

Abstract
Preeclampsia remains a significant obstetric risk worldwide. The pathophysiology of preeclampsia is complex, with multiple stages involving maladaptations in both placental and maternal physiology. The placenta links the pre-clinical stage of impaired remodeling of the uterine vasculature, occurring in early pregnancy, to the later clinical stages characterised by the maternal syndrome of hypertension and proteinuria. This review focuses on some of the recent candidates for the missing links in this process.
Keywords: Preeclampsia, Placenta, Inflammation, Endothelium, Coagulation, Syncytiotrophoblast, Microvesicles, Exosomes, Alarmins, Gasotransmitters, sFlt-1, sEng, PlGF, Tissue factor, Hypertension
Introduction
Preeclampsia (PE) occurs only during pregnancy or in the early postpartum period and is generally defined as new hypertension (diastolic blood pressure of >90 mm Hg or systolic blood pressure of >140mm Hg) and substantial proteinuria (≥300 mg in 24 h) at or after 20 weeks of gestation [1]. It affects approximately 2–8 % of pregnancies worldwide, disproportionately affecting women in developing countries [2, 3]. Despite decades of research, the mechanisms underlying the cause and progression of PE remain poorly understood. Risk factors include maternal and paternal family history of PE, nulliparity, ethnicity and existing disorders that feature vascular dysfunction, hypertension or inflammation such as diabetes, chronic hypertension, obesity, kidney disease, systemic lupus erythematosus and antiphospholipid syndrome [4]. Due to a lack of predictive biomarkers and effective pharmaceutical interventions, PE continues to be a serious obstetric complication causing increased maternal and fetal morbidity and mortality.
As PE is a syndrome, defined only by the presence of clinical symptoms, diagnosis is not robust and prediction, as yet, is not possible. Women present with a spectrum of symptoms that can be used to broadly classify disease severity from mild to severe or late to early onset. Women with severe PE may also develop HELLP syndrome (haemolysis, elevated liver enzymes, low platelets), which reflects disorders of the liver and the clotting system. Complications of PE that can lead to maternal death include placental abruption, hepatic rupture, pulmonary oedema and acute renal failure [5]. When the vascular dysfunction, which is one component responsible for the maternal symptoms (see Maternal pathology section), includes the vasculature of the brain, then eclampsia can develop, characterised by seizures. Here, stroke and cerebral hemorrhage can occur and are the major cause of eclampsia-related deaths [6]. Throughout the world, monitoring of blood pressure and proteinuria are used to screen for PE. Therefore, at present, the most effective control of PE comes from good intrapartum care [7, 8].
The placenta is central to the development of PE. Its removal remains the only effective treatment to halt disease progression. The foundations for severe disease are laid down early in pregnancy with maladaptation of the uterine vasculature to the ensuing pregnancy leading to altered placental function and, in severe cases, damage (see Placental pathology section). Preeclampsia can, however, occur without placental pathology and vice versa, illustrating the complex etiology of PE due to the involvement of two genomes (mother and fetus) influenced both by each other and external factors.
This review focuses on the pathophysiology of PE and some of the recent placental and maternal factors implicated in critical stages of development of the maternal syndrome.
Placental Pathology
Preeclampsia is believed to begin during the first trimester of pregnancy with inadequate remodeling of the distal portion of the uterine spiral arteries [9]. Successful invasion and remodeling of the spiral arteries requires accumulation of specialised uterine natural killer cells (uNK) and macrophages close to spiral arteries, which begins the process of disruption of the vascular smooth muscle and endothelium [10]. Invasive extravillous cytotrophoblasts (EVC) then infiltrate through the decidua into the myometrium and line the lumen of the vessel to form a pseudoendothelium. This completes the spiral artery transformation from narrow vasoreactive vessels into high-capacity, low-resistance vessels able to carry a continuous flow of maternal blood under low pressure to the placenta surface (reviewed in [11]). In PE, the failure of EVC to invade adequately is associated with incomplete remodelling of the spiral arteries, and as a consequence, the retention of the highly contractile distal portion of the vessels. This increases the incidence of interrupted blood flow and the risk of ischaemia/reperfusion insult, which are strong stimuli for oxidative stress [9]. The immunological processes involved in this have been extensively reviewed elsewhere [12].
The consequences of impaired uteroplacental blood flow are evident in the placentas from PE pregnancies. Intermittent blood flow can alter placental development in severe cases, as seen by reduced terminal villi volume and surface area [13]. It also leads to ischaemia/reperfusion injury. If the resultant endoplasmic reticulum stress response is unable to restore cellular homeostasis and prevent oxidative and inflammatory stress, apoptosis/necrosis of the syncytiotrophoblast layer occurs [9, 14]. Gross pathological changes, such as acute atherosis and infarction, are most evident, but not exclusive to severe PE occurring preterm [15].
With placental dysfunction comes the impairment of fetal development. Preeclampsia is a major cause of fetal malnutrition, morbidity and mortality. Fetal growth restriction and premature delivery also have lifelong consequences for the children born to PE mothers, including an increased risk of cardiovascular disease [16].
Maternal Pathology
Systemic inflammation, endothelial dysfunction and hypercoagulation underlie the maternal symptoms of hypertension, proteinuria and disordered clotting. Multiple cellular compartments of the vasculature are affected, including endothelium, inflammatory leucocytes and platelets [17]. The systemic nature of the maternal response means that in severe cases, multiple organs including liver, kidney and brain can be adversely affected. Preeclampsia also leaves a lasting legacy of increased future cardiovascular risk for the women affected [16]. Prolonged endothelial dysfunction is a strong pro-atherogenic risk factor. However, whether it is endothelial injury sustained during a PE pregnancy, or preexisting endothelial dysfunction that predisposes to both PE and subsequent cardiovascular risk is not clear.
In normal pregnancy, maternal innate immune responses are activated to bring about an inflammatory state. This may play a beneficial role throughout pregnancy, as inflammatory cytokines are involved in the processes of implantation and placentation, and may also help the mother to fight infection due to suppression of her T and NK cell-mediated immune responses [17]. However, in PE, the activation of the innate immune response is more extreme [18], and the same is true for the complement system [19].
Maternal hypertension in PE is characterised by peripheral vasoconstriction and reduced arterial compliance [20–24]. Biochemical markers of endothelial activation are elevated in PE [16], with evidence of endothelial dysfunction in conduit vessels before the onset of clinical disease [21], persisting long after the PE pregnancy [25, 26]. Fenestrated endothelia, which are densely perforated with transcellular fenestrae that allow rapid movement of water and small solutes, are particularly affected. Proteinuria reflects the PE specific renal lesion glomerular endotheliosis, characterised by swelling of the glomerular endothelial cells and loss of their fenestrations [27]. Podocytes (glomerular epithelial cells) can also detach and are found in the urine (podocyturia) of PE women prior to the onset of clinical symptoms [28•].
