

Abstract
Tumorigenesis is a multi-step process that reflects intimate reciprocal interactions between epithelia and underlying stroma. However, tumor-initiating mechanisms coordinating transformation of both epithelial and stromal components are not defined. In humans and mice, initiation of colorectal cancer is universally associated with loss of guanylin and uroguanylin, the endogenous ligands for the tumor suppressor guanylyl cyclase C (GUCY2C), disrupting a network of homeostatic mechanisms along the crypt-surface axis. Here, we reveal that silencing GUCY2C in human colon cancer cells increases Akt-dependent TGF-β secretion, activating fibroblasts through TGF-β type I receptors and Smad3 phosphorylation. In turn, activating TGF-β signaling induces fibroblasts to secrete hepatocyte growth factor (HGF), reciprocally driving colon cancer cell proliferation through cMET-dependent signaling. Elimination of GUCY2C signaling in mice (Gucy2c-/-) produces intestinal desmoplasia, with increased reactive myofibroblasts, which is suppressed by anti-TGF-β antibodies or genetic silencing of Akt. Thus, GUCY2C coordinates intestinal epithelial-mesenchymal homeostasis through reciprocal paracrine circuits mediated by TGF-β and HGF. In that context, GUCY2C signaling constitutes a direct link between the initiation of colorectal cancer and the induction of its associated desmoplastic stromal niche. The recent regulatory approval of oral GUCY2C ligands to treat chronic gastrointestinal disorders underscores the potential therapeutic opportunity for oral GUCY2C hormone replacement to prevent remodeling of the microenvironment essential for colorectal tumorigenesis.
Keywords: GUCY2C, TGF-β, desmoplasia, hepatocyte growth factor, colorectal cancer
Introduction
Tumorigenesis reflects an intimate collaboration between cancer cells and the supporting microenvironmental stroma (1-3). Genetic changes underlying malignant transformation corrupt circuits regulating epithelial-mesenchymal cross-talk that are essential for normal tissue organization, homeostasis, and regeneration (4). This reprogramming drives the co-evolution of epithelial cells and their supporting microenvironment, producing reactive stroma that allows tumor cells to acquire the hallmarks of cancer (2, 5, 6).
The dynamic changes in cancer-associated stroma resemble the desmoplastic reaction of wound-healing, characterized by extracellular mesenchymal remodeling with increased matrix deposition and matrix metalloproteinase (MMP) activity (4). Tumor-associated desmoplasia is supported primarily by the transformation of quiescent host fibroblasts into myofibroblasts (1, 2, 7, 8). While normal stroma contains few fibroblasts, there is an increase in activated myofibroblasts within the reactive stroma surrounding tumors (8). Induction of the myofibroblastic phenotype in quiescent fibroblasts is supported by the secretion of TGF-β by cancer cells (1, 3, 9). Reciprocally, myofibroblasts play an essential role in solid tumors by producing paracrine and juxtacrine factors, including HGF, that drive normal epithelial cells toward acquiring the malignant phenotype (1, 5, 6, 10-14).
Paracrine cross-talk between epithelia and mesenchyme is essential to cancer progression and metastasis (15), but the precise signaling mechanisms linking tumor-initiating events to desmoplastic induction remain incompletely defined. For example the cell type coordinating epithelial and mesenchymal reprogramming at the initiation of transformation remains unknown (2, 8, 16). Thus, dissecting the individual stromal and epithelial contributions would define the paracrine signaling loops initiating and supporting carcinogenesis and provide new targets for tumor prevention and therapy.
Guanylyl cyclase C (GUCY2C) is the intestinal isoform from the family of guanylyl cyclase transmembrane receptor enzymes that signal by producing cGMP (17). GUCY2C was first identified as the intestinal epithelial receptor regulating fluid and electrolyte transport in the secretory diarrhea induced by bacterial enterotoxins (ST) (17). Discovery of the endogenous paracrine hormones uroguanylin, in the small intestine, and guanylin, in the colorectum (18), revealed a role for GUCY2C in epithelial cell dynamics and the homeostatic balance of proliferation, metabolism, and differentiation that organizes the crypt-surface axis in the intestine (19-23).
Notably, guanylin and uroguanylin are the most commonly lost gene products in intestinal tumorigenesis in animals and humans, and their expression is universally silenced at the earliest stages of transformation (24-27). Further, eliminating GUCY2C amplifies the tumorigenesis induced by carcinogens or gene mutations in mice (20, 21). Indeed, silencing GUCY2C activates the AKT-mTOR signaling axis, which imparts cell-autonomous characteristics of transformation to intestinal epithelial cells, encompassing accelerated proliferation, reprogramming of metabolism from oxidative phosphorylation to aerobic glycolysis, and impaired DNA damage repair (19-23). Moreover, oral administration of GUCY2C hormone attenuates the colorectal tumorigenesis induced by APC mutations in mice (25). GUCY2C has emerged as a novel intestinal tumor suppressor whose silencing through hormone loss universally contributes to the initiation and progression of colorectal cancer.
In the heart, the natriuretic peptide receptor guanylyl cyclase A isoform (NPR1) is expressed in cardiomyocytes and establishes a homeostatic balance with the mesenchymal compartment (28, 29). Silencing cardiomyocyte NPR1 and cGMP signaling produces cardiac hypertrophy and elevated interstitial fibrosis (28, 29). In fact, increases in growth and fibrosis are disproportionate to and perhaps even independent of increases in blood pressure or volume, suggesting that NPR1 acts through local, paracrine pathways to suppress hypertrophy and desmoplasia (28, 29). The paracrine mechanisms mediating this cardiomyocyte-mesenchymal cross-talk remain incompletely characterized. However, based on this role of NPR1 and cGMP signaling in the heart, the present study explored whether the tumor suppressor GUCY2C and cGMP had homologous functions in the intestine, namely the maintenance of epithelial-mesenchymal homeostasis whose corruption contributes to tumor-associated desmoplasia.
Materials and Methods
Animal models
Gucy2c-/- wild type littermate mice on the C57BL/6 background (generation 14) were bred, maintained, genotyped, and functionally characterized in accordance with the Thomas Jefferson University Animal Care and Use guidelines (19-21). Akt1+/-Gucy2c+/+ and Akt1+/-Gucy2c-/-were bred from Akt1+/-Gucy2c+/-, genotyped and functionally characterized as described previously in accordance with the Thomas Jefferson University Animal Care and Use guidelines (21). Where indicated, animals received an IP dose (0.5 mg/kg) of IgG (Innovative Research, MI) or TGF-β-specific monoclonal antibody purified from the 1D11.16.8 hybridoma cell line (ATCC, VA) twice weekly for 10 weeks. This regimen inhibits downstream canonical TGF-β signaling in intestine in C57BL/6 mice (30).
