

Abstract
The processes that control aging remain poorly understood. We have exploited mutants in the nematode, C. elegans, that compromise mitochondrial function and scavenging of reactive oxygen species (ROS) to understand their relation to lifespan. We discovered unanticipated roles and interactions of the mitochondrial superoxide dismutases (mtSODs), SOD-2 and SOD-3. Both SODs localize to mitochondrial supercomplex I:III:IV. Loss of SOD-2 specifically: 1) decreases the activities of complexes I and II; complexes III, and IV remain normal, 2) increases the lifespan of animals with a complex I defect, but not the lifespan of animals with a complex II defect, and kills an animal with a complex III defect, 3) induces a presumed pro-inflammatory response. Knockdown of a molecule that may be a pro-inflammatory mediator very markedly extends lifespan and health of certain mitochondrial mutants. The relationship between the electron transport chain, ROS and lifespan is complex, and defects in mitochondrial function have specific interactions with ROS scavenging mechanisms. We conclude that mtSODs are embedded within the supercomplex I:III:IV, and stabilize or locally protect it from reactive oxygen species (ROS) damage. The results call for a change in the usual paradigm for the interaction of electron transport chain function, ROS release, scavenging and compensatory responses.
Keywords: lifespan, superoxide dismutase, reactive oxygen species, electron transport chain, supercomplexes, heat shock protein
Introduction
In 1956, Denham Harman first proposed that the accumulation of oxidative damage from reactive oxygen species (ROS), by-products of normal metabolism, is responsible for aging [1]. Multiple studies have brought this theory into question [2-5], such that the roles of ROS in aging are not yet fully resolved. The main site of production of ROS in the cell is the electron transport chain (ETC) within the mitochondrial inner membrane [6]. Studies in the model organism C. elegans have tested the role of oxidative damage in the process of aging by chemical manipulation of ROS levels (paraquat or juglone), or manipulation of ROS production by inhibiting electron transport [7-9]. More recently, the power of this organism as a genetic model has been applied to alter the expression of components of the ETC [10-12] or of scavengers of ROS [2, 5, 13, 14]. In particular, scavengers both within the mitochondrion (e.g. mitochondrial superoxide dismutase (mtSOD) and glutathione reductase) and within the cytosol (e.g. cytoplasmic SOD and catalase), have been studied in detail [2, 5, 15, 16].
Defects in the ETC are linked to ROS generation, and can decrease respiration rates, alter oxidative damage to cells, delay development, and impair fertility [11, 12, 17]. Initial theories linked the amount of oxidative damage and lifespan in several mitochondrial mutants. For example, several short-lived mutants (e.g. gas-1, a complex I mutant; mev-1, a complex II mutant) have increased oxidative damage to mitochondrial proteins [18, 19], while the long-lived mutant clk-1 (clk-1 codes for an enzyme required for ubiquinone synthesis) displays significantly less oxidative damage to mitochondrial proteins than wildtype [19].
If ROS play a major role in aging, one would expect that defects in ROS scavenging should adversely affect lifespan. C. elegans has five different genes that encode different superoxide dismutases (sod-1-5), which catalyze the enzymatic conversion of the toxic superoxide molecule to the less reactive species, hydrogen peroxide [13]. sod-1 encodes the major cytosolic superoxide dismutase (SOD), while sod-2 encodes the primary SOD found in the mitochondrion [5]. Expression of sod-1 or sod-2 was increased in C. elegans under conditions that increased lifespan [20].
Keaney et al. showed that when C. elegans was exposed to superoxide generators [21] addition of superoxide dismutase mimetics lengthened lifespans, though not back to normal. Under normal conditions these mimetics did not affect lifespans. However, the role of ROS in aging is disputed [2, 3, 13, 14]. Hekimi reported that a deletion of sod-2 lengthened lifespan, and that clk-1;sod-2 lived longer than the long-lived clk-1, despite increased oxidative damage in mitochondrial protein [5]. In fact, Van Raamsdonk eliminated all of the superoxide dismutases in C. elegans without shortening lifespan [22]. The overall conclusion was that superoxide dismutase had little relevance to normal lifespan in C. elegans[2, 3, 5, 13, 22].
Several authors have attempted to clarify the role of SODs in determining lifespan by the use of other longevity mutants. The well-studied and long-lived mutant daf-2 initiates a longevity pathway involving the insulin/IGF-I signaling pathway that ultimately causes nuclear localization of DAF-16, a FOXO transcription factor. The activation of daf-16 increased expression of sod-3, an mtSOD normally expressed in very low levels in wild type worms [20]. However, elimination of both mitochondrial sod-2 and sod-3 failed to suppress the long life span of daf-2[5, 23]. These studies suggested that upregulation of sod-3 (and presumably decreased superoxide damage to mitochondria) did not significantly contribute to the long life span of daf-2. However, others have shown that superoxide levels may be decreased by non-SOD mechanisms involving activation of the insulin/IGF-I signaling pathway, implying that superoxide may still play a role in aging in daf-2[24, 25]. The protein product of gst-10 scavenges the toxic ROS-generated product, 4-hydroxynonenal (HNE) [26]; its overexpression extends the lifespan of N2 while the gst-10 null reduces lifespan [27, 28], in support of a role for ROS damage in aging.
In the majority of studies that implicate ROS damage or mitochondrial function in the animal’s phenotype, these parameters are generally inferred, and seldom measured. Difficulties intrinsic to measuring ROS production/damage or mitochondrial function have limited the rigorous interpretation of data. Currently, the data concerning ROS and aging are complicated, even in a simple animal model. In this study, our goal was to determine the relationship between mitochondrial function, oxidative damage, and lifespan in C. elegans by studying the effect of mtSOD mutants on the ETC, as well as determining the effect of eliminating mtSOD activity in mitochondrial mutants. To date, there have been few reports of the effects of the mitochondrial sods on mitochondrial function [29], their role in minimizing ROS damage to the mitochondrial machinery, or their relative importance, if any, in the face of mitochondrial dysfunction. Our results show that the mitochondrial SODs are bound to components of the ETC, and that effects of loss of mtSODs can counteract each other, thereby complicating earlier simple interpretations of the roles of mtSODs in health and aging.
Results
Specific extension of lifespan by sod-2 and sod-3
As previously reported, gas-1(fc21) (a complex I mutant), and mev-1(kn1) (a complex II mutant), are short-lived in comparison to wild type (N2) (Figure 1) [11, 12]. sod-2(gk257) had a normal lifespan in agreement with Gems and colleagues [2, 21]. However, the loss of sod-2 in three mutant backgrounds that share common mitochondrial phenotypes, including sensitivity to oxidative stress, had very different results. sod-2;gas-1 lived longer than both gas-1(fc21) and the wild type, N2. sod-2;mev-1 was short-lived and not different from mev-1 (Figure 1). sod2;isp-1 (isp-1 is a complex III mutant) was a synthetic lethal.
