct from the 72-96 loop. These viruses also contained changes in regions that control interactions within μ1 trimers. Viruses containing these second-site changes displayed restored plaque phenotypes and were capable of undergoing ISVP-to-ISVP* conversion in a regulated manner. These findings highlight regions of μ1 that stabilize the reovirus capsid and demonstrate that an enhanced propensity to form ISVP*s in an unregulated manner compromises viral fitness.
INTRODUCTION
Capsids of mammalian orthoreovirus (reovirus) are composed of two concentric protein shells (1). The inner capsid, or core, contains 10 segments of double-stranded RNA (dsRNA) along with enzymes required for transcription and modification of viral mRNA (1). Although the viral genome is protected within the core, the core is unable to initiate infection when outside the host cell. This lack of infectivity is because the core is incapable of engaging cellular receptors and because it is devoid of the machinery to bypass the topological barrier posed by the host membrane (1). The capacity for cell attachment and membrane penetration to the reovirus particle is conferred by the viral outer capsid (1). Thus, although the reovirus outer capsid does not directly serve a protective function, it is critical for maintaining the infectivity of reovirus particles. Therefore, like capsids of all nonenveloped viruses, the reovirus outer capsid must be stable in the environment.
The μ1 protein, a major component of the reovirus outer capsid, renders reovirus capsids resistant to inactivation by denaturing agents such as ethanol, phenol, and heat (2–7). In addition, the resistance of some reovirus strains to inactivation by protease digestion is also a property of μ1 (8, 9). In the native state, the μ1 protein forms a structure that places a jelly-roll β-barrel atop an α-helical pedestal (10). Each monomer is intertwined with two others to form a trimer, and two hundred μ1 trimers form a lattice around the viral core (10–12) (Fig. 1A). The lattice is held together by interaction between trimers and between the trimer and the core. It is anticipated that intra- and intersubunit interactions that allow formation and maintenance of this lattice are important in allowing μ1 to control the environmental stability of the particle.
Fig 1.
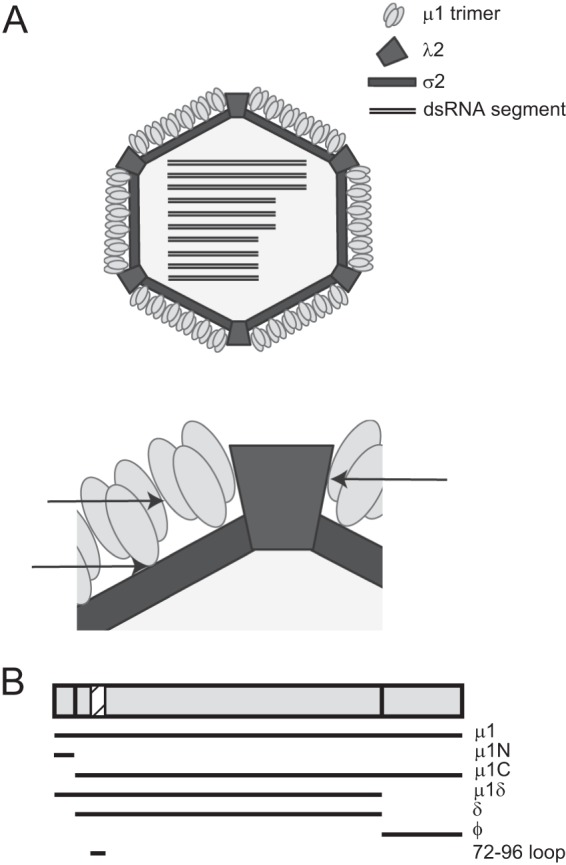
Schematic of the μ1 protein and its interactions within the virus particle. (A) A broad (top) and zoomed-in (bottom) schematic of the cutaway view of an ISVP showing the arrangement of the μ1 protein surrounding the core. Arrows indicate regions of interaction between μ1 trimers and between trimers and the core that could be mediated by the 72-96 loop. (B) The various fragments of μ1 generated during cell entry of reovirus are shown. The location of the μ1 72-96 loop is shown as a hatched box.
In addition to its role in preserving viral infectivity, the μ1 protein is also required for delivery of the viral core into the cytosol of host cells (13). Following attachment to proteinaceous and carbohydrate receptors on the host membrane, reovirus particles are endocytosed into clathrin-coated vesicles and delivered to Rab7-positive late endosomes via events that are dependent on β1 integrins, Src kinases, and microtubules (14–20). Within endosomes, the σ3 protector protein is digested by cathepsin proteases to expose the proteolytically digested μ1 fragments, μ1δ and ϕ, to the viral entry intermediate known as the infectious subvirion particle (ISVP) (16, 17, 21–26). ISVPs can also be formed extracellularly by luminal proteases found in the gastrointestinal or respiratory tract. These mucosal surfaces serve as portals for entry of reovirus into the mammalian host (27–30).
The loss of σ3 and exposure of μ1 on the surface of the ISVPs are not sufficient for cell entry because the exposed μ1 protein in the ISVPs adopts a structure identical to that found in the virion (12). In this configuration of μ1, the membrane-active peptides, which are required for cell entry, are buried proximal to the core. Penetration of host cells by reovirus requires a dramatic conformational change in μ1 that releases these peptides (31, 32). This conformational change is triggered by interaction of μ1 with membrane components and results in formation of ISVP*s (31, 33). Formation of ISVP*s is accompanied by autocleavage of μ1δ to form μ1N and δ (34) (Fig. 1B). Unwinding of the μ1 trimer is thought to constitute at least one type of conformational change that is required to release the buried μ1 N- and C-terminal fragments μ1N and ϕ, respectively (10, 35). The released fragments facilitate the delivery of the viral core across the membrane to initiate infection (36–38). Thus, mutually antagonistic functions of μ1 in preserving viral infectivity and in delivering the genomic payload are analogous to those of capsid proteins from simple nonenveloped viruses that function in directly protecting and delivering the genome.