Cell culture and fibroblast activation
T84 and Caco2 human colon cancer cells were maintained and cultured in DMEM-F12 and DMEM (Cellgro, VA) media supplemented with 10% FBS (21). CCD-18Co normal human intestinal fibroblasts were cultured and maintained in Eagle's Minimum Essential Medium (ATCC, VA). In studies of fibroblast induction, T84 or Caco2 cells were cultured in MEM Minimum Essential Medium (Cellgro) without FBS for 48 h. Then, completed growth media were conditioned by cancer cells for another 48 h, including either the GUCY2C ligand, ST (1 μM), or vehicle, PBS (21, 22). Conditioned media were centrifuged at maximum speed for 5 min to precipitate cancer cells and then diluted with 2 parts of Opti-MEM® GlutaMAX™ (Invitrogen, NY) to obtain a 1:3 dilution before applying to fibroblasts for 24 h. Notably, T84 and Caco2 cells express GUCY2C which mediates canonical downstream cGMP signaling upon ligand binding (19-21). In contrast, CCD-18Co do not express GUCY2C and are unresponsive to ligands targeting that receptor.
Antibody treatment, TGF-β receptor inhibitors, and siRNA transfection
In experiments exploring the role of cancer cell TGF-β in fibroblast activation, T84 or Caco2 cell-conditioned media was treated with a neutralizing antibody to TGF-β (30 ng/μL) or control IgG (30 ng/μL). In some experiments, TGF-β produced by colon cancer cells was depleted from conditioned media with neutralizing antibody to TGF-β (30 ng/μL) followed by immunoprecipitation with Dynabeads® Protein G (Invitrogen, VA). Cancer cell-derived TGF-β removed by immunoprecipitation was replaced with recombinant purified TGF-β (rTGF-β; R&D Systems, MN) at a concentration (10 ng/ml) corresponding to that produced by T84 cells, quantified by ELISA (R&D systems, MN). In experiments employing TGF-β receptor inhibitors, colon cancer cells were treated with PBS or 1 μM ST for 48 h before the conditioned media was transferred to CCD-18Co cells pretreated for 0.5 h with the TGF-β type I receptor inhibitor SB-505124 (50 nM, Sigma Aldrich). SB-505124 treatment continued in the conditioned media for the indicated times. In studies employing siRNA, colon cancer cells were transfected with TGF-β-targeted or control siRNA (100 nM; ABI) by Amaxa® Electroporation (Lonza, NJ) 48 h before use in conditioned media transfer experiments, to permit adequate time for suppression of TGF-β expression.
Akt activation and inhibition in cancer cells
T84 were infected with adenovirus-expressing siAKT1 and GFP control for inhibition experiments, or adenovirus-expressing wtAKT1 and myrAKT1 for activation experiments, at 50% confluence using MOI's of 100 at 48 h before GUCY2C activation during starvation (21). Then, cells were washed and incubated in complete media with 1 μM ST or PBS (control) for 48 h. The conditioned media was transferred to fibroblasts after dilution as described above.
HGF production by fibroblasts and cMET activation
Media conditioned by T84 cells with and without activation of GUCY2C signaling by 1 μm ST or PBS (control) for 48 h were used to induce fibroblast activation of CCD-18Co cells following incubation for 24 h. Then, fibroblast-conditioned media were transferred to HCT116 human colon cancer cells, which do not express GUCY2C and possess inactivating mutations in the TGF-β receptor (21, 31), and proliferation was quantified employing 3H-thymidine (21-23). HCT116 cells are particularly useful for quantifying the effects of fibroblast-derived HGF on colon cancer cell proliferation in the absence of confounding by GUCY2C or TGF-β ligands. In some experiments, media sequentially conditioned by human cancer cells and then by fibroblasts were treated with a neutralizing antibody to HGF or control IgG (20 μg/mL; R&D Systems, MN). Where indicated, HGF produced by fibroblasts was depleted from conditioned media with neutralizing antibody to HGF (20 μg/mL) followed by immunoprecipitation with Dynabeads® Protein G (Invitrogen, VA). Fibroblast-derived HGF depleted by immunoprecipitation was replaced with recombinant purified HGF (rHGF; 2.5 ng/ml; R&D Systems, MN). In some experiments, HCT116 cells were pre-treated with cMet Kinase Inhibitor III (500 nM;EMD Millipore, Germany), an inhibitor of cMET, the canonical receptor for HGF (32), for 5 min before incubations with conditioned media.
Immunoblot analyses and immunofluorescence
Protein was extracted from cells and tissues in M-PER reagent supplemented with protease and phosphatase inhibitors (Thermal Scientific, TX) and quantified by immunoblot analysis employing antibodies to procollagen I (Sigma-Aldrich, MO); α-SMA and MMP-9 (Biomol, NY); and GAPDH and prolyl hydroxylase-β (Abcam, MA). Other antibodies were from Cell Signaling (MA). Anti-α-smooth muscle actin (α-SMA; Sigma-Aldrich, MO), anti-collagen I and anti-β-catenin (Santa Cruz Biotechnology, CA) antibodies were used to stain fibroblasts and epithelial cells, respectively (1, 33). Expression levels were quantified by estimating the integrated density of the protein of interest and normalizing to the integrated density of the nucleus (DAPI). Stained tissues were quantified in 5-15 crypt units in 5-10 sections in each mouse.
Masson's trichrome stain
Tissue collected from mice were deparaffinized and stained with Weigert's iron hematoxylin solution and Biebrich scarlet-acid fuchsin, placed in phosphomolybdic/ polyphosphotungstic acid solution, and counterstained with Aniline blue (Dako, CA) or Masson's Trichrome Stain Kit (Sigma-Aldrich, MO). The bottom of crypts was identified by the presence of Paneth cells. In Figure 1, the interstitial thickness was calculated as: area of the blue stain divided by the length of the underlying muscle layer, quantified in Image J. In Figure 6, the interstitial thickness displays the shortest distance between the bottom of the crypt and the muscle layer. In both cases, pixels were converted back to metric dimensions. Quantification represents data from 5-10 crypt units in 5-10 sections per mouse.
Fig. 1. Silencing epithelial GUCY2C promotes intestinal desmoplasia and stromal fibroblast activation in mice.