Figure 1. Lifespans, effects of sod-2.
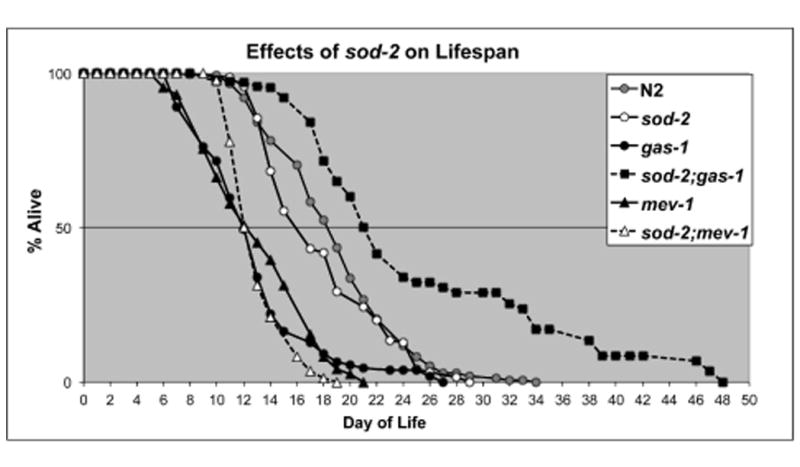
Lifespan studies started on day 0 defined as the day of hatching. Lifespans of wild type (N2), gas-1, mev-1, sod-2, sod-2;mev-1 and sod-2;gas-1. sod2;gas-1 lives longer than N2 (median lifespans, N2 19.2 ±0.5d, sod-2, 18.1 ±0.8d, gas-1, 11.4 ±0.4d, sod-2;gas-1, 24.2 ±0.9d).
In agreement with previous studies [2, 13], the lifespan of sod-3 was not different from N2 (Figure S1). However, the mutation sod-3 differed from sod-2 in its interactions with gas-1, mev-1 and isp-1. The lifespan of gas-1sod-3 was not different than that of gas-1. mev-1;sod-3 had a significantly shorter lifespan than did mev-1 (Figure S1). The lifespan of isp-1;sod-3 was significantly longer than either N2, isp-1 or sod-3 alone (Figure S1). sod-2;sod-3 had a lifespan similar to N2 as previously reported [13] (Data not shown).
Effects of sod-2 and sod-3 on reproductive capacity and post-embryonic development
We measured times of postembryonic development (Figure 2A) and rates of egg-laying (Figure 2B) in all mutants as measures of health. Compared to N2, post-embryonic times (from egg to adult) of sod-2, gas-1 and mev-1 were increased and fecundities were decreased. However, lengths of the reproductive periods (from adult to sterile), were decreased in sod-2 and mev-1, but increased in gas-1 (Figure 2A). sod-2 did not alter the times of post-embryonic development of gas-1 and mev-1 but did decrease their reproductive capacities (Figure 2B). sod-3 did not affect the development or fecundity of gas-1, mev-1, or isp-1 (data not shown).
Figure 2. Developmental Traits.
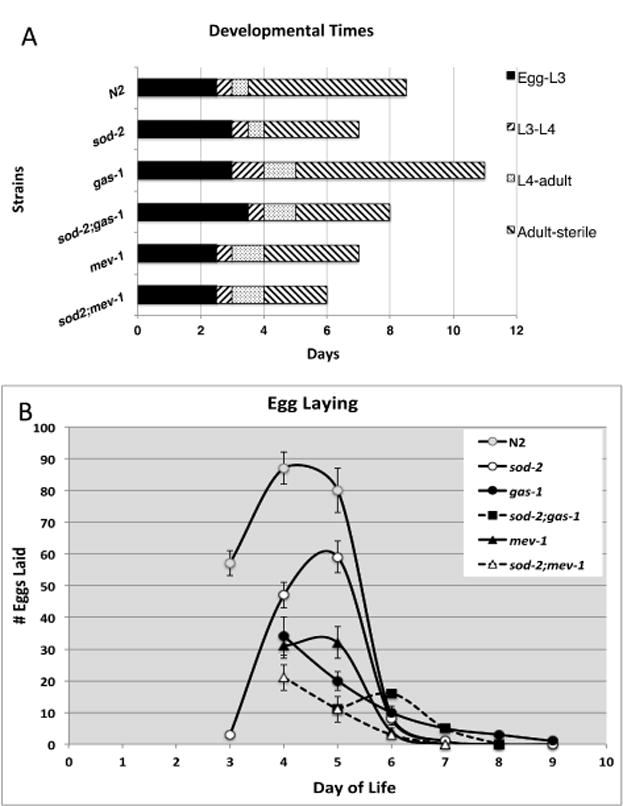
A. Post-embryonic Times. Rates of post-embryonic development of the animals were determined from synchronized cultures beginning at day 0 as eggs. Adult animals were scored until cessation of egg laying. Adult-sterile refers to the time from first day of adulthood to ceasing to lay eggs. At least 100 eggs were followed in each experiment and repeated three independent times. B. Egg Laying. Fecundity of the animals was measured by the number of eggs lain per worm per day. Day 0 was the day that parental animals were eggs. Data are represented as mean ± SEM from three independent worm cultures (totaling 20-30 adult animals for each strain).
Effects of sod-2 and sod-3 on respiration and electron transport chain function
Compared to N2, the complex I-dependent state 3 oxidative phosphorylation capacity was significantly decreased in sod-2 but not in sod-3 animals (Figures 3A, B). No additional effect on respiration was seen in sod-2;sod-3 compared to sod-2. Complex I respiratory capacity was less in sod-2;gas-1 than either single mutation alone (Figure 3A). Complex II-dependent respiration was unchanged in the sod mutants (3B). Respiration in other mitochondrial mutants was as previously described [11, 30]. State 4 respirations were identical to N2 in all mutants (data not shown). Because of the specific decreased complex I-dependent respiratory capacity of sod-2, we next examined specific enzymatic activities of the electron transport chain.
Figure 3. Oxidative Phosphorylation Studies.