In this study, we sought to determine how conformational changes in μ1 that allow release of peptides required for membrane penetration are influenced by structural elements that are involved in formation of the μ1 lattice that preserves viral infectivity. For our studies, we focused on a μ1 loop comprised of residues 72 to 96 (termed the 72-96 loop). This loop is proposed to mediate interactions between μ1 trimers and between μ1 and the core (10, 11). Using reverse genetics, we generated viruses with mutations at charged residues within this loop. Among these, substitutions at Glu89 (E89) produced viruses with a dramatically reduced plaque size in the presence of proteases, suggesting a diminishment in the capacity of the virus to complete its replication cycle. This reduction in replicative fitness was likely related to the capacity of mutant viruses to undergo ISVP-to-ISVP* conformational changes more readily under less stringent conditions. Passaging of the E89 mutants allowed us to select revertants with restored plaque morphology. The revertants contained second-site substitutions in μ1 at positions that can affect interactions within or between trimers. Regeneration of viruses with either type of μ1 second-site change restored regulated ISVP-to-ISVP* conversion. These findings indicate that an enhanced capacity for conformational changes renders reovirus capsids susceptible to proteolytic digestion and compromises reovirus infectivity. Our results also highlight how different types of interactions between structural units that form the reovirus capsid modulate the capacity of stable virus particles to undergo changes required for cell entry.
MATERIALS AND METHODS
Cells.
Murine L929 (L) cells were maintained in Joklik's minimum essential medium (MEM; Lonza) supplemented to contain 5% fetal bovine serum (FBS) (Atlanta Biological), 2 mM l-glutamine (Invitrogen), 100 U/ml of penicillin (Invitrogen), 100 mg/ml streptomycin (Invitrogen), and 25 ng/ml of amphotericin B (Sigma-Aldrich). BHK-T7 cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented to contain 10% FBS, 2 mM l-glutamine, 2% MEM amino acid solution (Invitrogen), and 1 mg/ml Geneticin (Invivogen) in alternate passages.
Viruses.
To generate plasmids with mutations in the M2 gene segment, pT7-M2T3D was subjected to QuikChange site-directed mutagenesis (Stratagene). To generate viruses with mutations in Glu73 or E73 (Table 1), 4 plasmids carrying 9 T1L gene segments (all excluding M2) were mixed with a pool of mutant pT7-M2T3D plasmids containing E73A, E73S, E73L, E73Q, and E73K. The mixture was transfected into BHK-T7 cells for recovering viruses as described previously (39, 40). Instead of 3.5 μg of pT7-M2T3D plasmid used for generating wild-type virus in this system, 0.7 μg of each mutant plasmid was combined for this pool. To generate viruses with mutations in Asp80 or D80 (Table 1), a pool of mutant pT7-M2T3D plasmids containing D80A, D80S, D80L, D80N, and D80K was similarly used. To generate viruses with mutations in Glu89 or E89 (Table 1), a pool of mutant pT7-M2T3D plasmids containing E89A, E89S, E89L, E89Q, and E89K was used. Mutant E89S, E89L, and E89K viruses could not be recovered after 3 independent attempts. For each pool, approximately 25 plaques were selected and amplified. To confirm sequences of mutant viruses, viral RNA was extracted from infected cells and subjected to reverse transcription-PCR (RT-PCR) using primers specific for the M2 gene segment. PCR products were resolved on Tris-acetate-EDTA agarose gels, purified, and subjected to sequence analysis (7).
Table 1.
Viruses with mutations in μ1 72-96 loop
| Residue in T3D μ1 | Mutants recovered |
|---|---|
| E73 | A, S, L, Q, K |
| D80 | A, S, L, N, K |
| E89 | A, Q |
To regenerate viruses with μ1 mutations identified in revertant viruses, pT7-M2T3D, pT7-M2T3D E89A, and pT7-M2T3D E89Q plasmids were mutagenized. To generate viruses with mutations in λ2, a 6-plasmid mixture was used. pT7-L2T1L was QuikChange mutagenized and used in place of parental pT7-L2T1L for generation of λ2 mutants. Sequences of primers used for mutagenesis, RT-PCR, and sequencing are available upon request.
Purification of viruses.
Purified reovirus virions were generated using second- or third-passage L-cell lysate stocks of reovirus. Viral particles were Vertrel-XF (Dupont) extracted from infected cell lysates, layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 187,183 × g for 4 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) (41). The concentration of reovirus virions in purified preparations was determined from an equivalence of one unit of optical density at 260 nm (OD260) being 2.1 × 1012 virions/ml (42). Viral titer determination and assessment of plaque morphology were done by plaque assay using L cells. Most plaque assays included N-p-tosyl-l-lysine chloromethyl ketone (TLCK)-treated chymotrypsin (CHT) in the overlay, fixed with 10% formaldehyde, and stained using crystal violet (3). Plaque assays that did not include CHT on the overlay were stained with neutral red (41).
Selection of revertants.
Passage 1 stocks of μ1 E89A and E89Q mutant viruses were serially passaged 5 times in L cells. For each passage, 1 ml of inoculum from the previous passage was used to infect L cells in 60-mm dishes and incubated at 37°C until all cells were detached. Frozen and thawed lysate from each passage was used for assessment of plaque morphology and for further passaging. Revertants with large plaques were selected by plaque purification and amplified in L cells to obtain viral lysate stocks. All revertants characterized in this study were picked after a single passage. After amplification, the viruses were replaqued to confirm the large-plaque phenotype. To confirm sequences of plaque size revertant viruses, viral RNA was extracted from infected cells and subjected to RT-PCR using S2, M2, and L2 gene segment-specific primers. PCR products were resolved on Tris-acetate-EDTA agarose gels, purified, and subjected to sequence analysis to cover the entire length of the S2, M2, and L2 open reading frames. Primer sequences are available upon request.