(A1) Masson's Trichrome stain reveals submucosal hypertrophy in Gucy2c-/-, compared to wild type Gucy2c+/+, mice, associated with (A2) increased interstitial thickness and (B) matrix deposition, including (B1) tenascin C, (B2) fibronectin, quantified by immunofluorescence (IF) [Green, fibronectin; Red, β-catenin; Blue, DAPI; bottom of crypt is determined by presence of β-catenin in nucleus (arrow)] and (B3) collagen, quantified by hydroxyproline analysis, and (B4) direct visualization by electron microscopy (n≥3 mice). (C1) Fibroblasts were visualized by α-smooth muscle actin (α-SMA) immunohistochemistry and (C2) quantified in 10 sections/mouse and 10 crypts/section (n=5 mice). Fibroblast activation in Gucy2c-/-, compared to Gucy2c+/+ mice, was associated with increased expression of (D) mRNA [Col, collagen I; HGF, hepatocyte growth factor] and (E) protein [ProCol I, procollagen I; TIMP-1, tissue inhibitor of metalloproteinase-1; α-SMA, α-smooth muscle actin; MMP-9, matrix metalloproteinase-9] markers of fibroblasts. Unless otherwise indicated, data represent means of n=5 ± SEM. In (E), statistics reflect comparisons to Gucy2c+/+. *, p<0.05, **, p<0.01.
Fig. 6. Myofibroblast HGF production is controlled by epithelial cell GUCY2C and mediates afferent circuits regulating colon cancer cell proliferation.

(A) TGF-β induces CCD-18Co human fibroblasts to produce paracrine substances in conditioned media driving T84 human colon cancer cell proliferation in a concentration-dependent fashion, quantified by 3H-thymidine incorporation. (B) Media conditioned by T84 human colon cancer cells incubated with PBS (control) or ST (1 μM, 48 h) were treated with control IgG or neutralizing anti-TGF-β monoclonal antibody, with or without replacement with rTGF-β. These media conditioned by T84 cells and processed as described were incubated with CCD-18Co human fibroblasts. Media sequentially conditioned by T84 cells and fibroblasts were then incubated with HCT116 human colon cancer cells which do not express GUCY2C or TGF-β receptors, followed by quantification of proliferation by 3H-thymidine incorporation. [Prior to conditioning by fibroblasts, T84 cell-conditioned media were treated with: IgG, control; TGF-β Ab, anti-TGF-β antibody (10 μg/mL); rTGF-β, depletion of TGF-β by immunoprecipitation followed by replacement with rTGF-β (5 ng/mL)]. (C) HGF induces HCT116 human colon cancer cell proliferation in a concentration-dependent fashion, quantified by 3H-thymidine incorporation. (D) ST (1 μM, 48 h) pretreatment attenuates media conditioned by T84 colon cancer cells to induce HGF mRNA (left) and protein (right) expression in CCD-18Co human fibroblasts, quantified by RT-PCR and ELISA, respectively. (E) HGF secreted by CCD-18Co human fibroblasts activated by media conditioned by T84 human colon cancer cells treated with PBS or ST (1 μM, 48 h) activates cMET in HCT116 human colon cancer cells. cMET activation was quantified by immunoblotting for phosphorylation of cMET at Tyr1234/1235. Before addition to HCT116 cells, CCD-18Co cell-conditioned media were treated with: IgG, control; HGF neutralizing antibody (HGF nAb, 20 μg/mL); rHGF, depletion of HGF by immunoprecipitation followed by replacement with rHGF (1 ng/mL)]. (F) HGF secreted by CCD-18Co human fibroblasts activated by media conditioned by T84 human colon cancer cells treated with PBS or ST (1 μM, 48 h) induces HCT116 colon cancer cell proliferation through cMET. Before addition to HCT116 colon cancer cells, conditioned media were treated with: IgG, control; HGF Ab, anti-HGF neutralizing antibody (20 μg/mL); rHGF, depletion of HGF-anti-HGF complexes by immunoprecipitation followed by replacement with recombinant HGF (2.5 ng/mL)]. In some experiments, cMET was inhibited by cMet Kinase Inhibitor III (500 nM; cMET Inhibitor). Processed media were then incubated with HCT116 human colon cancer cells, followed by quantification of proliferation by 3H-thymidine incorporation. Data in 6A, 6B, 6C, and 6F are representative of ≥3 experiments. Data in 6D and 6E represent means of n 3≥experiments ± SEM. p<0.05; **, p<0.01, or n.s. p>0.05.
Hydroxyproline assay
Tissue was freeze-dried, homogenized, and hydrolyzed in 0.5 mL of 5 M HCl for 16 h at 116° C. Samples were dried, dissolved in distilled water, and hydroxyproline quantified using a colorimetric demethylmethylene blue assay (Sigma-Aldrich, MO) (34).
Collagen contractility assay
CCD-18Co cells and Type I Collagen (Sigma-Aldrich, MO) dissolved in acetic acid (0.1%) was used to access functional differences in contractility between quiescent fibroblasts and fibroblasts (35). After solutions were neutralized with NaOH (1 μM), cells were suspended in the collagen, plated, and cancer cell-conditioned media was added (35).
Quantitative RT-PCR
Total RNA was subjected to one-step RT-PCR using TaqMan® EZ RT-PCR Core Reagents and specific primer/probes for TaqMan® Gene Expression Assays in an ABI 7000 (Applied Biosystems, CA) (36). Relative expression was calculated using the 2-ΔΔCT method with GAPDH or β-actin as reference (36).
Electron microscopy
Intestinal samples were collected, processed, fixed and embedded as previously described (21). Ninety nm sections were imaged with a JEOL-1010 electron microscope employing 80 kV of acceleration.
Statistical analyses
Statistical significance was determined by unpaired two-tailed Student's t test. Unless otherwise indicated, results represent means ± SEM from ≥3 animals or ≥3 experiments performed in triplicate.
Results
GUCY2C in epithelial cells opposes desmoplasia
The absence of epithelial GUCY2C signaling in Gucy2c knockout mice (Gucy2c-/-) produced submucosal hypertrophy (Fig. 1A1, 1A2), with canonical characteristics of desmoplasia (1, 2, 5, 7). Specifically, stromal expansion was associated with increased deposition of the matrix components tenascin C (Fig. 1B1), fibronectin (Fig. 1B2), and collagen I (Fig. 1B3, 1B4), which is a hallmark of malignancy-associated desmoplasia (37). Also, absence of epithelial GUCY2C signaling increased submucosal fibroblast content, particularly in the pericryptal sheath (Fig. 1C1, 1C2), an anatomical location driving mesenchymally-directed epithelial cell proliferation (8, 38). Further, these cells were identified as fibroblasts, reflected by increased transcription (Fig. 1D) and translation (Fig. 1E) of canonical markers of fibroblast activation (1, 2, 5, 7). Thus, this submucosal hypertrophy in Gucy2c-/- mice recapitulated the desmoplastic reaction in human colorectal cancer.