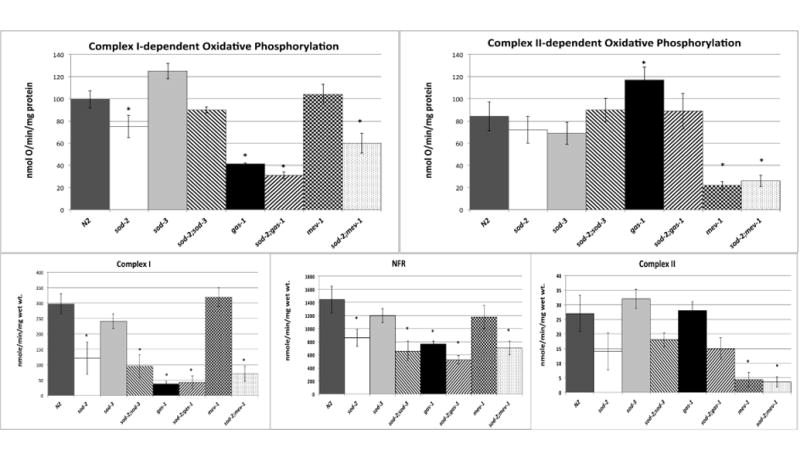
A. Complex I. State 3 rates of oxidative phosphorylation of wild type, gas-1, mev-1, sod-2, sod-3, sod-2;sod-3, sod-2;gas-1 and sod2;mev-1 using malate as a substrate. gas-1, sod-2, sod-2;gas-1 and sod-2;mev-1 each have decreased complex I dependent respiratory capacities. B. Complex II. State 3 Complex II dependent rates using succinate as a substrate. No change in complex II-dependent rates were noted in sod-2 or sod-3. C-E. Electron transport chain function. Respiratory chain enzymatic activities of wild type, gas-1, mev-1, sod-2, sod-3, sod-2;sod-3, sod-2;gas-1 and sod-2;mev-1. Enzymatic activity of: C. CI. D. NFR. and E. CII are shown. Complex I rates were decreased in sod-2 and sod-2 containing double mutants. CIII and CIV rates were normal in all mutants and are not shown. Data in all panels are represented as mean ± SEM from three to eight independent worm cultures. * indicates statistical significance as p < 0.05 in comparison to wild type (with a Bonferroni correction for n=8 resulting in p<0.00625 for significance as described in Statistical Methods).
The respiratory chain mutants had ETC enzymatic rates as previously described (Figures 3C-E) [9, 11, 12, 30]. Compared to N2, sod-2 significantly reduced complex I-related activities; the change in complex II rate did not reach significance (p=0.01) (Figures 3C-E). NADH ferricyanide reductase (NFR), which measures activity through the very proximal part of complex I and is taken as a measure of the amount of the complex [31], was decreased in sod-2. Complex I activity, which measures flow of electrons through the entire complex, was decreased to a greater extent in sod-2 than was NFR (Figures 3C,D). Similar to gas-1, sod2;gas-1 exhibited decreased activities of complex I (Figures 3C,D) and I-III (not shown). sod2;mev-1 exhibited decreased complex I and complex II function (Figures 3C,E). Complex II function of sod-2;mev-1 was not significantly different than that of mev-1 (Figures 3E). sod-3 showed no differences from N2 in complex I or II activities (data not shown). Complex III and IV activities of all the mutants remained normal (data not shown).
Changes in the formation of mitochondrial supercomplexes
Since sod-2 exhibited complex I respiratory dysfunction (Figure 3A), we determined whether the formation of mitochondrial I:III:IV supercomplexes was affected in sod-2(0) animals. BNGs revealed a 28% decrease in formation of supercomplexes I:III and I:III:IVn in sod-2(0) mitochondria by complex I in-gel activity (CI-IGA) (Figures 4A,B). The amount of complex I in the supercomplexes was decreased in all other mutants except mev-1 as also determined by CI-IGA (Figure 4B). sod-2;gas-1 had less complex I containing supercomplexes than either sod2 or gas-1 (30% of that in gas-1 and 10% of that in sod-2). The loss of complex I in sod-2 was further shown by a decrease in the complex I protein NDUFS2 in the supercomplexes (Figure 4C). sod-3 BNGs were normal (Figure 5E).
Figure 4. Supercomplex formation.
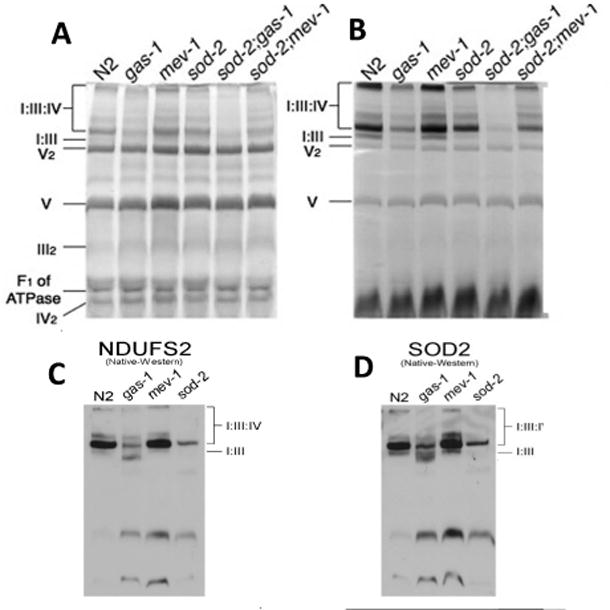
Digitonin-based blue native gels (BNGs) from frozen mitochondrial pellets of N2 and the mutants as indicated. A. Coomassie staining of BNGs. Identification of supercomplex I:III (I:III), supercomplex I:III:IV (I:III:IV), dimeric complex V (V2), monomeric complex V (V), dimeric complex III (III2), unbound form of the F1 part of complex V (F1 of ATPase) and dimeric complex IV (IV2) are inferred from previously published proteomic data and molecular weights [45]. B. Complex I in-gel-activity (CI-IGA) staining from the same samples as in A in a parallel gel. C. A Western blot of a BNG probed with anti-NDUFS2, a complex I subunit. NDUFS2 is the mammalian homologue of GAS-1. This blot confirms the decrease in the amount of complex I in supercomplexes of gas-1 and sod-2. D. Western blot from BNGs of mitochondrial mutants probed with anti-SOD-2. Note that SOD-2 is localized to the I:III:IV supercomplex. All gels were repeated three independent times. Figures are representative gels or blots.
Figure 5. Oxidative damage to proteins.