Normal pregnancy is a procoagulant state, characterised by an increase in many procoagulant factors and markers of thrombin generation [29]. In PE, this physiological activation of coagulation is exaggerated and includes excessive platelet activation, increased fibrin degradation products, and intervillous fibrin deposition in the placenta [30]. Clinically, women can present with a spectrum of procoagulant phenotypes, from the subtle changes seen in mild PE to the disseminated intravascular coagulation seen in severe cases. Platelets isolated from women prior to developing clinical symptoms of PE also show signs of activation [31], suggestive of platelet hyper-responsiveness in women who subsequently develop PE.
Missing Links?
The placenta is key to the pathogenesis of PE. This is evidenced by the removal of the placenta abating the maternal symptoms, and the occurrence of PE in molar pregnancies, demonstrating the requirement for the placenta and not the fetus for disease development. The risk of developing PE also rises with increases in the mass of placental tissue, such as with multiple pregnancies.
Poor placental perfusion due to failure of adequate adaptation of the uterine vasculature provokes the release of a factor or multiple factors by the placenta into the maternal circulation, linking the placenta with maternal systemic inflammation, endothelial dysfunction and activation of the clotting system (Figure 1). Several placental and maternal factors recently implicated in the pathophysiology of PE are outlined below. This is by no means an exhaustive list, as new factors are continuously reported.
Fig 1.
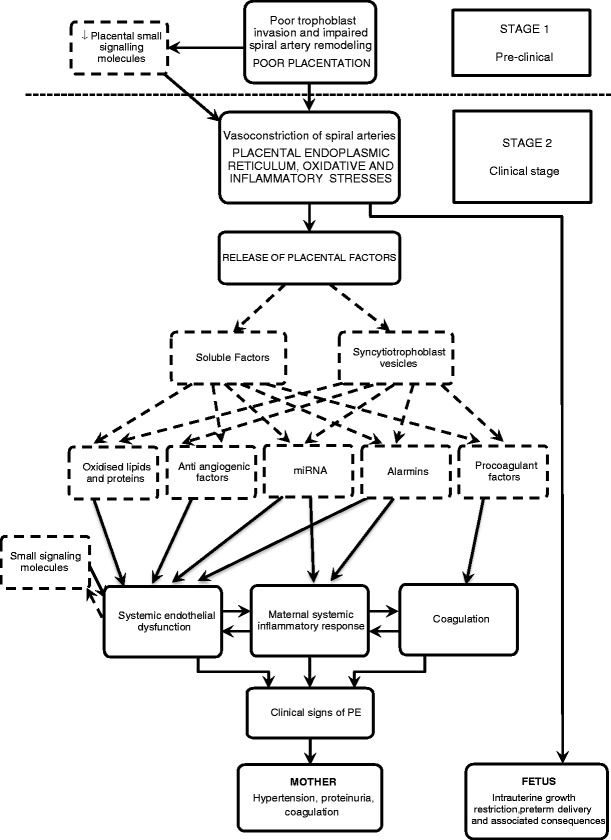
Schematic diagram illustrating the pathogenesis of preeclampsia and the possible involvement of factors recently implicated in disease progression. Dashed boxes and arrows denote the candidate factors and their source.
Maternal Consequences of Placental and Systemic Oxidative Stress
Both placental and systemic oxidative types of stress are increased in PE [14]. This is a consequence of poor placental perfusion, increased inflammation and endothelial cell activation. Oxidative stress modifies both proteins and lipids, bestowing harmful biological activities. Oxidatively-modified proteins are present in normal placentas, but significantly increased in PE placentas [32]. On the maternal side, circulating low-density lipoprotein (LDL) can also be converted to oxidised LDL, which binds to Lectin like oxidised LDL receptor-1 (LOX-1), a cell-surface receptor involved in endothelial dysfunction, and stimulation of increased oxidative stress. Expression of LOX-1 is increased in the syncytiotrophoblast and systemic vasculature in PE, suggesting a contribution to both placental and systemic oxidative stress (Fig 1) [33, 34].
Small Signaling Molecules H2S, CO, NO
Endogenously generated small signaling molecules or ‘gasotransmitters’, such as nitric oxide (NO), carbon monoxide (CO) and hydrogen sulphide (H2S), have been implicated in the pathophysiology of PE. All three molecules are short-lived, locally acting regulators of multiple physiological functions. Evidence suggests that a reduction in their cellular levels plays a role in multiple pathologies, including ischemia/reperfusion injury, hypertension and inflammation [35].
Nitric oxide, CO and H2S are produced in the placenta and systemically through the activities of nitric oxide synthases (NOS), heme oxygenases (HO) and cystathionine γ-lyase (CSE), respectively [36]. Nitric oxide is a major signalling molecule for endothelium-dependent regulation of vascular tone through its production by the endothelial NOS (eNOS) isozyme. It also has other important functions, including antioxidant and anticoagulant activity [37, 38]. In normal pregnancy, NO contributes to the decrease in vascular resistance [37] and actively participates in trophoblast invasion and placental development [39]. Smoking during pregnancy is one of the few factors that protects against PE [40]. One explanation for this is that smokers have higher circulating levels of the small signaling molecule CO [41]. Carbon monoxide protects against ischemia/reperfusion injury through its anti-inflammatory and anti-apoptotic activity [42].
In PE, placental and maternal production of NO, CO and H2S are decreased [43–45, 46••]. For NO, the situation is exacerbated by increased circulating levels of asymmetric dimethylarginine (ADMA), an endogenous eNOS inhibitor, and by the uncoupling of NOS, leading to the generation of harmful superoxides if the substrate L-arginine or the cofactor tetrahydrobiopterin are deficient [47, 48].
Given that CO, NO and H2S, are produced by the placenta and the maternal vasculature, these molecules may play a role in key stages of both the placental and maternal pathophysiology of PE (Fig 1). A better understanding of whether reduced expression and activity of the gasotransmitter generating enzymes is intrinsic to the placental and maternal tissues, or is a consequence of poor placental perfusion and the resultant release and systemic effects of harmful placental factors, will improve strategies for their use as therapeutic targets. However, direct delivery of these gasotransmitters is problematic, as they are highly unstable and in certain situations can also be toxic. The race is therefore on to find appropriate delivery systems that allow their targeted and controlled release [35].
Pro-inflammatory Mediators
Many of the harmful factors released by the placenta in PE can be considered to be damage-associated molecular pattern molecules (DAMPs or alarmins). These are intracellular molecules that, when released after cell stress or injury, are intensely pro-inflammatory. Alarmins include high-mobility group box protein 1 (HMGB1), heat shock protein 70 (HSP70), S100B, proteins modified by nonenzymatic glycation and oxidation in chronic inflammation to form “advanced glycation end products” (AGEs) and cell free ATP, actin, haemoglobin, and fetal DNA [49–54]. They all recruit and activate innate immune cells through interaction with receptors, such as toll-like receptors (TLRs) and the receptor for advanced glycation end products (RAGE) [52].