Generation of ISVPs.
ISVPs were generated by incubation of 2 × 1012 virions with 200 μg/ml of TLCK-treated CHT in a total volume of 0.1 ml (43) at 28 or 37°C for 20 min. Proteolysis was terminated by addition of 2 mM phenylmethylsulfonyl fluoride and incubation of reactions on ice. Generation of ISVPs was confirmed by SDS-PAGE and Coomassie brilliant blue staining.
Analysis of ISVP* generation.
ISVPs (2 × 1011), generated at 28°C for 20 min, were incubated at various temperatures for 20 min, transferred to ice for 20 min, and incubated with 100 μg/ml trypsin at 4°C for 30 min. Trypsin digestion was terminated by addition of SDS-PAGE loading buffer and transfer of the samples to dry ice. Generation of ISVP*s was confirmed by SDS-PAGE and Coomassie brilliant blue staining.
Hemolysis to measure ISVP* formation.
Citrated calf red blood cells (RBCs) (Colorado Serum Company) were washed extensively with chilled phosphate-buffered saline (PBS) supplemented to contain 2 mM MgCl2 (PBS-Mg) and resuspended at a concentration of 30% (vol/vol) in PBS-Mg. Hemolysis efficiency was analyzed by mixing a 2.22-μl aliquot of resuspended RBCs with ISVPs in a total volume of 22.22 μl, followed by incubation at 22, 29.5, and 37°C for 20 min. RBCs mixed with virion storage buffer without ISVPs or with 1% Triton X-100 (TX-100) were used as the control for 0 and 100% lysis, respectively. Samples were placed on ice for 30 min to prevent further hemolysis and centrifuged at 500 × g at 4°C for 3 min. The extent of hemoglobin release was quantified by measuring the A405 of a 1:5 dilution of the supernatant in a microplate reader (Molecular Devices). Percent hemolysis was calculated using the following formula: percent hemolysis = 100 × (Asample − Abuffer)/(ATX-100 − Abuffer).
RESULTS
Viruses with mutations in μ1 72-96 loop display small-plaque morphology.
Structural studies indicate that a loop comprised of residues 72 to 96 within μ1 is in a position to mediate interactions between μ1 trimers and could interact with the core proteins σ2 and λ2 (10, 11). We hypothesized that the strength of these interactions would be inversely proportional to the capacity of the particles to undergo ISVP-to-ISVP* conformational changes. To test this idea, we generated recombinant reoviruses (Table 1) with mutations at three charged residues (E73, D80, and E89) within this loop of T3D μ1 (Fig. 1B) using the plasmid-based reverse-genetics system (39, 40). All other genes in the recombinant viruses were isogenic and derived from reovirus strain T1L. To increase the possibility of recovering viable mutant viruses, we attempted to rescue viruses with five different amino acid substitutions at each residue. For E73 and D80, all possible viruses were retrieved (Table 1). For E89, only viruses with A and Q substitutions could be recovered, even after three independent attempts, perhaps suggesting the importance of E89 within the loop in maintaining viral infectivity.
The plaque morphologies of all E73 and D80 mutant viruses were similar to that of the parental strain (a T1L virus containing T3D M2) (Fig. 2), here referred to as the wild-type virus. The phenotypes of all E73 and D80 mutants were also similar in all subsequent experiments; therefore, only data for E73A, E73Q, D80A, and D80N are shown. In contrast, the majority of plaques for E89A and E89Q viruses were significantly smaller than those of the parental virus (Fig. 2). These data suggest that mutations at residue E89 within the 72-96 loop negatively impact the viral replication cycle. To expedite the plaque assay, we had included CHT in the overlay (3). Thus, it is possible that the observed plaque morphology reflects the importance of E89 in regulating viral replication efficiency in the presence of proteases. Indeed, virions would encounter such an environment within the intestinal and respiratory tracts or in cellular endosomes. To determine the effect of E89 on plaque morphology in the absence of protease, we compared the plaques formed by the wild-type and E89 mutant viruses in the absence of CHT (data not shown). We found that both E89A and E89Q viruses have plaque morphologies similar to that of the wild-type virus. These findings suggest that E89 influences the susceptibility of μ1 to proteolytic digestion.
Fig 2.

Plaque morphology of viruses with mutations in the μ1 72-96 loop. L cells were infected with cell lysate stocks of viruses and overlaid with agar-medium mix containing CHT. At 4 days postinfection, cells were fixed with 10% formaldehyde and stained with crystal violet.
The 72-96 loop of μ1 regulates conformational changes during reovirus disassembly.
The μ1 protein is a component of the viral outer capsid (1). Upon purification of wild-type and mutant virions, we detected no differences in the stoichiometry of the major capsid proteins (Fig. 3). These data suggested that the mutations in μ1 did not cause gross changes in the assembly of the virus particle. In keeping with the observation that E89 might influence the protease susceptibility of the particle, we first tested the capacity of the mutant viruses to form ISVPs (Fig. 3). For these experiments, purified virions of each virus strain were incubated with 200 μg/ml of CHT at 37°C for 20 min, the routinely used conditions for ISVP formation (43). As expected, following incubation at 37°C with CHT, the σ3 protein for the wild-type virus was proteolytically degraded and the μ1 protein was cleaved to the μ1δ and ϕ fragments to form ISVPs. The E73, D80, and E89A mutants showed a digestion pattern identical to that demonstrated by wild-type virions. In contrast, E89Q μ1 was completely digested to form cores. ISVPs can also be generated by digestion of particles at lower temperatures (44). To determine if the E89Q mutant outer capsid can be spared from complete digestion at a lower temperature, we incubated virions with 200 μg/ml CHT at 28°C for 20 min. We found that under these conditions, virions of all viruses, including E89 mutants, formed ISVPs. These findings suggest that E89 mutants are capable of ISVP formation, but the outer capsid of the E89Q mutant is susceptible to proteolysis at physiological temperature due to increased protease activity or a greater propensity of the E89 mutant μ1 to undergo structural changes that expose protease cleavage sites at higher temperatures.