Moreover, in vitro studies with human colon cancer cells and fibroblasts further demonstrated that fibroblast activation is modulated by GUCY2C signaling. Media conditioned by T84 human colon cancer cells in the presence or absence of GUCY2C-activating ligand (ST) were collected and used to treat CCD-18Co human fibroblasts. T84 cells express GUCY2C, which mediates canonical downstream cGMP signaling upon ligand binding (20-22). In contrast, CCD-18Co cells do not express GUCY2C and are unresponsive to ligands targeting that receptor. As quantified by activated fibroblast numbers (Fig. 2A), matrix synthesis (Fig. 2B), and contraction of collagen (Fig. 2C) (1, 7, 8, 35), conditioned media from T84 cells without ST treatment produced fibroblast activation, which was markedly reduced by ST.
Fig. 2. Activation of GUCY2C signaling prevents fibroblast activation.

CCD-18Co human fibroblasts were incubated with media conditioned by T84 or Caco 2 human colon cancer cells, which both express GUCY2C, pretreated with or without 1 μM ST for 48 h and fibroblast activation was quantified by (A) enumeration of fibroblast activation by α-SMA immunofluorescence [red, α-SMA; blue, DAPI], (B) immunoblot analysis of fibroblast markers [PHD, prolyl hydroxylase; Pro-Col I, procollagen I], (C) fibroblast contraction of collagen gels [Red circle indicates contracted gel area]. Data represent means of n=3 experiments ± SEM in (A-C). Data in (C) represent the percent reduction in gel surface area induced by contraction. * p<0.05, ** p<0.01.
Epithelial GUCY2C regulates fibroblast activation through TGF-β secretion
TGF-β is an established mediator of fibroblast activation and desmoplasia whose secretion characterizes the entire developmental continuum of solid tumors in animals and humans (1, 2, 7, 8, 11). GUCY2C activation by ST did not alter transcription of TGF-β, but markedly increased TGF-β retention and decreased TGF-β secretion, by cancer cells (Fig. 3A, B). In a series of conditioned-media experiments, media conditioned by T84 human colon cancer cells were collected and used to treat CCD-18Co human fibroblasts. Reducing TGF-β mRNA expression and protein secretion in cancer cells with siRNA (Fig.3A) mimicked ST-induced GUCY2C signaling and opposed fibroblast activation, including collagen contractility and translation of biomarkers of activation, by conditioned media (Fig. 3C-D). Inhibiting fibroblast TGF-β type I receptors with TGF-β receptor inhibitor SB-505124 also mimicked ST-induced GUCY2C signaling and opposed fibroblast activation by conditioned media (Fig. 3E). Further, cancer cell-conditioned media activated fibroblast TGF-β receptor signaling through phosphorylation of the TGF-β signaling target Smad-3, and GUCY2C signaling in cancer cells blocked that effect (Fig. 3F).
Fig. 3. TGF-β mediates GUCY2C-dependent colon cancer cell regulation of fibroblast activation in vitro.

(A) Effect of GUCY2C signaling on (A1) TGF-β mRNA expression, quantified by RT-PCR, or (A2) protein secretion, quantified by ELISA, by T84 human colon cancer cells. (A) Effect of TGF-β1-specific siRNAs (siTGF1 and siTGF2) on TGF-β1 mRNA (qRT-PCR) expression and secretion (ELISA) by T84 cells. (B) Effect of GUCY2C, induced by ST (1 μM ST, 48 h), to regulate TGF-β secretion, quantified by ELISA, in T84 human colon cancer cell. (C) Effect of TGF-β1-specific siRNAs on the ability of T84 cells pretreated with PBS or ST (1 μM ST, 48 h) to condition media quantified by (C1) fibroblast activation by confocal microscopy of α-SMA immunocytochemistry (activated fibroblasts) and (C2) fibroblast contraction of collagen gels (relative contractility). (D) Effect of TGF-β1-specific siRNA on the ability of T84 cells pretreated with PBS or ST (1 μM ST, 48 h) to condition media, quantified by fibroblast PHD or α-SMA expression by immunoblot analysis [None, no siRNA; CTR (control), Scrambled siRNA; siTGF, TGF-β1 siRNA]. (E) Effect of SB-505124, a selective inhibitor of TGF-β type I receptors, on the ability of CCD-18Co human fibroblasts to be activated by media conditioned by T84 human colon cancer cells pretreated with PBS (control) or the GUYC2C ligand ST. (F) Smad3 phosphorylation in fibroblasts activated by colon cancer cell-conditioned media [tSmad3, total Smad 3; pSmad3, phosphorylated Smad3]. Data represent means of n≥3 experiments ± SEM. *, p<0.05; **, p<0.01, or n.s. p>0.05.
Additionally, a neutralizing antibody to secreted TGF-β reduced both the fibroblast activation (Fig. 4A) and the Smad-3 phosphorylation (Fig. 4B) induced by cancer cell-conditioned media. The effects of neutralizing antibodies on fibroblast activation and TGF-β signaling were abrogated when TGF-β was re-introduced by adding recombinant purified TGF-β (rTGB-β) (Fig. 4A-B). To reinforce these in vitro findings, in vivo studies were conducted in which Gucy2c-/- mice were treated with TGF-β neutralizing antibodies. Antibody administration reversed the submucosal desmoplasia (Fig. 4C), reducing interstitial thickness (Fig. 4C), total collagen deposition (Fig. 4D), Smad-3 phosphorylation (Fig. 4E), and fibroblast activation (Fig. 4F). These data reveal that GUCY2C mediates an efferent mucosal paracrine limb which modulates epithelial TGF-β secretion and fibroblast activation through TGF-β receptors and Smad-3 phosphorylation, and thereby shapes the submucosal microenvironment to oppose the desmoplastic reaction.
Fig. 4. TGF-β eliminates fibroblast activation in vitro and desmoplasia induced by silencing epithelial cell GUCY2C in mice.

Media conditioned by T84 human colon cancer cells incubated with PBS (control) or ST (1 μM, 48 h) were treated with control IgG or neutralizing anti-TGF-β monoclonal antibody. In some experiments, T84 cell-derived TGF-β bound to neutralizing antibody was depleted using Protein G immunoaffinity beads and replaced with recombinant purified TGF-β (rTGF-β). Processed media were incubated with CCD-18Co human fibroblasts followed by quantification of (A) fibroblast activation by α-SMA immunocytochemistry and (B) phosphorylation of Smad3 by immunoblot analysis. Colon cancer cell-conditioned media were treated with: IgG, control; TGF-β Ab, anti-TGF-β antibody (30 μg/mL); rTGF-β, depletion of TGF-β-anti-TGF-β complexes with protein G immunoaffinity beads followed by replacement with recombinant TGF-β (10 ng/mL). In (C-F), Gucy2c+/+ and Gucy2c-/- mice were treated with IgG or anti-TGF-β monoclonal antibody (0.5 mg/kg) IP twice weekly for ten weeks to neutralize soluble TGF-β followed by quantification of intestinal (C) matrix deposition, (D) collagen content, (E) Smad3 phosphorylation, and (F) fibroblast activation. Data represent means of n≥4 experiments ± SEM *, p<0.05, **, p<0.01, or n.s., p>0.05.