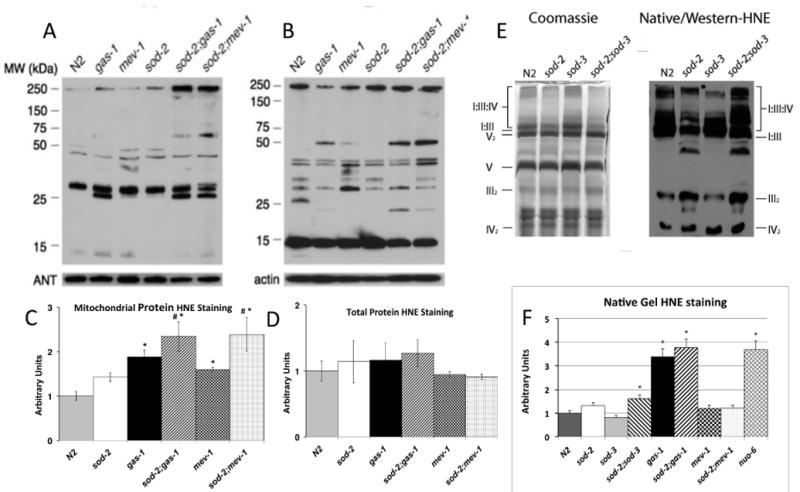
Western blots of A. mitochondrial protein and B. total worm protein probed with anti-HNE antibody. Adenine nucleotide translocase (ANT) and actin were used as loading controls as shown. C, D. Quantification of HNE-modified protein from A and B (C and D, respectively). Legend for strains applies to both C and D. Histogram fill patterns for strains are the same as for Figures 3 and 5F. Data shown in arbitrary units are normalized to wild type (N2) levels as mean ± SEM from three independent samples. * indicates different than amount in N2, p<0.05 in comparison to wild type (with a Bonferroni correction for n=6 resulting in p<0.0083 for significance as described in Statistical Methods). # indicates the amount of staining in sod-2;gas-1 and sod-2;mev-1 is similarly increased compared to gas-1 and mev-1, respectively. E. BNG and Western blot from BNGs of sod mutants (probed with anti-HNE). F. Ratios of HNE oxidative damage to Coomassie staining in a sister BNG normalized to N2 (taken as unity) in multiple mitochondrial mutants. The BNG and Western blot are shown in Figure S4, supplemental materials. The values are the averages from three gels. Strains containing complex I mutations (gas-1 and nuo-6) show the most increase in relative HNE staining. sod-2;sod-3 also showed a significant increase in HNE staining, compared to N2. Each gel was repeated at least three times.
We then studied the location of SOD-2 by BNG analysis. Western blots of BNGs indicated that SOD-2 co-localized with the I:III:IV supercomplex (Figure 4D). We have confirmed this finding in BNGs from mouse mitochondria as well (data not shown). The residual binding of anti-SOD-2 in the sod-2 mutant was much decreased in the sod-2;sod-3 mutant indicating that there was cross-specificity between SOD-2 and SOD-3 for this antibody (Figure S2). In addition, the results show that SOD-3 is also localized to the I:III:IV supercomplex.
HNE damage in mitochondrial protein and total worm protein
The loss of sod-2 increased HNE damage to mitochondrial protein in gas-1 and mev-1, but not total worm protein (Figures 5A-D). The extent of oxidative damage to supercomplex protein was assessed by normalizing HNE staining to total Coomassie staining. BNGs of mitochondria showed an increase in HNE staining in the sod-2;sod-3 double mutant compared to either mutant alone or N2 (Figure 5E). In those mutants containing complex I mutations (gas1 or nuo-6), HNE staining more than doubled compared to N2 (quantification shown in Figure 5F) in Western blots of native gels (Figure S3).
The response of antioxidant defense mechanisms
Our earlier studies comparing gene expression of gas-1 and mev-1 showed that sod-3, hsp-6 (involved in the mitochondrial unfolded protein response; mtUPR) and glutathione-S-transferase-14 (gst-14) were significantly increased in gas-1 but not mev-1[32]. Others have shown that the mtUPR plays an important role in determining lifespan [33, 34]. However, changes in the mtUPR did not explain the effects of sod-2(0) on gas-1 and mev-1 (Figures S4, S5). In order to further understand the role of antioxidant defense mechanisms in the lifespans of mitochondrial mutants, we studied the expression of genes encoding antioxidant proteins by real time PCR (Table 1). Expression of sod-1, a cytoplasmic superoxide dismutase, remained unchanged in all mutants in comparison to wild type. gas-1 and mev-1 did not alter the expression of sod-2 compared to wild type. Expression of sod-3 and gst-14 were increased in all strains except those containing mev-1. The highest levels of both were found in sod-2;gas-1. We also found that a triglyceride lipase (K04A8.5) (providing a potential alternate substrate source in a complex I mutant) was upregulated in gas-1 and sod-2;gas-1 (2.9(±1.3) and 7.06(±1.8) fold compared to N2). Since both sod-3 and K04A8.5 are known to be downstream from daf-16 (the FOXO transcription factor in the daf-2 pathway), we hypothesized that the lifespan extension in sod-2;gas-1 might be daf-16 dependent. As reported earlier by Kondo et al. [35], we found that DAF-16 was localized to the nucleus in gas-1 and mev-1 (not shown). However, we found that exposure of gas-1 or sod-2;gas-1 to daf-16 RNAi extended lifespan (mean lifespan gas-1:10 days vs. 19 days (grown on daf-16 RNAi); sod-2;gas-1: 17 days vs. 24 days (grown on daf-16 RNAi)). Thus, the extended lifespan of sod-2;gas-1 is not daf-16 dependent.
Table 1. Relative quantification of mRNA level of antioxidant genes.
Levels of expression of 4 antioxidant genes (sod-1, sod-2, sod-3and gst-14) in the mutants were normalized to that of the wild type. Data are represented as mean ± SEM from three to six day 1 adult cultures.
| Strains\Genes | sod-1 | sod-2 | sod-3 | gst-14 |
|---|---|---|---|---|
| N2 | 1 ± 0.1 | 1 ± 0.2 | 1 ± 0.1 | 1 ± 0.3 |
| gas-1 | 1.4 ± 0.3 | 0.8 ±0.2 | 7.8 ± 1.3* | 70.9 ± 1.2* |
| mev-1 | 0.9 ± 0.2 | 0.8 ±0.1 | 3.5 ± 0.8* | 0.04 ± 0.02* |
| sod-2 | 0.9 ±0.1 | 0 | 5.4 ± 0.8* | 19.7 ± 1.8* |
| sod-2;gas-1 | 1.4 ± 0.2 | 0 | 36.2 ± 2.1* | 305.9 ± 124.1* |
| sod-2;mev-1 | 1.4 ± 0.0 | 0 | 6.8 ± 0.7* | 2.3 ± 1.7 |
indicated statistical significance as p< 0.05 in comparison to wild type (with a Bonferroni correction for n=5 resulting in p<0.01 for significance as described in Statistical Methods).