Several alarmins (including HSP70, s100B, ATP and actin) have been shown to be expressed by the syncytiotrophoblast and to circulate at raised levels in PE [53, 55]. One action of ATP is to inhibit the activity of hemopexin, an acute-phase protein that scavenges heme and blocks its pro-oxidant activity [56], implicating ATP in the reduced antioxidant capacity in PE. Infusion of globular actin into rats, to replicate release from damaged tissues, induces intravascular coagulation and multiple organ dysfunction, as occurs in severe PE [57]. Increases in the levels of circulating cell-free fetal DNA have also been demonstrated in PE, showing a correlation with the severity of the disease, elevated circulating levels prior to the onset of maternal symptoms and a rapid fall following delivery of the placenta [58–60]. Its pro-inflammatory effects have been demonstrated in human peripheral blood mononuclear cells (PBMCs) and mice, where it binds to TLR9 and induces IL-6 secretion [61•].
In PE, fetal haemoglobin mRNA and protein are significantly increased in the placenta [62]. When released into the maternal circulation, fetal haemoglobin becomes a redox-reactive alarmin and a strong candidate for involvement in inflammation and oxidative damage in the maternal syndrome [63, 64]. Accumulation in the intervillous space also implicates free fetal haemoglobin in the oxidative stress and damage to the blood-placenta barrier associated with PE [65]. It also binds and inactivates NO, with subsequent induction of vasoconstriction [66].
Based on these findings, it is easy to envisage a degenerating syncytiotrophoblast releasing a toxic mix of pro-inflammatory material directly into the maternal circulation. Alarmins are therefore an important link between the placenta and maternal syndrome, contributing to the systemic inflammation and subsequent endothelial damage that are characteristic of PE (Fig 1).
Anti-angiogenic Factors
Since the first reports of the possible involvement of elevated soluble VEGF receptor, soluble FMS-like tyrosine kinase-1 (sFlt-1) and the soluble form of the transforming growth factorβ (TGFβ) co-receptor endoglin (sEng) in the pathobiology of PE [44, 67], further studies have strengthened the view of a central role of these placentally-derived anti-angiogenic factors in the endothelial dysfunction apparent in PE (reviewed in [68]). Soluble Flt-1 binds and neutralises vascular endothelial growth factor (VEGF) and placental growth factor (PlGF), which are required for the maintenance and integrity of vascular endothelium, in particular the fenestrated endothelium found in organs such as kidneys, liver and brain [69]. These organs are adversely affected in PE. Soluble Eng antagonises the biological effects of TGFβ, including maintenance of vascular endothelium, as illustrated by a pregnant rat model in which the co-administration of sEng with sFlt-1 increased severity of the PE-like symptoms, induced by raised sFlt-1 alone, to include the HELLP syndrome [44]. Antagonism of VEGF signaling by sFlt-1 in vitro also sensitised endothelial cells to pro-inflammatory cytokines, implicating sFlt-1 in the exaggerated maternal systemic inflammatory response [70].
As with sFlt-1 and sEng, circulating PlGF levels correlate with disease severity, with very low levels found in PE due to sequestering by sFlt-1. Indeed, sFlt-1/PlGF ratio improves the predictive value of adverse outcomes in women developing PE prior to 34 weeks of gestation compared to sFlt-1 alone [71]. Intriguingly, not all PE women have a high sFlt-1/PlGF ratio. These two different forms of PE have been termed angiogenic (high sFlt-1/PlGF) and non-angiogenic (low sFlt-1/PlGF), with only the former associated with adverse neonatal and maternal outcomes [72••]. This may also reflect what has previously been termed placental PE and maternal PE, to distinguish PE triggered predominantly by placental damage rather than maternal hypersensitivity to placental factors [73, 74].
Soluble Flt-1 and sEng are important contributors to the maternal syndrome. However, given that not all PE women have raised sFlt-1 and sEng levels, there must be subcategories of PE with different degrees of placental pathology and immune/endothelial dysfunction. Further analysis of the significance of elevated sFlt-1 and sEng is required to fully understand their role in the development of PE and their usefulness as biomarkers.
Procoagulant Factors
In PE, maternal plasma and placental levels of thrombomodulin, type 1 plasminogen activator inhibitor, tissue factor activity and procoagulant phospholipid levels are elevated compared to normal pregnancy, suggesting increased procoagulant potential both systemically and locally to the placenta, and a role in the increased thrombin formation and fibrin deposition apparent in PE [75–77].
Syncytiotrophoblast-derived Microvesicles and Exosomes
The numerous placental factors released into the maternal circulation in PE have previously been assumed to be soluble factors. However, it is now apparent that the syncytiotrophoblast releases an array of extracellular vesicles into the maternal circulation throughout pregnancy, which carry pro-inflammatory, anti-angiogenic and procoagulant factors including proteins, lipids and oxidised lipids and delivers them to target cells (leucocytes, endothelial cells and platelets) in the mother, altering their function (Fig 1). Levels of circulating placental vesicles increase with advancing pregnancy and labour, and are undetectable by ~48 hours post-delivery [60, 78]. Significantly more of these vesicles are found in the circulation of PE compared to normal pregnant women. They range in size from exosomes (30–100nm) to microvesicles (100–1μm) and apoptotic vesicles (1–5μm), together with necrotic debris. In PE, increased intracellular calcium, due to ER stress and oxidative stress, is a strong stimulus for microvesicle and apoptotic body release, while exosomes are secreted constitutively from intracellular multivesicular bodies that fuse with the plasma membrane as part of the endocytic pathway [79–81]. The composition of shed placental material may have an important bearing on their functional characteristics. Placental exosomes show immunosuppressive properties [82], while microvesicles are more pro-inflammatory [83] (Fig 2). Due to a lack of specific markers and sensitive detection systems, it is not yet possible to determine the pattern of release of individual vesicle types throughout pregnancy. However, increased modal size of vesicles prepared by perfusion of PE and normal placentas suggests increased release of larger pro-inflammatory microvesicles compared to immunoregulatory exosomes in PE [84•].
Fig 2.
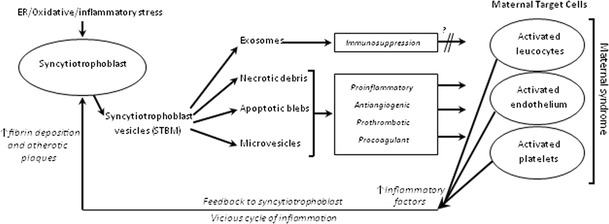
Proposed role of syncytiotrophoblast vesicles in the maternal syndrome of preeclampsia. ER = endoplasmic reticulum.