GUCY2C regulates epithelial TGF-β secretion and fibroblast activation through Akt
GUCY2C regulates epithelial cell homeostasis and suppresses tumorigenesis through the Akt signaling pathway (21). Notably, studies have also demonstrated that Akt regulates the secretion of TGF-β in mechanisms remodeling the stroma microenvironment (39). In vitro, elimination of AKT expression with siRNA (siAkt) mimicked ST-induced GUCY2C signaling, and replicated the effects of ST on cancer cell secretion of TGF-β (Fig. 5A) and fibroblast activation (Fig. 5B). Conversely, constitutive activation of AKT by myristoylation (myrAKT) (21) mimicked the absence of GUCY2C signaling, and blocked the reverting effects of ST, leading to the promotion of TGF-β secretion (Fig. 5C). Moreover, reducing AKT expression through gene disruption in vivo subdued the stromal remodeling produced by silencing GUCY2C (Fig. 5D). These observations support a model in which, as in other homeostatic processes like proliferation and metabolism (19-23), the regulation of epithelial TGF-β secretion, fibroblast activation, and stromal remodeling by GUCY2C is mediated through epithelial AKT signaling.
Fig. 5. GUCY2C regulates epithelial cell TGF-β secretion, fibroblast activation and desmoplasia through AKT.

(A) TGF-β protein secretion from colon cancer cells was quantified by ELISA. Cells were infected by adenovirus delivering GFP (control) or siAKT (siRNA targeting AKT). The effect of siAKT was confirmed by immunoblotting for AKT phosphorylated at ser473 (pAKT). (B) Effect of siAkt on the ability of T84 cells pretreated with PBS or ST (1 μM ST, 48 h) to condition media quantified by myofibroblast activation by α-SMA immunofluorescence [red, α-SMA; blue, DAPI; green, phospho-smad3]. (C) TGF-β protein secretion from cells infected with adenovirus delivering wild type AKT (wtAKT) or myristoylated AKT (myrAKT). Effect of myrAKT was confirmed by immunoblotting for pAKT. (D) Masson's Trichrome stain for quantification of submucosal matrix thickness. In (A-C), data represent means of n≥3 experiments ± SEM. In (D) each point represents one mouse. *, p<0.05, **, p<0.01, or n.s., p>0.05.
GUCY2C reciprocally regulates fibroblast-dependent epithelial cell hyperproliferation
Beyond the contribution of efferent mucosal signals to submucosal fibroblasts, mechanisms by which this microenvironmental re-programming reciprocally contributes to an afferent paracrine loop modulating epithelial hyperproliferation remain incompletely defined (1, 2, 7, 8). To investigate this afferent loop, we conducted a series of in vitro experiments employing sequentially conditioned media: media conditioned by human colon cancer cells were collected and used to activate human fibroblasts; and the media conditioned by these activated fibroblasts were collected and applied to human colon cancer cells.
In one study, CCD-18Co human fibroblasts were activated by TGF-β at different concentrations, and the conditioned media was collected. Application of this fibroblast-conditioned media drove proliferation of human colon cancer cells, as quantified by 3H-thymidine incorporation, in a TGF-β concentration-dependent manner (Fig. 6A). Similarly, colon cancer cell-conditioned media activated fibroblasts, which, in turn, produced conditioned media that reciprocally drove the proliferation of HCT116 human colon cancer cells, which do not express GUCY2C or TGF-β receptors (Fig. 6B). However, the ability of fibroblasts to drive epithelial proliferation was eliminated by pre-treating epithelial cell-conditioned media with TGF-β neutralizing antibodies (Fig. 6B). Further, when treated with media from cancer cells incubated with the GUCY2C ligand ST, fibroblasts failed to drive epithelial proliferation (Fig. 6B). Exogenous supplementation of fibroblasts with rTGF-β reconstituted the fibroblast-conditioned media's capacity to induce epithelial proliferation, abrogating the effects of TGF-β neutralizing antibody or GUCY2C signaling (Fig. 6B). These results reveal that GUCY2C signaling regulates the epithelial secretion of TGF-β, which not only activates fibroblasts, but also induces them to release factors that reciprocally feed back to drive epithelial cell proliferation.
Desmoplastic fibroblast activation is associated with expression of the mitogen HGF (Fig. 1D), a mediator of microenvironmental influences on epithelial cells, including stromal induction of epithelial proliferation (1, 2, 7, 8, 11, 40). In agreement with previous studies, HGF induced HCT116 human colon cancer cells to proliferate in a concentration-dependent fashion (Fig. 6C). Further, pretreatment of cancer cells with ST reduced HGF mRNA and protein production from fibroblasts that were activated by TGF-β secreted from colon cancer cells, (Fig. 6D). In turn, those fibroblast-conditioned media activated cMET, the canonical receptor for HGF (32), in HCT116 cells. This effect was eliminated by pre-treating fibroblast-conditioned media with HGF neutralizing antibody, an effect reversed by recombinant HGF (rHGF; Fig. 6E). Moreover, ST-activated GUCY2C signaling in epithelial cells reduced their ability to induce fibroblasts to drive colon cancer cell proliferation (Fig. 6B and F). This effect was replicated by adding HGF neutralizing antibodies or an inhibitor of cMET to the fibroblast-conditioned media and blocked by rHGF (Fig. 6F). These observations suggest a model in which GUCY2C mediates an efferent paracrine circuit regulating the epithelial secretion of TGF-β, and the consequent activation of fibroblasts. In turn, TGF-β ultimately induces a reciprocal afferent paracrine circuit in which the activated stromal fibroblasts produce HGF, which promotes cMET-dependent epithelial cell proliferation (Fig. 7).
Fig. 7. Model of GUCY2C signaling effects on tumor microenvironment.
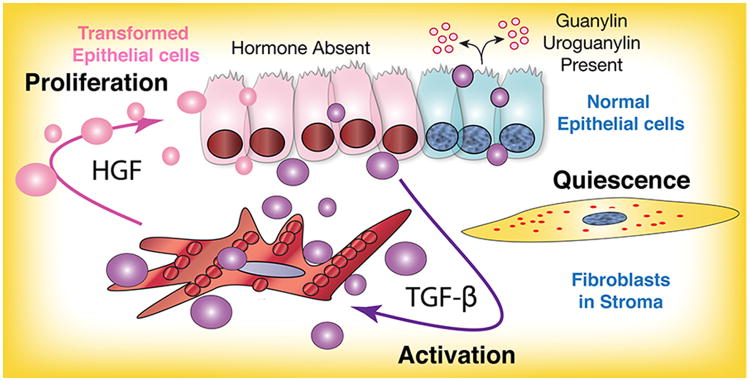
GUCY2C hormones, guanylin and uroguanylin, secreted by normal epithelial cells (blue) maintain a homeostatic balance between the mucosa and fibroblasts in underlying stroma, mediated by epithelial cell GUCY2C. Silencing of GUCY2C through loss of ligand expression, a universal early event in tumorigenesis, induces transforming epithelial cells (pink) to secrete TGF-β (purple bubbles) which activates fibroblasts. In turn, activated fibroblasts have the ability to promote tumor cell proliferation by HGF (pink bubbles).