If increased expression of sod-3 or gst-14 causes the prolonged lifespan of sod-2;gas-1, then interfering with their expression should shorten the lifespan of sod-2;gas-1. We found that the triple mutant sod-2;gas-1sod-3 was lethal, consistent with this hypothesis. In contrast, and contrary to this hypothesis, RNAi knockdown of gst-14 in sod-2;gas-1 animals more than doubled their lifespan on the same food source (HT115) (Figure 6). Development of those animals is accelerated compared to sod-2;gas-1 animals grown in the absence of gst-14 RNAi (data not shown). We obtained similar results for the effect of gst-14 RNAi on gas-1 animals (prolonged lifespan) (Figure 6). In both cases, the lifespan curves were biphasic, in that a subset of animals died very early in life (app. day 9) while others were long-lived. Exposure to gst-14 RNAi did not prolong the lifespan of mev-1 (Figure 6), nor the wildtype N2 (data not shown).
Figure 6. Compensatory responses of sod-2.Effects ofgst-14RNAi on lifespans of mitochondrial mutants.
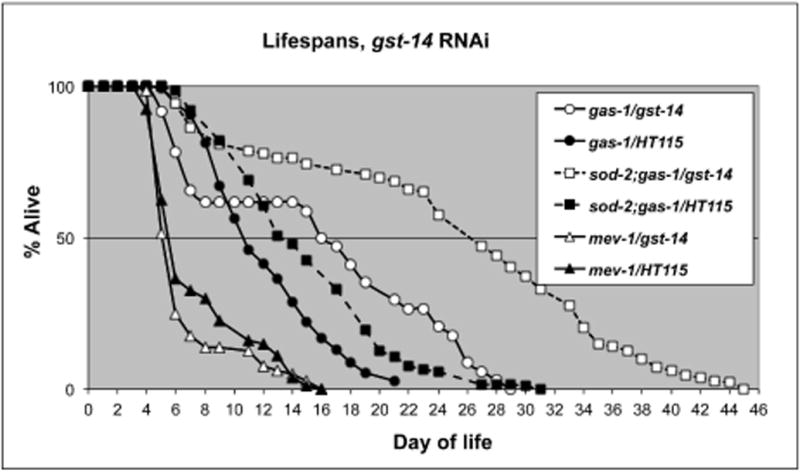
gst-14 RNAi caused a significant increase in the lifespan of gas-1 (mean lifespan 15.7 days with RNAi vs. 10.1 days without RNAi) and sod-2;gas-1 (mean lifespan 28.4 days with RNAi vs. 13.4 days without RNAi).
Discussion
We expected that combining the loss of SOD-2 with mitochondrial mutants known to increase oxidative stress [4, 12, 35] would be detrimental to lifespan. Loss of SOD-2 did in fact slow development and decrease fecundity compared to N2, and significantly worsened those phenotypes when coupled with gas-1 and mev-1. Surprisingly though, loss of SOD-2 specifically increased the lifespan of gas-1, but not that of mev-1. We could not attribute the lengthened lifespan of sod-2;gas-1 to any improvement of respiratory function of gas-1 or change in levels of ROS damage. In fact, no single component of mitochondrial physiology that we studied correlates simply with lifespan (Figure 7). However, loss of sod-2 directly decreased mitochondrial respiration, supercomplex formation, and ETC function. Therefore, the scavenging functions of the mtSODs must be considered in conjunction with their effects on respiration itself. Others have previously reported decreases in complex I and II-dependent function in SOD-2 deficient mice [29, 36]. This dependence of complex I function on SOD-2 extends to C. elegans as well, and as such implies an ancient conserved dependence of the ETC on this ROS scavenger.
Figure 7. Comparison of multiple aspects of mitochondrial physiology with lifespan in mitochondrial mutants.
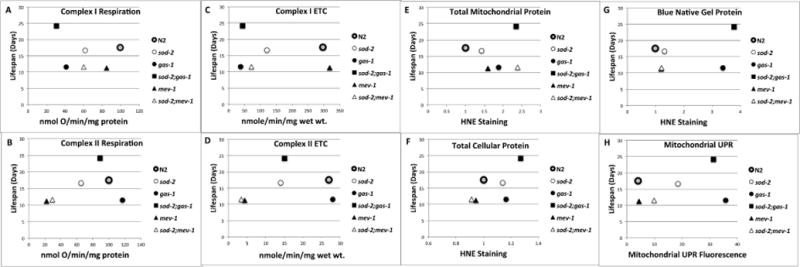
Correlation of several different aspects of mitochondrial physiology with lifespan of six strains of C. elegans. Lifespan values are from Figure 1; Respiration and ETC values are from Figure 3; HNE staining values are from Figure 5; Mitochondrial UPR fluorescence are from Figure S4. Symbols for the strains are the same as in Figure 1. The symbol for N2 is enlarged in all graphs for comparison to other strains. A. Complex I dependent oxygen consumption. B. Complex II dependent oxygen consumption. C. Complex I ETC activity. D. Complex II ETC activity. E. 4-hydroxynonenal (HNE) antibody staining of total worm protein. F. HNE antibody staining of total mitochondrial protein. G. HNE staining of complexes from blue native gels (BNGs). H. Mitochondrial UPR dependent fluorescence as measured by HSP-6::GFP expression normalized to that of N2 expressed in arbitrary units. No significant correlation was found with any measurement when compared to lifespan.
The surprising presence of SOD-2 in I:III:IV supercomplexes (Figure 4D) indicates that SOD-2 plays a pivotal role in local scavenging of superoxide at the site of its production. However, the lack of a large increase in ROS damage to supercomplexes in a sod-2 animal may indicate that SOD-3 is also capable of performing this function, especially when the amount of supercomplex I:III:IV is reduced. This possibility is strengthened by two observations: 1) ROS damage to the supercomplexes is relatively increased if both SOD-2 and SOD-3 are absent; and 2) in an animal that is sensitive to oxidative stress (gas-1), loss of both mtSODs is lethal. Our findings are consistent with the hypothesis that the mtSODs, which are associated with the I:III:IV supercomplex, protect components of the supercomplex from ROS damage; their loss is accompanied by damage to specific proteins in the mitochondrial ETC (Figures 5E,F).