Evidence that placental vesicles play a role in the maternal inflammatory response, endothelial dysfunction and activation of coagulation of PE continues to grow. They are rapidly taken up by monocytes in vitro [78, 85–87]. Increased production of several cytokines, chemokines and superoxides by PBMCs [88•] and neutrophils [89] has been shown. Placental vesicles also affect the function of endothelial cells, inhibiting proliferation in vitro [90–92] and relaxation of pre-constricted blood vessels ex vivo [90, 92, 93]. Furthermore, the conditioned media from placental vesicle-treated endothelial cells activates neutrophils, demonstrating the potential for a vicious cycle of inflammatory activation [94]. Finally, our recent work has also shown that vesicles prepared from PE placentas stimulate significantly higher levels of thrombin generation than those from normal placentas [95].
The challenge now is to identify the factors carried by these syncytiotrophoblast vesicles responsible for their functional effects. Prime candidates for pro-inflammatory molecules are alarmins, such as HSP70, HMGB1 and fetal DNA, [96, 97], together with syncytin 1, which stimulates the production of IL-1α and other pro-inflammatory cytokines by PBMCs [98]. We and others have shown that vesicles express both sFlt-1 and endoglin, which could contribute to endothelial dysfunction [84•, 97]. Plasminogen activator inhibitors (PAI-1/PAI-2), which regulate fibrinolysis, were also found, and could be responsible for the very high levels of fibrin deposition in the intervillous space and the placental infarction observed in these pregnancies [97]. We have also shown that the increased thrombin generation activity of PE placental vesicles is due to higher levels of functional tissue factor than those from normal placentas, which may account for the excessive activation of the clotting system seen in this disorder [95, 99]. Oxidation in the PE placenta may also amplify the inflammatory burden of the vesicles by modifying syncytiotrophoblast proteins and lipids that are not ordinarily pro-inflammatory [100]. Finally, the placenta is a rich source of microRNAs (miRNA)—small RNA species that regulate gene expression post-transcriptionally and are implicated in a growing number of diseases (reviewed in [101]). Trophoblasts release exosomes that contain miRNAs with functions relevant to PE pathobiology. Altered levels of placental miRNAs are also seen in PE, raising interest in their use as biomarkers (reviewed in [102]).
Conclusions
The factors highlighted in this review illustrate the multi-faceted pathophysiology of PE and the diverse molecule types that underlie the maternal symptoms. They also illustrate maladaptations throughout the course of disease progression and the interplay between the different affected cellular compartments. Preeclampsia is a complicated disease. Trying to unravel the etiology, as well as treat the disease, is confounded by its heterogeneity. Therefore, clarification of the different types of PE is a major challenge for researchers and clinicians. The use of circulating angiogenic factors to establish the degree of endothelial dysfunction is beginning to help identify those women at higher risk of complications and therefore requiring earlier interventions [103••].
At the heart of PE lies a compromised placenta. Understanding the response of the placenta to an adverse uterine environment is key to identifying the causative agent(s). Oxidative and inflammatory stresses are powerful stimuli for altered protein expression. As PE is specific to pregnancy, this suggests that the response of the placenta to an ischaemia/reperfusion insult is also unique. More comparisons of the effects of oxidative and inflammatory stress on placentae and other tissues may help to reveal the placenta-specific factors that are released into the maternal circulation. Results of a recent in vivo mouse study suggest that the timing of the placental insult may also alter the placental response to an ischemic insult [104].
A better understanding of the etiology of this multifactorial disease will allow the development of novel biomarkers for prediction and diagnosis, and potential treatments targeting key aspects of the pathophysiology. However, certain facets of the disease such as genetic predisposition and immune factors may prove difficult to overcome. Although treatment with antioxidant vitamins to ameliorate oxidative stress and prevent PE has shown no benefit in multiple trials, targeted used in subgroups of women with poor nutritional status may prove beneficial [105]. Alternative strategies such as the use of α1-microglobulin to counteract the damaging effects of free fetal haemoglobin have yet to be tried [66•]. We also await the results of trials into the use of statins (which, amongst other functions, upregulate heme-oxygenase-1) to ameliorate maternal symptoms, thereby prolonging pregnancy and reducing the incidence of fetal prematurity-associated disorders. If statin treatment works, it may well be due to multiple actions targeting the different pathologies involved in PE. Alternatively, a combination of treatments may also be more effective at targeting the different elements of the disease. In an animal model of vascular damage induced by diabetes, statins and antioxidant vitamins worked together to induce therapeutic angiogenesis [106]. Finally the removal of harmful placental factors from the circulation using apheresis has been investigated. In a pilot study, dextran sulphate cellulose apheresis columns were used to lower circulating sFlt-1 levels in women with early onset PE, with a resultant reduction in proteinuria, stabilization of blood pressure and prolonging of pregnancy with evidence of fetal growth [107••].
The outlook for the future impact of PE is poor. The rise in risk factors such as obesity and diabetes in the general population will fuel a continued increase in gestational complications such as PE, with increased healthcare expenditures and long-term health implications for both mother and offspring. In the short term, better triaging of presenting women based on risk factors and levels of circulating biomarkers of endothelial dysfunction will aid timely intervention. In the future, with advances in biomarkers, therapeutics and genetic profiling, an individualised treatment regime may be more effective at improving immediate and long-term outcomes for both mother and child.
Acknowledgments
This work was supported by a Medical Research Council Programme Grant Ref: MR/J003360/1.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Dionne Tannetta and Ian Sargent declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Dionne Tannetta, Phone: +44-1865-851064, Email: dionne.tannetta@obs-gyn.ox.ac.uk.
Ian Sargent, Phone: +44-1865-221019, Email: ian.sargent@obs-gyn.ox.ac.uk.