Fig 3.
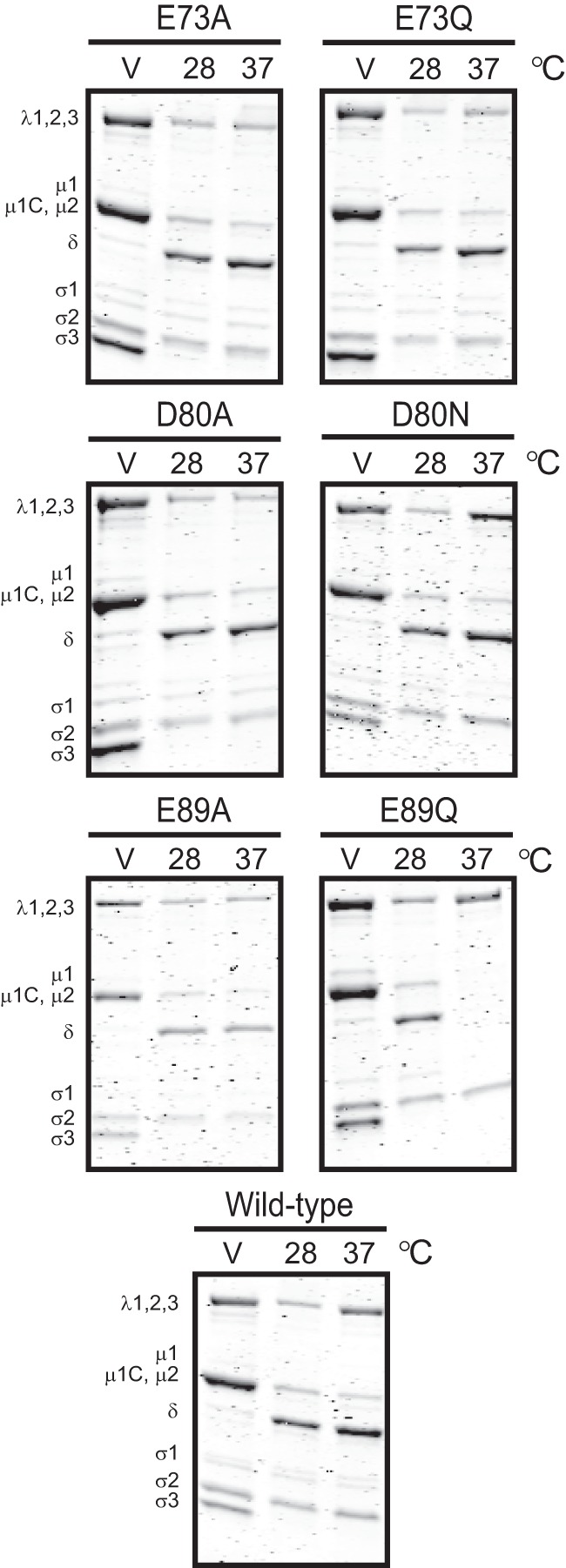
72-96 loop of μ1 regulates reovirus disassembly. Purified virions of the indicated viruses were treated with CHT at 28 or 37°C for 20 min. Samples were resolved by SDS-PAGE and stained using Coomassie brilliant blue. V denotes purified virions treated with CHT and transferred to ice immediately. The positions of reovirus capsid proteins are shown. μ1δ resolves as δ (34). ϕ is too small to resolve on the gel.
During conversion of ISVPs to ISVP*s, μ1 undergoes conformational changes that render it susceptible to proteolysis (31). To directly test whether changes in the 72-96 loop alter the efficiency with which the ISVP-to-ISVP* transition occurs, we generated ISVPs by CHT digestion at 28°C and subsequently treated ISVPs at temperatures ranging from 22 to 32°C for 20 min prior to assessment of trypsin sensitivity of the δ fragment. The δ fragment of the wild-type virus became sensitive to protease digestion at 32°C (Fig. 4). The δ fragments of E73A, E73Q, and D80N mutants displayed protease resistance and sensitivity to temperatures identical to those of the wild-type virus. The δ fragment of D80A became sensitive to the protease at 30°C. Most strikingly, however, the δ fragments of the E89A and E89Q mutants became protease sensitive at 24°C and at least as low as 22°C, respectively. These data indicate that mutations at E89 allow ISVP-to-ISVP* conversion to occur more readily than in the wild-type virus.
Fig 4.

ISVP-to-ISVP* conformational changes are regulated by the μ1 72-96 loop. ISVPs of the indicated viruses were generated by digestion of virions at 28°C. The ISVPs were heated at the indicated temperatures (°C) for 20 min and treated with trypsin at 4°C for 30 min. Samples were resolved by SDS-PAGE and stained using Coomassie brilliant blue. The positions of reovirus capsid proteins are shown. μ1δ resolves as δ (34). ϕ is too small to resolve on the gel.
E89 mutant viruses undergo reversion by generating second-site mutations.