A dependence of the amount of supercomplex I:III:IV on SOD-2 in supercomplexes has not been previously noted. Maranzana et al.[37] did report that the stability of the I:III:IV supercomplex was linked to superoxide production in bovine heart mitochondria. In contrast to our results, they found increased ROS when I:III:IV deteriorated. However, they measured ROS production from an intact complex I that had been acutely dissociated from the supercomplex with detergent. We measured accumulated ROS damage generated in vivo from animals that had lifelong complex I dysfunction due to the loss of SOD-2. These animals also upregulated expression of the inducible mtSOD in worms, sod-3. Lifelong decreased mitochondrial respiration, with increased SOD-3, partially accounts for the lack of increased ROS damage in gas-1 and sod-2;gas-1. We further postulate that the loss of SOD-2 (in sod-2;gas-1) locally increases ROS damage to the key components of the ETC causing a dissolution of supercomplex I:III:IV, which in a negative feedback loop, decreases complex I-dependent respiration and limits superoxide production from complex I. However, since complex I function is decreased in sod-2 mitochondria out of proportion to the amount of ROS damage, it is also possible that the mtSODs may directly serve as stabilizing factors in the I:III:IV supercomplex.
The loss of sod-2 did not result in global cellular ROS damage to proteins (Figures 5A-D), nor in a large increase in ROS damage to mitochondrial proteins. This is perhaps not surprising since loss of superoxide dismutase, by itself, will not increase ROS production nor decrease the ultimate conversion of ROS to non-harmful molecules. The increases in HNE staining seen in total mitochondrial proteins in sod-2;gas-1 and sod-2;mev-1 (Figure 5A) are in proteins not maintained in MRC complexes in native gels (Figure 5F). These proteins may be soluble matrix proteins, which normally dissociate from the MRC complexes under conditions used for BNGs, or damaged proteins that are eliminated from the MRC complexes. We favor the third possibility, though further study is necessary to confirm the model.
We also found that sod-3 expression was significantly upregulated in sod-2 animals (Table1), in contrast to Back et al[38]. However, their study did find a 1.5 fold increase in sod-3 expression that did not reach statistical significance. Our nematodes were grown on OP50, not K12 as in the Back study. It is possible that the decreased bacterial lawn of the uracil-requiring bacteria (OP50) introduced an extra level of stress resulting in more upregulation of sod-3.
How does sod-2 extend the lifespans of gas-1 and clk-1[5] but not mev-1 or isp-1? Both gas-1 and clk-1 have decreased complex I function as well as increased ROS production [18, 19, 30, 39]. The extended lifespans of sod-2;gas-1 and sod-2;clk-1[5] likely result from decreased metabolism coupled with an increase in expression of sod-3 (and perhaps other ROS scavengers). The slowing of complex I-dependent respiration can be viewed as a negative feedback mechanism to limit the production of ROS from the I:III:IV supercomplex in the face of defective superoxide scavenging. In addition, it appears that sod-2 function is necessary for the upregulation of complex II activity normally seen in gas-1. It is unclear why this is so, but this change may further decrease the metabolic rate in sod-2;gas-1 and also limit the production of superoxide. Although enzymatic activity of complex II is decreased in sod-2;mev-1, overall integrated function of the mitochondrion, as reflected in mitochondrial respiratory capacity, is not affected.
Loss of sod-3, which is upregulated in gas-1, does not change the lifespan of gas-1, yet it increases the lifespan of isp-1, and decreases the lifespan of mev-1. We do see a small increase in complex I activity staining in BNGs of sod-3 animals; perhaps this partially compensates for the loss of complex III in isp-1[31] and can extend its lifespan. If so, one might presume that this same phenomenon would operate in mev-1, yet loss of sod-3 in mev-1 decreases its lifespan. Studies are now being undertaken to characterize the interaction of sod3 with supercomplex I:III:IV formation.
The role of gst-14 as a response to complex I defects is also not clear. Several forms of GST proteins are known to scavenge HNE-damage proteins. The HSP-6 family of proteins (also upregulated in gas-1[32]) have been shown to modify GST proteins to cause mitochondrial targeting [40]. We anticipated, given the tremendous upregulation of this molecule in sod-2;gas-1, that gst-14 overexpression was linked to the extension of lifespan of sod-2;gas-1 compared to gas-1. However, reduction in gst-14 expression lengthened the lifespan of both gas-1 and sod2;gas-1. The protein GST-14 is most homologous to the mammalian pro-inflammatory protein, hematopoetic prostaglandin D2 synthase (HPGDS) [41, 42]. Given this homology, we hypothesize that GST-14 actually is responsible for an inflammatory response in nematodes, rather than ROS scavenging and are testing this possibility. It should be noted that there are several other gst genes in C. elegans that code for homologues of HPGDS; it is not known whether gst-14 is unique in its role in governing lifespan. Regardless of the specificity of gst-14, the lifespan of mitochondrial mutants is governed by an interaction of metabolic rate, ROS damage and an inflammatory response.
There are some differences in the lifespans in our laboratory compared with reports of others. First, we consistently find little to no increase in the lifespan of isp-1 compared to N2. This may result partially from our reluctance to remove worms from our lifespan measurements that contain one internal hatched offspring (a “bagged” worm) in adults exhibiting almost no mobility on the prior day (10-15% of the adults in our studies). In our opinion, these animals represent dead adults that have retained one hatched egg. However, this does not fully explain the differences between our results and those of others. Second, Van Raamsdonk reported a decrease in lifespan for the sod-2;isp-1 double mutant [13]. We were unable to obtain viable sod-2;isp-1 offspring and so report this as a lethal. We do not know the cause for our differences in isp-1 related results, though we suspect feeding differences may play a role. Finally, the finding that loss of DAF-16 increases the lifespan on gas-1 is extremely surprising. We suspect either that gst-14 expression is DAF-16 dependent or that the metabolic changes dependent on DAF-16 [43] interact with gas-1.
The effects of sod-2 and sod-3 mutants on the viability and lifespan of gas-1, mev-1 and isp-1 support the findings of others that there is not a simple correlation between mitochondrial respiration, superoxide scavenging, ROS damage and lifespan [2, 3, 5, 13, 19, 22] (Figure 7).Clearly the notion that loss of mtSODs simply increases ROS levels and damage is too simplistic. Superoxide dismutase does not actually remove potentially damaging ROS; rather it only speeds up the conversion from one form (superoxide) to another (hydrogen peroxide).