Discussion
Undergoing continuous cycles of regeneration, the intestinal crypt-surface axis is dynamic. GUCY2C signaling organizes the spatiotemporal tissue pattern, maintaining homeostasis along this axis (19-23). Specifically, GUCY2C controls epithelial cell proliferation by regulating the expression of critical mediators that slow the cell cycle (19-23). Further, GUCY2C and cGMP signaling establish cell fate by promoting the differentiation of epithelial cells in the secretory lineage, which includes Paneth and goblet cells (19). These effects define the balance between the relative sizes of proliferating and differentiated cell compartments along the crypt-surface axis (19-23). Moreover, GUCY2C is key in matching the appropriate metabolic program to the functional demands along this axis. Namely, low GUCY2C signaling in the crypt favors glycolysis to support rapidly proliferating transit-amplifying cells (21). Conversely, robust GUCY2C signaling in the villus/surface compartment shifts the metabolic program to mitochondrial oxidative phosphorylation, the canonical source for ATP generation in differentiated cells (21).
The present study expands the homeostatic role for GUCY2C beyond these cell-autonomous functions to coordinating the paracrine cross-talk between the epithelial and mesenchymal compartments that is essential for normal tissue patterning (1, 3, 40). Indeed, it suggests a model in which mucosal GUCY2C coordinates bi-directional epithelial-mesenchymal cross-talk in the intestine (Fig. 7). In this model, GUCY2C signaling in the epithelium opposes an efferent mucosal paracrine loop by suppressing the epithelial secretion of TGF-β, the activation of submucosal fibroblasts, and the desmoplastic reaction. In turn, the suppression of fibroblast activation by GUCY2C-regulated paracrine signaling reduces HGF, silencing an afferent loop that drives epithelial cell proliferation. Notably, this GUCY2C function in the intestinal mucosa recapitulates that of the related guanylyl cyclase isoform NPR1 in cardiomyocytes (28, 29). NPR1 coordinates mesenchymal homeostasis in the heart, and its silencing produces cardiac fibrosis that is independent of increases in blood pressure (28, 29). Targeted NPR1 knockout (Npr1-/-) produces local extracellular matrix remodeling in paracrine pathways involving increased TGF-β (28, 29).
The discovery that GUCY2C underlies spatiotemporal patterning along the crypt-surface axis expands its role in the homeostatic maintenance of the intestinal mucosae (19, 22). Moreover, it highlights the emergence of GUCY2C as a tumor suppressor universally silenced in the initiation and progression of colorectal cancer (20, 21, 23). The endogenous ligands for GUCY2C, guanylin and uroguanylin, are the most commonly lost gene products in intestinal tumorigenesis and these ligands are lost at the earliest stages of transformation (24-27). Loss of paracrine ligands silences GUCY2C which, in turn, activates AKT and mTOR, well-established oncogenes driving tumorigenesis in many tissues (21). Indeed, eliminating GUCY2C signaling amplifies intestinal tumorigenesis induced by carcinogens or gene mutations (20).
Silencing GUCY2C establishes a number of cell-autonomous mechanisms underlying epithelial cell tumorigenesis (6). Thus, the cell cycle is accelerated, expanding the proliferating crypt compartment (19-23). Further, metabolic programming is shifted from mitochondrial oxidative phosphorylation to aerobic glycolysis characterizing the Warburg metabolic phenotype (21). Moreover, genomic instability is accelerated by over-production of reactive oxygen species and exacerbated by a decrease in the ability to repair DNA damage (20, 21). Indeed, eliminating GUCY2C signaling induces Apc loss of heterozygosity in Apcmin mice and the acquisition of mutations in β-catenin in mice treated with the carcinogen AOM, encompassing the genomic changes underlying nearly all sporadic intestinal tumorigenesis in humans (20). The present study expands the role of GUCY2C as a tumor suppressor whose silencing not only corrupts cell-autonomous pathways regulating epithelial transformation, but also epithelial-mesenchymal paracrine circuits underlying fibroblast activation and desmoplasia, a process which universally contributes to tumorigenesis in most tissues (2, 5, 6).
While interactions between tumor cells and the desmoplastic stroma are essential for cancer progression, invasion, metastasis, and the evolution of therapeutic resistance, the relative contributions of each compartment to tumor initiation remains undefined (1, 2, 5, 16). The oncogenomic model of cancer suggests that tumors initiate and evolve by accumulating mutations, which alter critical signaling pathways regulating cell-autonomous homeostatic functions, which ultimately lead to genomic instability and acquisition of invasive and metastatic phenotypes (6, 41-43). In this model, the mesenchymal compartment is dependent and reactive, shaped by the epithelial tumor to support cancer evolution. An alternate hypothesis suggests that, in some cases, stroma could be the pathophysiological initiator of tumorigenesis in dependent normal epithelial cells. Mice in which TGF-β receptors are silenced selectively in stromal fibroblasts develop neoplasia in the prostate and stomach (10, 44). Similarly, genetic models of HGF over-expression in fibroblasts are characterized by mammary tumorigenesis (45). Additionally, mutations of tumor suppressor genes in stromal fibroblasts drive the development of prostate tumors (46). This mechanistic debate concerning which compartment “leads” transformation has not yet been resolved (2, 5). The present study, in the context of GUCY2C silencing as one of the earliest changes underlying intestinal neoplasia (24-27), suggests that epithelial cell transformation leads tumor initiation in intestine by corrupting cell-autonomous (19-23) and non-cell-autonomous homeostatic pathways, including the promotion of desmoplasia in colorectal cancer.
