

Abstract
Calcitonin gene-related peptide (CGRP) is a member of the calcitonin (CT) family of peptides. It is a widely distributed neuropeptide implicated in conditions such as neurogenic inflammation. With other members of the CT family, it shares an N-terminal disulphide-bonded ring which is essential for biological activity, an area of potential α-helix, and a C-terminal amide. CGRP binds to the calcitonin receptor-like receptor (CLR) in complex with receptor activity-modifying protein 1 (RAMP1), a member of the family B (or secretin-like) GPCRs. It can also activate other CLR or calcitonin-receptor/RAMP complexes. This 37 amino acid peptide comprises the N-terminal ring that is required for receptor activation (residues 1–7); an α-helix (residues 8–18), a region incorporating a β-bend (residues 19–26) and the C-terminal portion (residues 27–37), that is characterized by bends between residues 28–30 and 33–34. A few residues have been identified that seem to make major contributions to receptor binding and activation, with a larger number contributing either to minor interactions (which collectively may be significant), or to maintaining the conformation of the bound peptide. It is not clear if CGRP follows the pattern of other family B GPCRs in binding largely as an α-helix.
LINKED ARTICLES
This article is part of a themed section on Neuropeptides. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.170.issue-7
Keywords: calcitonin, CGRP, adrenomedullin, amylin, helix cap, receptor activation, domain, juxtamembrane domain, GPCR, RAMP, receptor activity-modifying protein
Introduction
Calcitonin gene-related peptide (CGRP) is a 37 amino acid neuropeptide belonging to the calcitonin (CT) family of peptides. This family also includes amylin, the calcitonin receptor-stimulating peptides, and the adrenomedullins (AMs), and they are found in vertebrates from fish to mammals (Ogoshi et al., 2006). Figure 1 provides an alignment of CGRP peptides from a representative selection of 19 species. There is a high degree of conservation across these species, with 23 of 37 residues absolutely conserved. The majority of sequence changes in the other positions can be considered to be conservative replacements.
Figure 1.

Sequence alignment of CGRP sequences from a selection of species generated using ClustalW and visualized using EsPript using the %Equivalence algorithm. Conserved regions are shown in red boxes with regions with similar physicochemical parameters in yellow. Species are listed by their common names: human-α (Homo sapiens), horse (Equus caballus), rat-α (Rattus norvegicus), mouse (Mus musculus), chicken (Gallus gallus), pig (Sus scrofa), sheep (Ovis aries), cow (Bos taurus), dog (Canis lupus familiaris), marmoset (Callithrix jacchus), opossum (Monodelphis domestica), Gekko (Gekko japonicas), frog (Phyllomedusa bicolor), pufferfish (Takifugu rubripes), flounder (Paralichthys olivaceus), goldfish (Carassius auratus), salmon (Salmo salar), medalea (Oryzias latipes), zebrafish (Danio rerio).
CGRP is found throughout the sensory nervous system; it is a potent vasodilator and a key mediator in neurogenic inflammation. CGRP antagonists have attracted considerable attention for the treatment of migraine; agonists are of potential interest to treat a range of cardiovascular disorders (Brain and Grant, 2004; Durham and Vause, 2010). There are two forms of CGRP in humans and rodents, α and β; unless otherwise stated in this review, it is the human α-form that is being considered. This is the most abundant form of CGRP for which most information is available. Generally, there seems little difference between the pharmacology of α- and β-CGRP (Bailey and Hay, 2006) or between rat and human forms (Poyner, 1992).
CGRP has several key structural features, most prominently a disulphide bond between residues 2 and 7 and a C-terminal amide. In common with other peptide ligands for family B GPCRs, it is believed to follow a two-step mechanism of action to activate its receptor. The C-terminus of the ligand binds to the extracellular domain (ECD) of the receptor and the N-terminus of the ligand (which incorporates the disulphide-bonded loop) binds to the transmembrane domain (TM) and extracellular loop (ECL) region of the receptor to give activation.
Many peptide and non-peptide CGRP antagonists have been described. These were initially based on peptide fragments lacking the N-terminal disulphide-bonded loop, of which the best characterized is CGRP8–37 (Chiba et al., 1989; Dennis et al., 1990). More recently, attention has focused on shorter peptides, which are often derivatives of CGRP27–37 (Yan et al., 2011). The non-peptide antagonists are typified by olcegepant and telcagepant (Negro et al., 2012). Crystal structures are available for the ECD of the CGRP receptor showing these two antagonists bound but no structure is available of any bound peptide or of the TM region (ter Haar et al., 2010).