In summary, we have found specific interactions between mitochondrial mutants and the mitochondrial superoxide dismutases. It is clear that losses of mitochondrial SODs have unanticipated effects on lifespan and mitochondrial function. Since they are associated with mitochondrial supercomplexes, they may regulate electron transport or supercomplex stability in a negative feedback loop, and protect specific proteins within the ETC from ROS damage. Paradoxically, loss of sod-2 may actually serve as a control point that limits maximal ROS production and keeps levels of ROS damage relatively constant as found by Yang [4]. Our data would indicate that this decrease is mediated by dissolution of supercomplexes, which are stabilized by the mtSODs, which in turn decreases mitochondrial respiration. Evolution would likely favor linkage of the presence of a ROS scavenger to the physical entity which generates the ROS.
Experimental Procedures
C. elegans strains and culture
Worms were maintained on NGM agar plates seeded with OP50 bacteria at 20°C. Strains obtained from the Caenorhabditis Genetics Center include; N2 Bristol (wild-type), gas-1(fc21), mev-1(kn1), isp-1(qm150), sod-2(gk257), sod-3(tm760) and SJ4100 (zcIs13[hsp-6p::GFP]). SJ4100 was kindly shared by Matt Kaeberlein (UW) and Andrew Dillin (UCSF). Double mutants were generated by standard methods when visible phenotypes were available. Cross-progeny were identified by PCR if there were no visible phenotypes. All double mutants were seuenced to confirm genotype and outcrossed a minimum of 4 times.
Lifespan analysis
All lifespan assays were conducted at 20°C on fresh OP50 NGM plates or HT115 plates in RNAi studies. Synchronized cultures of C. elegans were generated by seeding gravid young adults on plates to lay eggs for 4 hours on day 0. Individual plates were maintained with a maximum of 25 worms per plate. Worms were transferred to new plates every 2 days to avoid counting offspring of the original cohort. Worms that were not active were gently prodded by a platinum worm pick; if worms did not move when prodded 3X they were scored as dead. Nematodes that died abnormally (bagging, trapped on the side of the petri dish) were excluded from counting as previously described [12]. A minimum of 100 animals were followed for each lifespan experiment and each lifespan study was repeated at least three times.
Egg laying and hatching
Synchronous young adult worms were allowed to lay eggs for 4 hours on day 0. On day one, five L1 or L2 worms were transferred to fresh plates. Once these animals began to lay eggs, they were transferred daily until they no longer laid any eggs. The number of days until the first eggs were laid, the total number of eggs laid and the number that hatched were determined on each plate. All cultures were maintained at 20°C.
Oxidative Phosphorylation and Electron Transport Chain (ETC) assays
Mitochondria were isolated as previously outlined [30, 39]. Mitochondrial respiratory capacity was measured as previously described using freshly prepared mitochondria [30, 39]. The mitochondrial samples that were isolated for oxidative phosphorylation assays were also used as fresh samples for the ETC assays. ETC assays were carried out using techniques previously described [44]. All ETC assays were carried out on fresh mitochondrial samples that were kept on ice and repeated on three independent isolates.
Blue Native Gels (BNGs) and In-gel Activity (IGA)
BNGs were performed as previously described [45]. Western blots and in-gel activities (IGA) of BNGs were done as described by Suthammarak [31]. All gels were repeated on three independent samples.
Western Blots
For Western blots of SDS-PAGE gels and native gels, mitochondria were isolated as described previously [31, 45]. 50 ug of mitochondrial protein from each strain was loaded onto a 12% SDS-PAGE gel. The gel was run and transferred as previously described [31, 45]. Blots were probed with a primary antibody to either 4-hydroxy-2-nonenal (HNE) fluorophore (Calbiochem, San Diego, CA), NDUFS2 (Santa Cruz Biotechnology, Dallas, TX) or SOD-2 (Santa Cruz Biotechnology, Dallas, TX). The membranes were washed as previously described and incubated with horseradish peroxidase-conjugated secondary antibody [19] (Santa Cruz Biotechnology, Dallas, TX). Blots were incubated with SuperSignal® West Pico chemiluminescence substrate. Adenosine Nucleoside Transporter (ANT) antibody (Mitosciences, Eugene OR) was used as a loading control.
Statistical Analysis
Analysis of variance was used to analyze groups of data for significant differences. Unpaired Student's t test was then employed to calculate statistical significance of specific pairs if a difference was noted with analysis of variance. Significance was defined as p<0.05 with a Bonferroni correction for the number of strains evaluated with each test (generally 8 strains resulting in a p<0.05/8=0.00625). Error bars in the figures represent mean ± S.D. for each experiment.
Supplementary Material
Acknowledgments
The authors thank the technical help of Beatrice Predoi, Judith Preston, and Katrina Elsaesser. This work was supported, in whole or in part, by National Institutes of Health Grants GM58881 and AG026273 (MMS and PGM) and by the Royal Thai Government, Science and Technology Scholarship (WS), and the Northwest Mitochondrial Research Guild (WS, PGM, MMS).
Abbreviations
- HNE
4-hydroxy-nonenal
- IGA
in-gel activity
- mtSOD
mitochondrial superoxide dismutase
- ETC
electron transport chain
- MRC
mitochondrial respiratory chain
Footnotes
Author Contributions. WS designed and performed experiments, evaluated results, contributed to the writing of the manuscript. BHS designed and performed experiments, evaluated results. EO designed methods, performed experiments and contributed to the writing of the manuscript. MMS and PGM designed and performed experiments, evaluated results, primarily wrote the manuscript.
Contributor Information
Wichit Suthammarak, Email: wichit.sut@mahidol.ac.th.
Elyce Opheim, Email: elyce.opheim@seattlechildrens.org.
Margaret Sedensky, Email: sedenm@uw.edu.