Beyond this question of compartmental hierarchy, the relationship between canonical networks whose corruption initiates neoplasia, and reciprocal paracrine signaling axes organizing epithelial-mesenchymal cross-talk producing desmoplasia, remains undefined. For example, nearly all sporadic colorectal tumors harbor mutations in the WNT signaling pathway, primarily in APC (about 80%) or β-catenin (about 15%), which drive cell-autonomous changes in epithelia (41, 43, 47). However, the precise linkages between corrupted WNT signaling pathways underlying tumorigenesis and mechanisms coordinating desmoplasia characterizing colorectal cancer remain unknown. The present study offers a mechanistic explanation for coordination of epithelial transformation and the development of the desmoplastic reaction in colorectal cancer. Indeed, silencing GUCY2C in epithelial cells appears to be one of the earliest universal changes in colorectal carcinogenesis (24-27), with the ability to promote genomic instability and mutations in WNT signaling pathway intermediates (20). In this model, silencing the GUCY2C tumor suppressor early in transformation corrupts cell-autonomous functions that are essential for transformation, including proliferation, metabolism, and DNA damage and repair (19-23). Moreover, the present study reveals that silencing GUCY2C simultaneously initiates AKT-dependent paracrine signaling through increased epithelial cell TGF-β secretion, activating fibroblasts that reshape the mesenchymal matrix compartment, producing the desmoplastic reaction promoting colorectal tumor invasion and progression.
There continues to be intense interest in elucidating the precise compartmental contributions, and their sequence, underlying epithelial-mesenchymal cross-talk contributing to neoplasia (16). Deconvoluting this spatiotemporal signaling sequence could provide therapeutic targets that disrupt the mutually reinforcing intercompartmental cycle essential for tumor initiation, progression, invasion, and metastasis (1, 2, 4, 5, 8, 16). This challenge is underscored by the incompletely understood role of TGF-β in tumorigenesis, sometimes referred to as the “TGF-β Paradox” (11, 48, 49). On the one hand, TGF-β has potent anti-proliferative properties in epithelial cells, and silencing tumor cell TGF-β receptors by mutation is a key genomic event mediating progression of colorectal tumorigenesis (41). On the other hand, TGF-β is one of the most potent drivers of quiescent fibroblast transformation to the fibroblast phenotype, although the precise compartment producing this cytokine driving desmoplasia remains uncertain (1, 2, 5, 8). Moreover, the activity of TGF-β on the stroma can promote tumor initiation, progression, and metastasis (50). Similarly, although HGF, which drives epithelial proliferation through cMET, is produced by fibroblasts in desmoplasia, the events inducing that production remain uncharacterized in most tumors (1, 2, 5, 8). In that context, the present study provides insight into the spatiotemporal sequence of signaling interactions coordinating events that drive tumor initiation and progression in epithelia and stroma in colorectal cancer. It suggests a model in which the earliest stages of tumorigenesis initiate, at least in part, by silencing GUCY2C, reflecting loss of ligand expression (24-27). Eliminating cGMP signaling in intestinal epithelial cells not only drives cell-autonomous changes coincident with transformation (19-23), but also amplifies epithelial secretion of TGF-β, creating an efferent signal driving fibroblast activation and creating a reciprocal afferent cytokine circuit with the potential to signal back to the epithelial compartment through cMET. In more advanced stages of transformation, characterized by loss of TGF-β signaling and its cell cycle suppression in epithelia (41, 48, 49), the potential of this reciprocal afferent HGF circuit is realized, further amplifying proliferation driving the progression of tumorigenesis.
Epithelial cell GUCY2C signaling is one component of a program establishing and maintaining the intestinal crypt-surface axis through continuous waves of physiological regeneration. Beyond this role in normal tissue patterning, loss of guanylin and uroguanylin at the earliest stages of premalignancy suggests a novel model of intestinal tumorigenesis, initiating as a disease of tissue-specific paracrine hormone insufficiency (24-27). The resultant silencing of the GUCY2C tumor suppressor produces maladaptive amplification of survival circuits producing DNA damage and mutations contributing to the cell-autonomous basis of colorectal cancer (19-23). Moreover, these changes in epithelial transformation are coordinated with simultaneous changes in cytokine signaling mediating the desmoplastic reaction which, reciprocally, drives epithelial proliferation. This unique model of intestinal transformation as a paracrine hormone deficiency coordinating epithelial-mesenchymal transformation has substantial implications for the prevention and treatment of colorectal cancer. Indeed, GUCY2C is over-expressed by intestinal tumors compared to normal adjacent tissues (36). Early loss of hormones with compensatory over-expression of receptors offers a unique therapeutic window preceding the corruption of epithelial survival pathways and stromal reactivity to prevent maladaptive reprogramming and the evolution of cancer through oral GUCY2C hormone replacement therapy (25). These considerations highlight the potential translational opportunities for oral GUCY2C paracrine hormone supplementation as a novel strategy that could contribute to the prevention of colorectal cancer, underscored by the recent FDA approval and commercial availability of GUCY2C ligands to treat chronic constipation (51).
Acknowledgments
Financial Support: These studies were supported by grants from the National Institutes of Health (R01 CA75123, R01 CA95026, RC1 CA146033, P30 CA56036, R01 CA170533), the Pennsylvania Department of Health (SAP #4100059197, SAP #4100051723), and Targeted Diagnostic and Therapeutics Inc. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations or conclusions. A.V.G. was supported by an NIH Minority Supplement (R01-CA095026-04S1). J.E.L. was supported by NIH institutional award T32 GM08562 for Postdoctoral Training in Clinical Pharmacology and is the recipient of the Young Investigator Award from the American Society for Clinical Pharmacology and Therapeutics (ASCPT). G.W.K. is the recipient of the Graduate Award for Integrative Research in Pharmacology from the American Society for Pharmacology and Experimental Therapeutics (ASPET). P.L. was enrolled in the NIH-supported institutional K30 Training Program In Human Investigation (K30 HL004522) and was supported by NIH institutional award T32 GM08562 for Postdoctoral Training in Clinical Pharmacology. A.E.S. is the recipient of the Measey Foundation Fellowship. S.A.W. is the Samuel MV Hamilton Professor of Thomas Jefferson University.
Abbreviations
- α-SMA
α-smooth muscle actin
- cGMP
cyclic GMP
- Col I
collagen I
- NPR1
guanylyl cyclase A
- GUCY2C
guanylyl cyclase C
- HGF
hepatocyte growth factor
- IP
intraperitoneal
- MMP-9
matrix metalloproteinase-9
- NAT
normal adjacent tissue
- ProCol I
procollagen I
- pSmad3
phosphorylated Smad3
- rHGF
recombinant HGF
- rTGF-β
recombinant TGF-β
- siTGF
TGF-β1 siRNA
- ST
bacterial heat-stable enterotoxin
- TIMP-1
tissue inhibitor of metalloproteinase-1
- tSmad3
total Smad 3
Footnotes
Disclosures: S.A.W. is the Chair of the Data Safety Monitoring Board for the C-Cure Trial™ sponsored by Cardio Biosciences, and the Chair (uncompensated) of the Scientific Advisory Board of Targeted Diagnostics & Therapeutics, Inc. which provided research funding that, in part, supported this work and has a license to commercialize inventions related to this work.