The small plaques formed by E89A and E89Q mutants suggested that these changes rendered one or more events in the viral replication cycle inefficient (Fig. 2). We hypothesized that serial passage of such viruses would yield revertants with restored replicative fitness. Mapping the genetic changes in the capsid proteins of revertants would provide insight into the mechanisms by which the balance between viral capsid stability and conformational flexibility to generate intermediates required for cell entry is maintained. Following a single passage, revertants of E89A and E89Q were selected based on the striking difference in plaque morphology between the mutant and wild-type viruses (Fig. 2 and data not shown). Interestingly, because these revertants were produced even though the viruses were passaged in the absence of CHT, these data suggest that mutations at E89 also are unfavorable without the addition of exogenous proteases. Viruses with large plaques were selected for amplification and further analysis. The 72-96 loop of μ1 potentially interacts with μ1 in neighboring trimers or with core proteins σ2 and λ2 (10, 11). Therefore, we sequenced the μ1-encoding M2 gene, the λ2-encoding L2 gene, and the σ2-encoding S2 gene segments for each revertant. No reversions of the original mutation were identified. Among the revertants sequenced, none contained changes in the S2 gene segment. The E89A revertants sequenced contained a second-site change in μ1 at one of four residues (A276V, D371N, Q456R, or P497S). Among the E89Q revertants sequenced, one contained an A276V mutation in μ1 and the other contained two alterations, an L185S change in μ1 and an N1245D change in λ2 (Table 2). The second-site mutations in μ1 were at locations where they could mediate interactions between μ1 trimers (D371N) or between monomers within a trimer (L185S, A276V, Q456R, and P497S) (Fig. 5). The mutation in λ2 was not in a position where it can modulate interactions with μ1 (data not shown). Rather, the mutated λ2 residue was located near the base of the turret at the 5-fold vertices, where it may interact with the fiber formed by the N terminus of the σ1 attachment protein (11, 45).
Table 2.
Second-site changes identified in E89 mutants
| Introduced change | Second-site change |
|---|---|
| E89A | μ1 A276V |
| μ1 D371N | |
| μ1 Q456R | |
| μ1 P497S | |
| E89Q | μ1 A276V |
| μ1 L185S + λ2 N1245D |
Fig 5.

Second-site mutations in growth-impaired E89 mutant viruses. Side view (A) and bottom view (B) of a μ1 trimer were rendered using UCSF chimera from the crystal structure of μ1 (PDB entry 1JMU) (10) showing the locations of second-site mutations that are in positions to mediate intratrimer μ1 interactions. Side view (C) and top view (D) of two adjacent μ1 trimers were rendered using UCSF chimera from the crystal and cryoelectron microscopy structures of μ1 (PDB entries 1JMU and 2CSE) (10, 11) showing the locations of second-site mutations that are in positions to mediate intertrimer μ1 interactions. The three monomers within a μ1 trimer are shown as gray-, pink-, and green-colored ribbons. Residue 97 is labeled as residue 89, as the 72-96 loop was not resolved in the crystal structure and is not in the PDB file.
To determine whether the amino acid substitutions found in the revertant viruses restore wild-type virus-like plaque morphology, we regenerated viruses with the changes found in the revertants in the E89A or E89Q mutant background on which they were identified. Additionally, to determine if the residues present in the revertants influence the properties of the virus independently of the amino acid present at E89, viruses with changes identified in the plaque size revertants were also incorporated into the wild-type μ1 background. Regeneration of revertants was also needed to rule out the possibility that genetic alterations present in the seven remaining gene segments of the revertants that were not sequenced contributed to the altered phenotype.
As an initial measure of the effect of introduced second-site change in μ1, we compared the plaque morphology of the regenerated revertants to that of the original E89 mutants and to the wild-type virus (Fig. 6). The regenerated E89A and E89Q revertants that contained an additional mutation in the μ1 protein showed plaque morphology similar to that of the wild-type virus. These results suggest that second-site changes identified in the μ1 protein contribute to restoration of the plaque phenotype in the revertant viruses. In the case of the E89Q virus containing the λ2 site reversion (N1245D), we observed an ∼2:1 mixture of small and large plaques. We have determined that the large plaque variants within this mixture have acquired at least one more substitution mutation in μ1 (data not shown). The triple mutant, which contains an additional μ1 change (L185S), exhibits a wild-type plaque phenotype. Because the μ1 L185S change alone is sufficient to alter the small-plaque morphology of E89Q, these results suggest that like the other revertants, plaque size restoration in the triple mutant is also a property of μ1. Based on these results, we think that the λ2 mutation does not suppress the small-plaque phenotype of E89Q. Biochemical characterization of λ2 mutant viruses described below also corroborates this inference (Fig. 7 and 8). Introduction of second-site changes in the wild-type μ1 background either did not affect or increased the size of the plaques formed by the wild-type virus. Thus, mutations found in reversions do not have a negative impact on the fitness of the wild-type virus. These findings indicate that the restoration of the large plaque phenotype is due to introduction of the intragenic substitutions within μ1.
Fig 6.

Plaque morphology of viruses containing second-site reversions. L cells were infected with cell lysate stocks of viruses with second-site reversions alone or in combinations in E89A (A), E89Q (B), or wild-type (C) μ1 backgrounds and overlaid with agar-medium mix containing CHT. At 4 days postinfection, cells were fixed with 10% formaldehyde and stained with crystal violet.
Fig 7.

Efficiency of heat-induced ISVP-to-ISVP* transition is restored in viruses containing second-site reversions. ISVPs of viruses with second-site reversions alone or in combination in E89A (A), E89Q (B), or wild-type (C) μ1 backgrounds were generated by digestion of virions at 28°C. The ISVPs were heated at the indicated temperatures (°C) for 20 min and treated with trypsin at 4°C for 30 min. Samples were resolved by SDS-PAGE and stained using Coomassie brilliant blue. The positions of reovirus capsid proteins are shown. μ1δ resolves as δ (34). ϕ is too small to resolve on the gel.
Fig 8.

Restored ISVP-to-ISVP* conversion efficiency in viruses containing second-site mutations. ISVPs of viruses with second-site reversions alone or in combination in E89A (A), E89Q (B), or wild-type (C) μ1 backgrounds were generated by digestion of virions at 28°C. A 3% (vol/vol) solution of bovine RBCs was incubated with ISVPs in virion storage buffer at the indicated temperatures (°C) for 20 min. Hemolysis was quantified by determining absorbance of the supernatant at 405 nm. Results are expressed as mean percent hemolysis for triplicate samples. Error bars indicate SD.