References
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
- 1.Milne F, Redman C, Walker J, Baker P, Bradley J, et al. The pre-eclampsia community guideline (PRECOG): how to screen for and detect onset of pre-eclampsia in the community. BMJ. 2005;330(7491):576–580. doi: 10.1136/bmj.330.7491.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376(9741):631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- 3.Duley L. The global impact of pre-eclampsia and eclampsia. Seminars in perinatology. 2009;33(3):130–137. doi: 10.1053/j.semperi.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Duckitt K, Harrington D. Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. Brit Med J. 2005;330(7491):565–567. doi: 10.1136/bmj.38380.674340.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isler CM, Rinehart BK, Terrone DA, Martin RW, Magann EF, et al. Maternal mortality associated with HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. Am J Obstet Gynecol. 1999;181(4):924–928. doi: 10.1016/s0002-9378(99)70343-1. [DOI] [PubMed] [Google Scholar]
- 6.Coppage KH, Sibai BM. Treatment of hypertensive complications in pregnancy. Curr Pharm Design. 2005;11(6):749–757. doi: 10.2174/1381612053381864. [DOI] [PubMed] [Google Scholar]
- 7.Campbell OMR, Graham WJ, Series LMS. Maternal survival 2 - Strategies for reducing maternal mortality: getting on with what works. Lancet. 2006;368(9543):1284–1299. doi: 10.1016/S0140-6736(06)69381-1. [DOI] [PubMed] [Google Scholar]
- 8.Nicolaides KH. Turning the Pyramid of Prenatal Care. Fetal Diagn Ther. 2011;29(3):183–196. doi: 10.1159/000324320. [DOI] [PubMed] [Google Scholar]
- 9.Khong TY, Dewolf F, Robertson WB, Brosens I. Inadequate Maternal Vascular-Response to Placentation in Pregnancies Complicated by Preeclampsia and by Small-for-Gestational-Age Infants. Brit J Obstet Gynaec. 1986;93(10):1049–1059. doi: 10.1111/j.1471-0528.1986.tb07830.x. [DOI] [PubMed] [Google Scholar]
- 10.Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for Immune Cell Involvement in Decidual Spiral Arteriole Remodeling in Early Human Pregnancy. Am J Pathol. 2009;174(5):1959–1971. doi: 10.2353/ajpath.2009.080995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knofler M, Pollheimer J. IFPA Award in Placentology Lecture: Molecular regulation of human trophoblast invasion. Placenta. 2012;33:S55–S62. doi: 10.1016/j.placenta.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erlebacher A. Immunology of the Maternal-Fetal Interface. Annu Rev Immunol. 2013;31:387–411. doi: 10.1146/annurev-immunol-032712-100003. [DOI] [PubMed] [Google Scholar]
- 13.Egbor M, Ansari T, Morris N, Green CJ, Sibbons PD. Morphometric placental villous and vascular abnormalities in early- and late-onset pre-eclampsia with and without fetal growth restriction. Bjog-Int J Obstet Gy. 2006;113(5):580–589. doi: 10.1111/j.1471-0528.2006.00882.x. [DOI] [PubMed] [Google Scholar]
- 14.Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained Intrauterine Growth Restriction and Early Onset Preeclampsia. Placenta. 2009;30:S43–S48. doi: 10.1016/j.placenta.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts DJ, Post MD. The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol. 2008;61(12):1254–1260. doi: 10.1136/jcp.2008.055236. [DOI] [PubMed] [Google Scholar]
- 16.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365(9461):785–799. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- 17.Redman CWG, Sacks GP, Sargent IL. Preeclampsia: An excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. 1999;180(2):499–506. doi: 10.1016/s0002-9378(99)70239-5. [DOI] [PubMed] [Google Scholar]
- 18.Sargent IL, Borzychowski AM, Redman CWG. Immunoregulation in normal pregnancy and pre-eclampsia: an overview. Reprod Biomed Online. 2006;13(5):680–686. doi: 10.1016/s1472-6483(10)60659-1. [DOI] [PubMed] [Google Scholar]
- 19.Lynch AM, Salmon JE. Dysregulated Complement Activation as a Common Pathway of Injury in Preeclampsia and Other Pregnancy Complications. Placenta. 2010;31(7):561–567. doi: 10.1016/j.placenta.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noori M, Donald AE, Angelakopoulou A, Hingorani AD, Williams DJ. Prospective Study of Placental Angiogenic Factors and Maternal Vascular Function Before and After Preeclampsia and Gestational Hypertension. Circulation. 2010;122(5):478–487. doi: 10.1161/CIRCULATIONAHA.109.895458. [DOI] [PubMed] [Google Scholar]
- 21.Savvidou MD, Hingorani AD, Tsikas D, Frolich JC, Vallance P, et al. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre-eclampsia. Lancet. 2003;361(9368):1511–1517. doi: 10.1016/S0140-6736(03)13177-7. [DOI] [PubMed] [Google Scholar]
- 22.Cockell AP, Poston L. Flow-mediated vasodilatation is enhanced in normal pregnancy but reduced in preeclampsia. Hypertension. 1997;30(2):247–251. doi: 10.1161/01.hyp.30.2.247. [DOI] [PubMed] [Google Scholar]
- 23.Bowyer L, Brown MA, Jones M. Forearm blood flow in pre-eclampsia. Bjog-Int J Obstet Gy. 2003;110(4):383–391. [PubMed] [Google Scholar]
- 24.Robb AO, Mills NL, Din JN, Smith IBJ, Paterson F, et al. Influence of the Menstrual Cycle, Pregnancy, and Preeclampsia on Arterial Stiffness. Hypertension. 2009;53(6):952–U115. doi: 10.1161/HYPERTENSIONAHA.109.130898. [DOI] [PubMed] [Google Scholar]
- 25.Chambers JC, Fusi L, Malik IS, Haskard DO, De Swiet M, et al. Association of maternal endothelial dysfunction with preeclampsia. Jama-J Am Med Assoc. 2001;285(12):1607–1612. doi: 10.1001/jama.285.12.1607. [DOI] [PubMed] [Google Scholar]
- 26.Meher S, Duley L. (2007) Nitric oxide for preventing pre-eclampsia and its complications. Cochrane Db Syst Rev(2). [DOI] [PMC free article] [PubMed]
- 27.Stillman IE, Karumanchi SA. The glomerular injury of preeclampsia. J Am Soc Nephrol. 2007;18(8):2281–2284. doi: 10.1681/ASN.2007020255. [DOI] [PubMed] [Google Scholar]
- 28.Craici IM, Wagner SJ, Bailey KR, Fitz-Gibbon PD, Wood-Wentz CM, et al. Podocyturia Predates Proteinuria and Clinical Features of Preeclampsia Longitudinal Prospective Study. Hypertension. 2013;61(6):1289–U343. doi: 10.1161/HYPERTENSIONAHA.113.01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenkranz A, Hiden M, Leschnik B, Weiss EC, Schlembach D, et al. Calibrated automated thrombin generation in normal uncomplicated pregnancy. Thromb Haemostasis. 2008;99(2):331–337. doi: 10.1160/TH07-05-0359. [DOI] [PubMed] [Google Scholar]
- 30.Bonnar J, Mcnicol GP, Douglas AS. (1971) Coagulation and Fibrinolytic Systems in Pre-Eclampsia and Eclampsia. Brit Med J.2(5752):12-&. [DOI] [PMC free article] [PubMed]