References
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Doonan R, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22(23):3236–41. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gems D, Doonan R. Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle. 2009;8(11):1681–7. doi: 10.4161/cc.8.11.8595. [DOI] [PubMed] [Google Scholar]
- 4.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8(12):e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang W, Li J, Hekimi S. A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics. 2007;177(4):2063–74. doi: 10.1534/genetics.107.080788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koopman WJ, et al. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal. 2010;12(12):1431–70. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 7.Heidler T, et al. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010;11(2):183–95. doi: 10.1007/s10522-009-9239-x. [DOI] [PubMed] [Google Scholar]
- 8.Hartwig K, et al. Feeding a ROS-generator to Caenorhabditis elegans leads to increased expression of small heat shock protein HSP-16.2 and hormesis. Genes Nutr. 2009;4(1):59–67. doi: 10.1007/s12263-009-0113-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senoo-Matsuda N, et al. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J Biol Chem. 2001;276(45):41553–8. doi: 10.1074/jbc.M104718200. [DOI] [PubMed] [Google Scholar]
- 10.Yang W, Hekimi S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell. 2010;9(3):433–47. doi: 10.1111/j.1474-9726.2010.00571.x. [DOI] [PubMed] [Google Scholar]
- 11.Ishii N, et al. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394(6694):694–7. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- 12.Kayser EB, Sedensky MM, Morgan PG. The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegans. Mech Ageing Dev. 2004;125(6):455–64. doi: 10.1016/j.mad.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5(2):e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabreiro F, et al. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic Biol Med. 2011;51(8):1575–82. doi: 10.1016/j.freeradbiomed.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McElwee JJ, et al. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J Biol Chem. 2004;279(43):44533–43. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- 16.Sampayo JN, Olsen A, Lithgow GJ. Oxidative stress in Caenorhabditis elegans: protective effects of superoxide dismutase/catalase mimetics. Aging Cell. 2003;2(6):319–26. doi: 10.1046/j.1474-9728.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- 17.Grad LI, Lemire BD. Mitochondrial complex I mutations in Caenorhabditis elegans produce cytochrome c oxidase deficiency, oxidative stress and vitamin-responsive lactic acidosis. Hum Mol Genet. 2004;13(3):303–14. doi: 10.1093/hmg/ddh027. [DOI] [PubMed] [Google Scholar]
- 18.Hartman PS, et al. Mitochondrial mutations differentially affect aging, mutability and anesthetic sensitivity in Caenorhabditis elegans. Mech Ageing Dev. 2001;122(11):1187–201. doi: 10.1016/s0047-6374(01)00259-7. [DOI] [PubMed] [Google Scholar]
- 19.Yang YY, et al. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech Ageing Dev. 2009;130(6):370–6. doi: 10.1016/j.mad.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honda Y, Tanaka M, Honda S. Modulation of longevity and diapause by redox regulation mechanisms under the insulin-like signaling control in Caenorhabditis elegans. Exp Gerontol. 2008;43(6):520–9. doi: 10.1016/j.exger.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Keaney M, et al. Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic Biol Med. 2004;37(2):239–50. doi: 10.1016/j.freeradbiomed.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A. 2012;109(15):5785–90. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honda Y, Honda S. Oxidative stress and life span determination in the nematode Caenorhabditis elegans. Ann N Y Acad Sci. 2002;959:466–74. doi: 10.1111/j.1749-6632.2002.tb02117.x. [DOI] [PubMed] [Google Scholar]
- 24.Yanase S, Ishii N. Hyperoxia exposure induced hormesis decreases mitochondrial superoxide radical levels via Ins/IGF-1 signaling pathway in a long-lived age-1 mutant of Caenorhabditis elegans. J Radiat Res. 2008;49(3):211–8. doi: 10.1269/jrr.07043. [DOI] [PubMed] [Google Scholar]
- 25.Yazaki K, et al. Supplemental cellular protection by a carotenoid extends lifespan via Ins/IGF-1 signaling in Caenorhabditis elegans. Oxid Med Cell Longev, 2011. 2011:596240. doi: 10.1155/2011/596240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohn JA, et al. Chemical characterization of a protein-4-hydroxy-2-nonenal cross-link: immunochemical detection in mitochondria exposed to oxidative stress. Arch Biochem Biophys. 1996;328(1):158–64. doi: 10.1006/abbi.1996.0156. [DOI] [PubMed] [Google Scholar]
- 27.Ayyadevara S, et al. Life span and stress resistance of Caenorhabditis elegans are differentially affected by glutathione transferases metabolizing 4-hydroxynon-2-enal. Mech Ageing Dev. 2007;128(2):196–205. doi: 10.1016/j.mad.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ayyadevara S, et al. Lifespan extension in hypomorphic daf-2 mutants of Caenorhabditis elegans is partially mediated by glutathione transferase CeGSTP2-2. Aging Cell. 2005;4(6):299–307. doi: 10.1111/j.1474-9726.2005.00172.x. [DOI] [PubMed] [Google Scholar]
- 29.Melov S, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96(3):846–51. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kayser EB, et al. Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J Biol Chem. 2001;276(23):20551–8. doi: 10.1074/jbc.M011066200. [DOI] [PubMed] [Google Scholar]
- 31.Suthammarak W, Morgan PG, Sedensky MM. Mutations in mitochondrial complex III uniquely affect complex I in Caenorhabditis elegans. J Biol Chem. 2010;285(52):40724–31. doi: 10.1074/jbc.M110.159608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falk MJ, et al. Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Mol Genet Metab. 2008;93(4):388–97. doi: 10.1016/j.ymgme.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker BM, et al. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8(6):e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo M, et al. Effect of oxidative stress on translocation of DAF-16 in oxygen-sensitive mutants, mev-1 and gas-1 of Caenorhabditis elegans. Mech Ageing Dev. 2005;126(6-7):637–41. doi: 10.1016/j.mad.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 36.Van Remmen H, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;281(3):H1422–32. doi: 10.1152/ajpheart.2001.281.3.H1422. [DOI] [PubMed] [Google Scholar]
- 37.Maranzana E, et al. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex I. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Back P, et al. Effects of sod gene overexpression and deletion mutation on the expression profiles of reporter genes of major detoxification pathways in Caenorhabditis elegans. Exp Gerontol. 2010;45(7-8):603–10. doi: 10.1016/j.exger.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 39.Kayser EB, et al. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J Biol Chem. 2004;279(52):54479–86. doi: 10.1074/jbc.M403066200. [DOI] [PubMed] [Google Scholar]
- 40.Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. 2011;278(22):4243–51. doi: 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanaoka Y, Urade Y. Hematopoietic prostaglandin D synthase. Prostaglandins Leukot Essent Fatty Acids. 2003;69(2-3):163–7. doi: 10.1016/s0952-3278(03)00077-2. [DOI] [PubMed] [Google Scholar]
- 42.Mohri I, et al. Inhibition of prostaglandin D synthase suppresses muscular necrosis. Am J Pathol. 2009;174(5):1735–44. doi: 10.2353/ajpath.2009.080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robida-Stubbs S, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15(5):713–24. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoppel CL, et al. The malonyl-CoA-sensitive form of carnitine palmitoyltransferase is not localized exclusively in the outer membrane of rat liver mitochondria. J Biol Chem. 1998;273(36):23495–503. doi: 10.1074/jbc.273.36.23495. [DOI] [PubMed] [Google Scholar]
- 45.Suthammarak W, et al. Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J Biol Chem. 2009;284(10):6425–35. doi: 10.1074/jbc.M805733200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.