Second-site changes in μ1 restore efficiency of ISVP* formation.
Our initial characterization of the E89 mutant viruses indicated that E89A and E89Q viruses converted to ISVP* at a much lower temperature than viruses expressing wild-type μ1 (Fig. 4). To identify second-site suppressors of E89A and E89Q mutants that restore ISVP-to-ISVP* conversion efficiency to that displayed by the wild-type virus, ISVPs generated from each virus were incubated at 0, 28, or 32°C for 20 min. Our results from initial characterization of the E89 mutants indicated that these three temperatures are sufficient to differentiate between wild-type and mutant ISVP-to-ISVP* conversion efficiency (Fig. 4). Conformational changes in particle-associated δ were examined by assessing its sensitivity to trypsin digestion (31, 33). Consistent with our data shown in Fig. 4 and described above, the δ fragment of the wild-type virus was resistant to trypsin digestion following incubation at 0 and 28°C and became sensitive to digestion at 32°C (Fig. 7C). In contrast, the δ fragments of E89A and E89Q were protease sensitive at both 28 and 32°C (Fig. 7A and B). The regenerated E89A and E89Q revertants that contain a μ1 second-site change rendered the δ fragment of these viruses resistant to protease digestion at 28°C. Notably, with the exception of D371N, these secondary μ1 mutations conferred protease resistance to the δ fragment even at 32°C. Incorporation of the second-site substitution in λ2 did not affect the protease sensitivity of E89Q, and the δ fragment of this virus remained susceptible to digestion at both 28 and 32°C. These findings indicate that second-site changes in μ1 diminish the enhanced propensity of E89A and E89Q μ1 proteins to undergo conformational changes that resemble ISVP-to-ISVP* conversion. Incorporation of the μ1 second-site changes in the wild-type background not only maintained the protease resistance of the δ fragment at 0 and 28°C but also rendered the δ fragment of the virus resistant to protease digestion at 32°C (Fig. 7C). These data suggest that amino acid changes in μ1, selected during passage of E89 mutants, protect the viral capsid against proteolysis irrespective of the presence or absence of mutations at E89. These changes may lead to a decrease in exposure of protease-sensitive regions of δ and thereby generate larger plaques as shown in Fig. 6 above.
During infection of cells, ISVP-to-ISVP* conversion is triggered by membrane interaction (31). Membranes of red blood cells (RBCs) serve as useful models for assessing the efficiency of ISVP-to-ISVP* conversion under physiologic conditions (31, 33). ISVP* formation in the presence of RBCs results in osmotic lysis of RBCs as a consequence of pore formation by the released μ1N and ϕ fragments (36–38). Therefore, hemolysis is a facile measure of ISVP* formation. Incubation of RBCs with wild-type ISVPs promotes hemolysis at 37°C but not at lower temperatures of 22 and 29.5°C (Fig. 8C). In contrast, the E89A and E89Q mutants could hemolyze RBCs at all three temperatures, with E89A showing slightly lower hemolysis at 22°C (Fig. 8A and B). The capacity of these viruses to hemolyze at 22 and 29.5°C is consistent with their propensity to form ISVP*s more readily than the wild-type virus. To define how the second-site changes present in the revertant viruses influence membrane-triggered ISVP-to-ISVP* conversion, we tested the hemolysis capacity of each regenerated revertant. ISVPs of viruses with the second-site change in λ2 displayed hemolysis efficiency analogous to that of the original E89Q virus at all three temperatures. In contrast, ISVPs of all variants that contain a second-site change in μ1 along with an E89A or E89Q change failed to hemolyze RBCs at 22 and 29.5°C. With the exception of the ISVPs containing a D371N and L185S second-site change in μ1, these ISVPs also failed to hemolyze RBCs at 37°C. Introduction of the μ1 second-site changes in a wild-type μ1 background also prevented hemolysis at 37°C (Fig. 8C). The observed effects of μ1 second-site mutations on hemolysis are consistent with the effect of μ1 changes on the capacity of the viral capsid to undergo conformational changes at elevated temperatures (Fig. 7). These data indicate that at each temperature, the capacity for hemolysis is directly related to the efficiency with which the μ1 protein is capable of undergoing conformational changes. Together, our findings indicate that second-site changes in μ1 within sites of inter- and intratrimer interactions restore regulated, trigger-dependent ISVP* formation.
DISCUSSION
In this study, we evaluated the role of a μ1 loop comprised of residues 72 to 96. This loop is proposed to be involved in forming a stabilizing lattice on the reovirus particle. We found that two different mutations at E89 within this loop produced viruses that display a small-plaque morphology in the presence of CHT. These mutations allowed ISVP-to-ISVP* conversion to occur under less stringent conditions. The decreased replication efficiency of the E89 mutants engendered reversions. The revertants contained mutations at additional sites within μ1 and also within the λ2 protein. Viruses with second-site changes in μ1 (but not λ2) displayed restored replicative fitness, as assessed by plaque morphology. The capsids of such viruses were capable of ISVP-to-ISVP* conversion only when the appropriate trigger was provided. These findings indicate that tipping the balance between capsid stability and conformational flexibility toward enhanced flexibility diminishes the fitness of the virus.
Our data show that deregulated conversion of ISVPs to ISVP*s correlates with a small-plaque phenotype. Conversion of ISVPs to ISVP*s results in release of the reovirus membrane penetration peptides μ1N and ϕ from the particle (34, 37). These peptides localize to membranes, recruit viral particles, and facilitate their translocation across the membrane (37). If these peptides were released in the absence of membranes, a viral intermediate lacking its membrane penetration machinery would be generated. Such particles would be unable to cross membranes and therefore be rendered noninfectious. Thus, it would be anticipated that the relative level of infectivity of a preparation of such a mutant would be less than that of the wild-type virus. In keeping with this, titers of mutant viruses that we identified to have higher propensity of ISVP-to-ISVP* conversion were consistently lower than that of the wild-type virus or those of viruses with lower efficiency of ISVP-to-ISVP* transition (data not shown).