- 31.Janes SL, Goodall AH. Flow cytometric detection of circulating activated platelets and platelet hyper-responsiveness in pre-eclampsia and pregnancy. Clin Sci (Lond) 1994;86(6):731–739. doi: 10.1042/cs0860731. [DOI] [PubMed] [Google Scholar]
- 32.Zusterzeel PL, Rutten H, Roelofs HM, Peters WH, Steegers EA. Protein carbonyls in decidua and placenta of pre-eclamptic women as markers for oxidative stress. Placenta. 2001;22(2–3):213–219. doi: 10.1053/plac.2000.0606. [DOI] [PubMed] [Google Scholar]
- 33.Lee H, Park H, Kim YJ, Kim HJ, Ahn YM, et al. Expression of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in human preeclamptic placenta: Possible implications in the process of trophoblast apoptosis. Placenta. 2005;26(2–3):226–233. doi: 10.1016/j.placenta.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Sankaralingam S, Xu Y, Sawamura T, Davidge ST. Increased Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 Expression in the Maternal Vasculature of Women With Preeclampsia Role for Peroxynitrite. Hypertension. 2009;53(2):270–U310. doi: 10.1161/HYPERTENSIONAHA.108.122630. [DOI] [PubMed] [Google Scholar]
- 35.Bannenberg GL, Vieira HLA. Therapeutic applications of the gaseous mediators carbon monoxide and hydrogen sulfide. Expert Opin Ther Pat. 2009;19(5):663–682. doi: 10.1517/13543770902858824. [DOI] [PubMed] [Google Scholar]
- 36.Yang GD, Wu LY, Jiang B, Yang W, Qi JS, et al. H(2)S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Demir B, Demir S, Pasa S, Guven S, Atamer Y, et al. The role of homocysteine, asymmetric dimethylarginine and nitric oxide in pre-eclampsia. J Obstet Gynaecol. 2012;32(6):525–528. doi: 10.3109/01443615.2012.693985. [DOI] [PubMed] [Google Scholar]
- 38.Gielen S, Sandri M, Erbs S, Adams V. Exercise-Induced Modulation of Endothelial Nitric Oxide Production. Curr Pharm Biotechno. 2011;12(9):1375–1384. doi: 10.2174/138920111798281063. [DOI] [PubMed] [Google Scholar]
- 39.Huang LT, Hsieh CS, Chang KA, Tain YL. Roles of Nitric Oxide and Asymmetric Dimethylarginine in Pregnancy and Fetal Programming. Int J Mol Sci. 2012;13(11):14606–14622. doi: 10.3390/ijms131114606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conde-Agudelo A, Althabe F, Belizan JM, Kafury-Goeta AC. Cigarette smoking during pregnancy and risk of preeclampsia: A systematic review. Am J Obstet Gynecol. 1999;181(4):1026–1035. doi: 10.1016/s0002-9378(99)70341-8. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed A. New insights into the etiology of preeclampsia: identification of key elusive factors for the vascular complications. Thrombosis research. 2011;127:S72–S75. doi: 10.1016/S0049-3848(11)70020-2. [DOI] [PubMed] [Google Scholar]
- 42.Tsuchihashi S, Fondevila C, Ma J, Ke BB, Zhai Y, et al. HO-1 cytoprotective and anti-inflammatory effects in hepatic ischemia/reperfusion injury are Toll-like receptor-4 independent. Am J Transplant. 2004;4:434–435. [Google Scholar]
- 43.Brennecke SP, Gude NM, DiIulio JL, King RG. Reduction of placental nitric oxide synthase activity in pre-eclampsia. Clin Sci. 1997;93(1):51–55. doi: 10.1042/cs0930051. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nature medicine. 2006;12(6):642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura M, Sekizawa A, Purwosunu Y, Okazaki S, Farina A, et al. Cellular mRNA expressions of anti-oxidant factors in the blood of preeclamptic women. Prenatal Diag. 2009;29(7):691–696. doi: 10.1002/pd.2278. [DOI] [PubMed] [Google Scholar]
- 46.Wang KQ, Ahmad S, Cai M, Rennie J, Fujisawa T, et al. Dysregulation of Hydrogen Sulfide Producing Enzyme Cystathionine gamma-lyase Contributes to Maternal Hypertension and Placental Abnormalities in Preeclampsia. Circulation. 2013;127(25):2514–2522. doi: 10.1161/CIRCULATIONAHA.113.001631. [DOI] [PubMed] [Google Scholar]
- 47.Laskowska M, Laskowska K, Terbosh M, Oleszczuk J. A comparison of maternal serum levels of endothelial nitric oxide synthase, asymmetric dimethylarginine, and homocysteine in normal and preeclamptic pregnancies. Med Sci Monitor. 2013;19:430–437. doi: 10.12659/MSM.883932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roe ND, Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vascular pharmacology. 2012;57(5-6):168–172. doi: 10.1016/j.vph.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 49.Holmlund U, Wahamaa H, Bachmayer N, Bremme K, Sverremark-Ekstrom E, et al. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology. 2007;122(3):430–437. doi: 10.1111/j.1365-2567.2007.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barut F, Barut A, Dogan Gun B, Kandemir NO, Aktunc E, et al. Expression of heat shock protein 70 and endothelial nitric oxide synthase in placental tissue of preeclamptic and intrauterine growth-restricted pregnancies. Pathology, research and practice. 2010;206(9):651–656. doi: 10.1016/j.prp.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Marinoni E, Di Iorio R, Gazzolo D, Lucchini C, Michetti F, et al. Ontogenetic localization and distribution of S-100beta protein in human placental tissues. Obstetrics and gynecology. 2002;99(6):1093–1099. doi: 10.1016/s0029-7844(02)01996-8. [DOI] [PubMed] [Google Scholar]
- 52.Yang H, Wang HC, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukocyte Biol. 2005;78(1):1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 53.Schmidt AP, Tort ABL, Amaral OB, Schmidt AP, Walz R, et al. Serum S100B in pregnancy-related hypertensive disorders: A case-control study. Clin Chem. 2004;50(2):435–438. doi: 10.1373/clinchem.2003.027391. [DOI] [PubMed] [Google Scholar]
- 54.Chekir C, Nakatsuka M, Noguchi S, Konishi H, Kamada Y, et al. Accumulation of advanced glycation end products in women with preeclampsia: possible involvement of placental oxidative and nitrative stress. Placenta. 2006;27(2–3):225–233. doi: 10.1016/j.placenta.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 55.Fukushima A, Kawahara H, Isurugi C, Syoji T, Oyama R, et al. Changes in serum levels of heat shock protein 70 in preterm delivery and pre-eclampsia. The journal of obstetrics and gynaecology research. 2005;31(1):72–77. doi: 10.1111/j.1447-0756.2005.00244.x. [DOI] [PubMed] [Google Scholar]
- 56.Bakker WW, Donker RB, Timmer A, van Pampus MG, van Son WJ, et al. Plasma hemopexin activity in pregnancy and preeclampsia. Hypertens Pregnancy. 2007;26(2):227–239. doi: 10.1080/10641950701274896. [DOI] [PubMed] [Google Scholar]
- 57.Haddad JG, Harper KD, Guoth M, Pietra GG, Sanger JW. Angiopathic consequences of saturating the plasma scavenger system for actin. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(4):1381–1385. doi: 10.1073/pnas.87.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong XY, Gebhardt S, Hillermann R, Tofa KC, Holzgreve W, et al. Parallel assessment of circulatory fetal DNA and corticotropin-releasing hormone mRNA in early- and late-onset preeclampsia. Clin Chem. 2005;51(9):1730–1733. doi: 10.1373/clinchem.2005.053959. [DOI] [PubMed] [Google Scholar]