References
- 1.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 2.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 4.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Rasanen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316:2713–2722. doi: 10.1016/j.yexcr.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 8.Worthley DL, Giraud AS, Wang TC. Stromal fibroblasts in digestive cancer. Cancer Microenviron. 2010;3:117–125. doi: 10.1007/s12307-009-0033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 10.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 12.Guo X, Oshima H, Kitmura T, Taketo MM, Oshima M. Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer. J Biol Chem. 2008;283:19864–19871. doi: 10.1074/jbc.M800798200. [DOI] [PubMed] [Google Scholar]
- 13.Hasebe T, Sasaki S, Imoto S, Ochiai A. Proliferative activity of intratumoral fibroblasts is closely correlated with lymph node and distant organ metastases of invasive ductal carcinoma of the breast. Am J Pathol. 2000;156:1701–1710. doi: 10.1016/S0002-9440(10)65041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noma K, Smalley KS, Lioni M, Naomoto Y, Tanaka N, El-Deiry W, King AJ, Nakagawa H, Herlyn M. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology. 2008;134:1981–1993. doi: 10.1053/j.gastro.2008.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Littlepage LE, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell. 2005;7:499–500. doi: 10.1016/j.ccr.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 16.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 17.Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375–414. [PubMed] [Google Scholar]
- 18.Michell AR, Debnam ES, Unwin RJ. Regulation of renal function by the gastrointestinal tract: potential role of gut-derived peptides and hormones. Annu Rev Physiol. 2008;70:379–403. doi: 10.1146/annurev.physiol.69.040705.141330. [DOI] [PubMed] [Google Scholar]
- 19.Li P, Lin JE, Chervoneva I, Schulz S, Waldman SA, Pitari GM. Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am J Pathol. 2007;171:1847–1858. doi: 10.2353/ajpath.2007.070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P, Schulz S, Bombonati A, Palazzo JP, Hyslop TM, Xu Y, Baran AA, Siracusa LD, Pitari GM, Waldman SA. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology. 2007;133:599–607. doi: 10.1053/j.gastro.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 21.Lin JE, Li P, Snook AE, Schulz S, Dasgupta A, Hyslop TM, Gibbons AV, Marszlowicz G, Pitari GM, Waldman SA. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology. 2010;138:241–254. doi: 10.1053/j.gastro.2009.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pitari GM, Di Guglielmo MD, Park J, Schulz S, Waldman SA. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc Natl Acad Sci U S A. 2001;98:7846–7851. doi: 10.1073/pnas.141124698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitari GM, Zingman LV, Hodgson DM, Alekseev AE, Kazerounian S, Bienengraeber M, Hajnoczky G, Terzic A, Waldman SA. Bacterial enterotoxins are associated with resistance to colon cancer. Proc Natl Acad Sci U S A. 2003;100:2695–2699. doi: 10.1073/pnas.0434905100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Notterman DA, Alon U, Sierk AJ, Levine AJ. Transcriptional gene expression profiles of colorectal adenoma, adenocarcinoma, and normal tissue examined by oligonucleotide arrays. Cancer Res. 2001;61:3124–3130. [PubMed] [Google Scholar]
- 25.Shailubhai K, Yu HH, Karunanandaa K, Wang JY, Eber SL, Wang Y, Joo NS, Kim HD, Miedema BW, Abbas SZ, Boddupalli SS, Currie MG, Forte LR. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 2000;60:5151–5157. [PubMed] [Google Scholar]
- 26.Steinbrecher KA, Tuohy TM, Heppner Goss K, Scott MC, Witte DP, Groden J, Cohen MB. Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem Biophys Res Commun. 2000;273:225–230. doi: 10.1006/bbrc.2000.2917. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]
- 28.Holtwick R, van Eickels M, Skryabin BV, Baba HA, Bubikat A, Begrow F, Schneider MD, Garbers DL, Kuhn M. Pressure-independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest. 2003;111:1399–1407. doi: 10.1172/JCI17061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vellaichamy E, Khurana ML, Fink J, Pandey KN. Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A. J Biol Chem. 2005;280:19230–19242. doi: 10.1074/jbc.M411373200. [DOI] [PubMed] [Google Scholar]
- 30.McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol. 2005;174:5774–5780. doi: 10.4049/jimmunol.174.9.5774. [DOI] [PubMed] [Google Scholar]
- 31.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 32.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 33.Simmons JG, Pucilowska JB, Keku TO, Lund PK. IGF-I and TGF-beta1 have distinct effects on phenotype and proliferation of intestinal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;283:G809–818. doi: 10.1152/ajpgi.00057.2002. [DOI] [PubMed] [Google Scholar]
- 34.Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem. 1996;29:225–229. doi: 10.1016/0009-9120(96)00003-6. [DOI] [PubMed] [Google Scholar]
- 35.Ngo P, Ramalingam P, Phillips JA, Furuta GT. Collagen gel contraction assay. Methods Mol Biol. 2006;341:103–109. doi: 10.1385/1-59745-113-4:103. [DOI] [PubMed] [Google Scholar]
- 36.Schulz S, Hyslop T, Haaf J, Bonaccorso C, Nielsen K, Witek ME, Birbe R, Palazzo J, Weinberg D, Waldman SA. A validated quantitative assay to detect occult micrometastases by reverse transcriptase-polymerase chain reaction of guanylyl cyclase C in patients with colorectal cancer. Clin Cancer Res. 2006;12:4545–4552. doi: 10.1158/1078-0432.CCR-06-0865. [DOI] [PubMed] [Google Scholar]
- 37.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neal JV, Potten CS. Description and basic cell kinetics of the murine pericryptal fibroblast sheath. Gut. 1981;22:19–24. doi: 10.1136/gut.22.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dey-Guha I, Wolfer A, Yeh AC, J GA, Darp R, Leon E, Wulfkuhle J, Petricoin EF, 3rd, Wittner BS, Ramaswamy S. Asymmetric cancer cell division regulated by AKT. Proc Natl Acad Sci U S A. 2011;108:12845–12850. doi: 10.1073/pnas.1109632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 43.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 44.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 45.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, Richardson A, Weinberg RA. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 47.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 48.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian M, Schiemann WP. The TGF-beta paradox in human cancer: an update. Future Oncol. 2009;5:259–271. doi: 10.2217/14796694.5.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, Sevillano M, Nadal C, Jung P, Zhang XH, Byrom D, Riera A, Rossell D, Mangues R, Massague J, Sancho E, Batlle E. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lembo AJ, Schneier HA, Shiff SJ, Kurtz CB, MacDougall JE, Jia XD, Shao JZ, Lavins BJ, Currie MG, Fitch DA, Jeglinski BI, Eng P, Fox SM, Johnston JM. Two randomized trials of linaclotide for chronic constipation. N Engl J Med. 2011;365:527–536. doi: 10.1056/NEJMoa1010863. [DOI] [PubMed] [Google Scholar]