Based on the potential of the μ1 72-96 loop in mediating interactions between μ1 trimers and between μ1 and the core, we anticipated that revertant viruses would contain secondary mutations within this loop or in regions that interact with this loop (10, 11). However, none of the revertants that we isolated and sequenced contained mutations within such regions. It is possible that constraints faced by μ1 and core proteins in maintaining its different functions prevented recovery of mutations at such sites. It also cannot be ruled out that this loop is involved in maintaining the stability of the virus by a mechanism that does not require it to directly interact with μ1 in neighboring trimers or with core proteins. Instead, one suppressor mutation in a revertant virus was present in a distinct region of μ1 intertrimer contact near the top of the trimers. Somewhat surprisingly, the second-site changes in the other revertants were in regions of contact between monomers within a trimer. The areas where second-site changes were identified have previously been implicated in maintaining the stability of reovirus capsids. Reovirus variants that are resistant to the presence of ethanol or high temperature contain mutations within these regions (3, 5, 6). Such stabilizing mutations diminish efficiency of ISVP-to-ISVP* conversion and prevent inactivation of reovirus by these denaturing agents (7, 32), likely by preventing the release of membrane-active peptides or the attachment moiety, σ1.
Remarkably, some of the mutations identified in hyperstable viruses described previously were found at sites identical to those that we have found in our revertant viruses. For example, a P497S change that we found in an E89A revertant also has been found in an ethanol-resistant mutant of reovirus strain T3A (5). Likewise, the D371N change identified in an E89A revertant is similar to the D371A change in a thermostable virus (3). Interestingly, a labilizing reversion of D371 bears an E89K mutation (46). Other changes that render D371A capsids labile occur at A184 and P277 (46), which are adjacent to L185 and A276 residues, which we have found to control the efficiency of conversion to ISVP*s. Based on the current and previous studies, we think that μ1 intertrimer interaction both at the base (likely mediated by the 72-96 loop) and the top of the trimers, along with regions that hold together μ1 monomers within a trimer, are key to maintaining the thermal stability of the reovirus outer capsid. The repeated isolation of mutations that alter viral stability in the same residues or structural domains of μ1 point to a limited number of solutions the virus has available to maintain the balance between stability and flexibility.
Structural predictions (10, 11), backed by experimental evidence gathered in this study, support the idea that the 72-96 loop is important in maintaining viral stability. This loop is missing from the μ1 homolog of aquareoviruses, VP5 (47). Therefore, it would be anticipated that capsids of these viruses are relatively less stable and consequently are able to undergo conformational changes required for cell entry more readily. Consistent with this idea, aquareoviruses are capable of initiating infection at significantly lower temperatures (47). We speculate that the absence of this loop, along with other changes within the capsid, is an evolutionary adaptation to allow capsids of aquareoviruses to maintain infectivity and transmissibility in the environment of their hosts. The avian reovirus homolog of μ1, μB, also lacks the 72-96 loop (48). Since avian reoviruses likely infect their hosts at higher temperatures, these viruses must have evolved an alternative mechanism to maintain greater capsid stability.
In this study, we disrupted the balance between capsid stability and flexibility by generating virus particles that convert ISVPs to ISVP*s readily. An enhanced capacity for ISVP* formation compromised viral replication and yielded revertants with efficiency of ISVP* formation similar to that of the wild-type virus. Revertants that generate ISVP*s less efficiently than the wild-type virus also were produced. These data suggest that decreases in ISVP* formation efficiency, a correlate of enhanced thermal stability of the capsid (3, 4, 46), are more favorable than increased conformational flexibility. In direct contrast with this observation, we have found that reoviruses with hyperstable μ1 proteins replicate inefficiently in the central nervous system (CNS) (7, 49). We think there are two possible explanations for this discrepancy. First, although viruses examined here and in our previous studies are more stable than the wild-type virus, they may not be equivalently hyperstable. It is conceivable that stability is favored but only to a certain extent, and further increase in stability outside that window would diminish the efficiency of virus infection. Second, it is possible that stability and flexibility are favored in different environments. In support of this idea and distinct from our findings in the CNS, viruses with enhanced stability replicate more efficiently in the murine intestinal tract than more flexible viruses (4, 31, 33, 50). Our ongoing studies are designed to assess the range of stabilities and flexibilities that can be tolerated and how altering viral stability and flexibility affects replication in different tissues infected by reovirus.
ACKNOWLEDGMENTS
This research was supported by startup funds from Indiana University.
We thank members of our laboratory, Karl Boehme, Bernardo Mainou, and Tuli Mukhopadhyay for helpful suggestions and reviews of the manuscript.