- 59.Zhong XY, Holzgreve W, Hahn S. The levels of circulatory cell free fetal DNA in maternal plasma are elevated prior to the onset of preeclampsia. Hypertens Pregnancy. 2002;21(1):77–83. doi: 10.1081/PRG-120002911. [DOI] [PubMed] [Google Scholar]
- 60.Reddy A, Zhong XY, Rusterholz C, Hahn S, Holzgreve W, et al. The effect of labour and placental separation on the shedding of syncytiotrophoblast microparticles, cell-free DNA and mRNA in normal pregnancy and pre-eclampsia. Placenta. 2008;29(11):942–949. doi: 10.1016/j.placenta.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 61.Scharfe-Nugent A, Corr SC, Carpenter SB, Keogh L, Doyle B, et al. TLR9 provokes inflammation in response to fetal DNA: mechanism for fetal loss in preterm birth and preeclampsia. Journal of Immunology. 2012;188(11):5706–5712. doi: 10.4049/jimmunol.1103454. [DOI] [PubMed] [Google Scholar]
- 62.Centlow M, Carninci P, Nemeth K, Mezey E, Brownstein M, et al. Placental expression profiling in preeclampsia: local overproduction of hemoglobin may drive pathological changes. Fertil Steril. 2008;90(5):1834–1843. doi: 10.1016/j.fertnstert.2007.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faivre B, Menu P, Labrude P, Vigneron C. Hemoglobin autooxidation/oxidation mechanisms and methemoglobin prevention or reduction processes in the bloodstream. Literature review and outline of autooxidation reaction. Artificial cells, blood substitutes, and immobilization biotechnology. 1998;26(1):17–26. doi: 10.3109/10731199809118943. [DOI] [PubMed] [Google Scholar]
- 64.Olsson MG, Centlow M, Rutardottir S, Stenfors I, Larsson J, et al. Increased levels of cell-free hemoglobin, oxidation markers, and the antioxidative heme scavenger alpha(1)-microglobulin in preeclampsia. Free radical biology & medicine. 2010;48(2):284–291. doi: 10.1016/j.freeradbiomed.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 65.May K, Rosenlof L, Olsson MG, Centlow M, Morgelin M, et al. Perfusion of human placenta with hemoglobin introduces preeclampsia-like injuries that are prevented by alpha1-microglobulin. Placenta. 2011;32(4):323–332. doi: 10.1016/j.placenta.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 66.Kim-Shapiro DB, Schechter AN, Gladwin MT. Unraveling the reactions of nitric oxide, nitrite, and hemoglobin in physiology and therapeutics. Arteriosclerosis, thrombosis, and vascular biology. 2006;26(4):697–705. doi: 10.1161/01.ATV.0000204350.44226.9a. [DOI] [PubMed] [Google Scholar]
- 67.Maynard SE, Min JY, Merchan J, Lim KH, Li J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. The Journal of clinical investigation. 2003;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a Disease of the Maternal Endothelium The Role of Antiangiogenic Factors and Implications for Later Cardiovascular Disease. Circulation. 2011;123(24):2856–2869. doi: 10.1161/CIRCULATIONAHA.109.853127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maharaj ASR, Saint-Geniez M, Maldonado AE, D'Amore PA. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168(2):639–648. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cindrova-Davies T, Sanders DA, Burton GJ, Charnock-Jones DS. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor-mediated signalling. Cardiovascular research. 2011;89(3):671–679. doi: 10.1093/cvr/cvq346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rana S, Powe CE, Salahuddin S, Verlohren S, Perschel FH, et al. Angiogenic Factors and the Risk of Adverse Outcomes in Women With Suspected Preeclampsia. Circulation. 2012;125(7):911–U199. doi: 10.1161/CIRCULATIONAHA.111.054361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rana S, Schnettler WT, Powe C, Wenger J, Salahuddin S, et al. Clinical characterization and outcomes of preeclampsia with normal angiogenic profile. Hypertension in Pregnancy. 2013;32(2):189–201. doi: 10.3109/10641955.2013.784788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soto E, Romero R, Kusanovic JP, Ogge G, Hussein Y, et al. Late-onset preeclampsia is associated with an imbalance of angiogenic and anti-angiogenic factors in patients with and without placental lesions consistent with maternal underperfusion. J Matern-Fetal Neo M. 2012;25(5):498–507. doi: 10.3109/14767058.2011.591461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 75.Rousseau A, Favier R, Van Dreden P. Elevated circulating soluble thrombomodulin activity, tissue factor activity and circulating procoagulant phospholipids: new and useful markers for pre-eclampsia? European journal of obstetrics, gynecology, and reproductive biology. 2009;146(1):46–49. doi: 10.1016/j.ejogrb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 76.Estelles A, Gilabert J, Grancha S, Yamamoto K, Thinnes T, et al. Abnormal expression of type 1 plasminogen activator inhibitor and tissue factor in severe preeclampsia. Thromb Haemostasis. 1998;79(3):500–508. [PubMed] [Google Scholar]
- 77.Teng Y, Jiang R, Lin Q, Ding C, Ye Z. The relationship between plasma and placental tissue factor, and tissue factor pathway inhibitors in severe pre-eclampsia patients. Thromb Res. 2010;126(1):e41–e45. doi: 10.1016/j.thromres.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 78.Germain SJ, Sacks GP, Sooranna SR, Sargent IL, Redman CW. Systemic inflammatory priming in normal pregnancy and preeclampsia: the role of circulating syncytiotrophoblast microparticles. Journal of Immunology. 2007;178(9):5949–5956. doi: 10.4049/jimmunol.178.9.5949. [DOI] [PubMed] [Google Scholar]
- 79.Yano Y, Shiba E, Kambayashi J, Sakon M, Kawasaki T, et al. The effects of calpeptin (a calpain specific inhibitor) on agonist induced microparticle formation from the platelet plasma membrane. Thromb Res. 1993;71(5):385–396. doi: 10.1016/0049-3848(93)90163-i. [DOI] [PubMed] [Google Scholar]
- 80.Ahn YS, Jy W, Jimenez JJ, Horstman LL. More on: cellular microparticles: what are they bad or good for? J Thromb Haemost. 2004;2(7):1215–1216. doi: 10.1111/j.1538-7836.2004.00797.x. [DOI] [PubMed] [Google Scholar]
- 81.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 82.Hedlund M, Stenqvist AC, Nagaeva O, Kjellberg L, Wulff M, et al. Human Placenta Expresses and Secretes NKG2D Ligands via Exosomes that Down-Modulate the Cognate Receptor Expression: Evidence for Immunosuppressive Function. Journal of Immunology. 2009;183(1):340–351. doi: 10.4049/jimmunol.0803477. [DOI] [PubMed] [Google Scholar]