Footnotes
Published ahead of print 2 October 2013
REFERENCES
- 1.Dermody TS, Parker JC, Sherry B. 2013. Orthoreoviruses. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Drayna D, Fields BN. 1982. Genetic studies on the mechanism of chemical and physical inactivation of reovirus. J. Gen. Virol. 63:149–160 [DOI] [PubMed] [Google Scholar]
- 3.Middleton JK, Agosto MA, Severson TF, Yin J, Nibert ML. 2007. Thermostabilizing mutations in reovirus outer-capsid protein μ1 selected by heat inactivation of infectious subvirion particles. Virology 361:412–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Middleton JK, Severson TF, Chandran K, Gillian AL, Yin J, Nibert ML. 2002. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein mu1. J. Virol. 76:1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hooper JW, Fields BN. 1996. Role of the μ1 protein in reovirus stability and capacity to cause chromium release from host cells. J. Virol. 70:459–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wessner DR, Fields BN. 1993. Isolation and genetic characterization of ethanol-resistant reovirus mutants. J. Virol. 67:2442–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J. Virol. 82:161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drayna D, Fields BN. 1982. Activation and characterization of the reovirus transcriptase: genetic analysis. J. Virol. 41:110–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madren JA, Sarkar P, Danthi P. 2012. Cell entry-associated conformational changes in reovirus particles are controlled by host protease activity. J. Virol. 86:3466–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. 2002. Structure of the reovirus membrane-penetration protein, μ1, in a complex with its protector protein, σ3. Cell 108:283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. 2005. Features of reovirus outer capsid protein μ1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure 13:1545–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell Biol. 122:1023–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Danthi P, Guglielmi KM, Kirchner E, Mainou B, Stehle T, Dermody TS. 2010. From touchdown to transcription: the reovirus cell entry pathway. Curr. Top. Microbiol. Immunol. 343:91–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulant S, Stanifer M, Kural C, Cureton DK, Massol R, Nibert ML, Kirchhausen T. 2013. Similar uptake but different trafficking and escape routes of reovirus virions and ISVPs imaged in polarized MDCK cells. Mol. Biol. Cell. 24:1196–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maginnis MS, Mainou BA, Derdowski A, Johnson EM, Zent R, Dermody TS. 2008. NPXY motifs in the beta1 integrin cytoplasmic tail are required for functional reovirus entry. J. Virol. 82:3181–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mainou BA, Dermody TS. 2011. Src kinase mediates productive endocytic sorting of reovirus during cell entry. J. Virol. 85:3203–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus infection. J. Virol. 86:8346–8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mainou BA, Zamora PF, Ashbrook AW, Dorset DC, Kim KS, Dermody TS. 2013. Reovirus cell entry requires functional microtubules. mBio 4:e00405–13. 10.1128/mBio.00405-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. 2006. Beta1 integrin mediates internalization of mammalian reovirus. J. Virol. 80:2760–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulz WL, Haj AK, Schiff LA. 2012. Reovirus uses multiple endocytic pathways for cell entry. J. Virol. 86:12665–12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borsa J, Copps TP, Sargent MD, Long DG, Chapman JD. 1973. New intermediate subviral particles in the in vitro uncoating of reovirus virions by chymotrypsin. J. Virol. 11:552–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CT, Zweerink HJ. 1971. Fate of parental reovirus in infected cell. Virology 46:544–555 [DOI] [PubMed] [Google Scholar]
- 23.Baer GS, Ebert DH, Chung CJ, Erickson AH, Dermody TS. 1999. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J. Virol. 73:9532–9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silverstein SC, Astell C, Levin DH, Schonberg M, Acs G. 1972. The mechanism of reovirus uncoating and gene activation in vivo. Virology 47:797–806 [DOI] [PubMed] [Google Scholar]
- 25.Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 277:24609–24617 [DOI] [PubMed] [Google Scholar]
- 26.Sturzenbecker LJ, Nibert ML, Furlong DB, Fields BN. 1987. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J. Virol. 61:2351–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amerongen HM, Wilson GAR, Fields BN, Neutra MR. 1994. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J. Virol. 68:8428–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. 1990. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J. Virol. 64:1830–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bodkin DK, Nibert ML, Fields BN. 1989. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J. Virol. 63:4676–4681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nygaard RM, Golden JW, Schiff LA. 2012. Impact of host proteases on reovirus infection in the respiratory tract. J. Virol. 86:1238–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J. Virol. 76:9920–9933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. 2003. The delta region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J. Virol. 77:13361–13375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar P, Danthi P. 2010. Determinants of strain-specific differences in efficiency of reovirus entry. J. Virol. 84:12723–12732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nibert ML, Odegard AL, Agosto MA, Chandran K, Schiff LA. 2005. Putative autocleavage of reovirus μ1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 345:461–474 [DOI] [PubMed] [Google Scholar]
- 35.Zhang L, Chandran K, Nibert ML, Harrison SC. 2006. Reovirus μ1 structural rearrangements that mediate membrane penetration. J. Virol. 80:12367–12376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agosto MA, Ivanovic T, Nibert ML. 2006. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc. Natl. Acad. Sci. U. S. A. 103:16496–16501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. 2008. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 27:1289–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang L, Agosto MA, Ivanovic T, King DS, Nibert ML, Harrison SC. 2009. Requirements for the formation of membrane pores by the reovirus myristoylated micro1N peptide. J. Virol. 83:7004–7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berard A, Coombs KM. 2009. Mammalian reoviruses: propagation, quantification, and storage. Curr. Protoc. Microbiol.. Chapter 15:Unit15C.11 [DOI] [PubMed] [Google Scholar]
- 42.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810 [DOI] [PubMed] [Google Scholar]
- 43.Nibert ML, Chappell JD, Dermody TS. 1995. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved σ1 protein. J. Virol. 69:5057–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wetzel JD, Wilson GJ, Baer GS, Dunnigan LR, Wright JP, Tang DS, Dermody TS. 1997. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J. Virol. 71:1362–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reinisch KM, Nibert ML, Harrison SC. 2000. Structure of the reovirus core at 3.6 Å resolution. Nature 404:960–967 [DOI] [PubMed] [Google Scholar]
- 46.Agosto MA, Middleton JK, Freimont EC, Yin J, Nibert ML. 2007. Thermolabilizing pseudoreversions in reovirus outer-capsid protein micro 1 rescue the entry defect conferred by a thermostabilizing mutation. J. Virol. 81:7400–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 2010. 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 141:472–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Tang J, Walker SB, O'Hara D, Nibert ML, Duncan R, Baker TS. 2005. Structure of avian orthoreovirus virion by electron cryomicroscopy and image reconstruction. Virology 343:25–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog. 4:e1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubin DH, Fields BN. 1980. Molecular basis of reovirus virulence: role of the M2 gene. J. Exp. Med. 152:853–868 [DOI] [PMC free article] [PubMed] [Google Scholar]