- 83.Southcombe J, Tannetta D, Redman C, Sargent I. The immunomodulatory role of syncytiotrophoblast microvesicles. PloS one. 2011;6(5):e20245. doi: 10.1371/journal.pone.0020245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tannetta DS, Dragovic RA, Gardiner C, Redman CW, Sargent IL. Characterisation of syncytiotrophoblast vesicles in normal pregnancy and pre-eclampsia: expression of Flt-1 and endoglin. PloS one. 2013;8(2):e56754. doi: 10.1371/journal.pone.0056754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Messerli M, May K, Hansson SR, Schneider H, Holzgreve W, et al. Feto-maternal interactions in pregnancies: placental microparticles activate peripheral blood monocytes. Placenta. 2010;31(2):106–112. doi: 10.1016/j.placenta.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 86.Atay S, Gercel-Taylor C, Suttles J, Mor G, Taylor DD. Trophoblast-derived exosomes mediate monocyte recruitment and differentiation. Am J Reprod Immunol. 2011;65(1):65–77. doi: 10.1111/j.1600-0897.2010.00880.x. [DOI] [PubMed] [Google Scholar]
- 87.Atay S, Gercel-Taylor C, Taylor DD. Human trophoblast-derived exosomal fibronectin induces pro-inflammatory IL-1beta production by macrophages. Am J Reprod Immunol. 2011;66(4):259–269. doi: 10.1111/j.1600-0897.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 88.Holder BS, Tower CL, Jones CJ, Aplin JD, Abrahams VM. Heightened pro-inflammatory effect of preeclamptic placental microvesicles on peripheral blood immune cells in humans. Biology of reproduction. 2012;86(4):103. doi: 10.1095/biolreprod.111.097014. [DOI] [PubMed] [Google Scholar]
- 89.Aly AS, Khandelwal M, Zhao J, Mehmet AH, Sammel MD, et al. Neutrophils are stimulated by syncytiotrophoblast microvillous membranes to generate superoxide radicals in women with preeclampsia. Am J Obstet Gynecol. 2004;190(1):252–258. doi: 10.1016/j.ajog.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 90.Smarason AK, Sargent IL, Starkey PM, Redman CW. The effect of placental syncytiotrophoblast microvillous membranes from normal and pre-eclamptic women on the growth of endothelial cells in vitro. Brit J Obstet Gynaec. 1993;100(10):943–949. doi: 10.1111/j.1471-0528.1993.tb15114.x. [DOI] [PubMed] [Google Scholar]
- 91.Gupta AK, Rusterholz C, Huppertz B, Malek A, Schneider H, et al. A comparative study of the effect of three different syncytiotrophoblast micro-particles preparations on endothelial cells. Placenta. 2005;26(1):59–66. doi: 10.1016/j.placenta.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 92.Hoegh AM, Tannetta D, Sargent I, Borup R, Nielsen FC, et al. Effect of syncytiotrophoblast microvillous membrane treatment on gene expression in human umbilical vein endothelial cells. BJOG : an international journal of obstetrics and gynaecology. 2006;113(11):1270–1279. doi: 10.1111/j.1471-0528.2006.01061.x. [DOI] [PubMed] [Google Scholar]
- 93.Cockell AP, Learmont JG, Smarason AK, Redman CW, Sargent IL, et al. Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. Brit J Obstet Gynaec. 1997;104(2):235–240. doi: 10.1111/j.1471-0528.1997.tb11052.x. [DOI] [PubMed] [Google Scholar]
- 94.von Dadelszen P, Hurst G, Redman CW. Supernatants from co-cultured endothelial cells and syncytiotrophoblast microvillous membranes activate peripheral blood leukocytes in vitro. Hum Reprod. 1999;14(4):919–924. doi: 10.1093/humrep/14.4.919. [DOI] [PubMed] [Google Scholar]
- 95.Gardiner C, Tannetta DS, Simms CA, Harrison P, Redman CW, et al. Syncytiotrophoblast microvesicles released from pre-eclampsia placentae exhibit increased tissue factor activity. PloS one. 2011;6(10):e26313. doi: 10.1371/journal.pone.0026313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Redman CW, Tannetta DS, Dragovic RA, Gardiner C, Southcombe JH, et al. (2012) Review: Does size matter? Placental debris and the pathophysiology of pre-eclampsia. Placenta.33 Suppl:S48-54. [DOI] [PubMed]
- 97.Guller S, Tang Z, Ma YY, Di Santo S, Sager R, et al. Protein composition of microparticles shed from human placenta during placental perfusion: Potential role in angiogenesis and fibrinolysis in preeclampsia. Placenta. 2011;32(1):63–69. doi: 10.1016/j.placenta.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Holder BS, Tower CL, Forbes K, Mulla MJ, Aplin JD, et al. Immune cell activation by trophoblast-derived microvesicles is mediated by syncytin 1. Immunology. 2012;136(2):184–191. doi: 10.1111/j.1365-2567.2012.03568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reverdiau P, Jarousseau AC, Thibault G, Khalfoun B, Watier H, et al. Tissue factor activity of syncytiotrophoblast plasma membranes and tumoral trophoblast cells in culture. Thromb Haemost. 1995;73(1):49–54. [PubMed] [Google Scholar]
- 100.Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacology & therapeutics. 2007;115(1):13–24. doi: 10.1016/j.pharmthera.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 101.Gommans WM, Berezikov E. Controlling miRNA regulation in disease. Methods Mol Biol. 2012;822:1–18. doi: 10.1007/978-1-61779-427-8_1. [DOI] [PubMed] [Google Scholar]
- 102.Sargent I, Dragovic RA, Tannetta D, Redman C. Extracellular vesicles in normal pregnancy and pre-eclampsia. In: Harrison P, Gardiner C, Sargent I, editors. Extracellular vesicles in health and disease. UK: Pan Stanford Publishing; 2014. [Google Scholar]
- 103.Staff AC, Benton SJ, von Dadelszen P, Roberts JM, Taylor RN, et al. Redefining Preeclampsia Using Placenta-Derived Biomarkers. Hypertension. 2013;61(5):932–942. doi: 10.1161/HYPERTENSIONAHA.111.00250. [DOI] [PubMed] [Google Scholar]
- 104.Banek CT, Bauer AJ, Gingery A, Gilbert JS. Timing of ischemic insult alters fetal growth trajectory, maternal angiogenic balance, and markers of renal oxidative stress in the pregnant rat. American journal of physiology Regulatory, integrative and comparative physiology. 2012;303(6):R658–R664. doi: 10.1152/ajpregu.00250.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Salles AMR, Galvao TF, Silva MT, Motta LCD, Pereira MG. (2012) Antioxidants for Preventing Preeclampsia: A Systematic Review. Sci World J. [DOI] [PMC free article] [PubMed]
- 106.El-Azab MF, Hazem RM, Moustafa YM. Role of simvastatin and/or antioxidant vitamins in therapeutic angiogenesis in experimental diabetic hindlimb ischemia: effects on capillary density, angiogenesis markers, and oxidative stress. European journal of pharmacology. 2012;690(1-3):31–41. doi: 10.1016/j.ejphar.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 107.Thadhani R, Kisner T, Hagmann H, Bossung V, Noack S, et al. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation. 2011;124(8):940–950. doi: 10.1161/CIRCULATIONAHA.111.034793. [DOI] [PubMed] [Google Scholar]