The mechanism by which the endogenous ligands for family B GPCRs, like CGRP, bind to and activate their receptors is of considerable interest (Neumann et al., 2008; Parthier et al., 2009; Watkins et al., 2012) because this can lead to the design of novel antagonists and agonists. Understanding the structure–activity relationships of natural peptides has led to the development of several stable and efficacious peptide-based agonists of family B GPCRs that have ultimately become therapeutics (Shimizu et al., 2000; Devigny et al., 2011; Garber, 2012). This review considers recent developments in the structure–activity relationship for CGRP and how these might be interpreted in the light of current knowledge of the binding of ligands to family B GPCRs.
CGRP-responsive receptors and their implications for pharmacological studies
The CGRP receptor is a complex between a family B GPCR known as the calcitonin receptor-like receptor (CLR) and a single-pass membrane protein, receptor activity-modifying protein 1 (RAMP1) (McLatchie et al., 1998). A third protein, receptor component protein, is needed for G protein coupling but is of little known relevance for ligand binding (Prado et al., 2002). CGRP also has high affinity at the AMY1 receptor, a receptor for amylin formed from the calcitonin receptor (CTR) and RAMP1 (Christopoulos et al., 1999; Poyner et al., 2002); indeed it exhibits higher potency than amylin at the receptor formed between RAMP1 and the Δ47 splice variant of the CTR (Qi et al., 2012). It is also possible that neuronally released CGRP may be able to activate the AMY1 receptor. At pharmacological concentrations, CGRP can also activate the AM2 and AMY2 and AMY3 receptors and has weak affinity for the AM1 receptor (Hay et al., 2008). This in turn poses problems for interpreting many studies where the pharmacology of CGRP or its derivatives have been examined on native tissues (where receptor subunit expression often remains undetermined), or cells known to express more than one type of receptor that can be activated by CGRP. There is a possibility that the apparent activity of some derivatives may be due to activation of one of the other CGRP-responsive receptors, not the CLR-RAMP1 complex. In Tables 1–4 where the activity of CGRP derivatives has been reported, the nature of the preparation has been noted; SK-N-MC and L6 cells are commonly used as models of a CGRP-receptor-expressing system (Hay et al., 2002). Although species differences can influence the potency of non-peptide antagonists, due to their mode of interaction with RAMP1 (Mallee et al., 2002), this seems of much less importance for studies with full-length peptides (Poyner et al., 2002).
Table 1.
Mutation of residues 1–7
| Residue | Mutation | Effect | Comment | Reference |
|---|---|---|---|---|
| Ala1 | [Tyr0]-CGRP | Threefold decrease in affinity | SK-N-MC cells | (van Valen et al., 1989) |
| Ala1 | [Tyr0]-CGRP | 12-fold increase in affinity | Rat brain membranes | (Dennis et al., 1989) |
| Ala1 | [Tyr0]-CGRP | 33-fold decrease in affinity | L6 myocytes | (Poyner et al., 1992) |
| Ala1 | Extension by biotin | 150-fold decrease in potency | L6 myocytes | (Howitt and Poyner, 1997) |
| Asp3 | Coupling to 4 azidoaniline | Twofold decrease in affinity | Solubilized rat cerebellum | (Stangl et al., 1993) |
| Asp3 | Long-chain acylation | Sevenfold decrease in potency | CGRP receptor-expressing cell line | Patent WO/2011/051312 |
| Thr4 | [Val4]-CGRP | Eightfold decrease in affinity, fivefold decrease in potency | Porcine iris ciliary body | (Heino et al., 1998) |
| Thr4 | Long-chain acylation | Sevenfold decrease in potency | CGRP receptor-expressing cell line | Patent WO/2011/051312 |
| Thr6 | [Val6]-CGRP | Fivefold decrease in affinity, no efficacy | Porcine iris ciliary body | (Heino et al., 1998) |
| Cys2, Cys7 | Removal of disulphide | No agonist activity | Rat atria | (Tippins et al., 1986) |
| Cys2, Cys7 | Removal of disulphide | Sixfold reduction in affinity | SK-N-MC cells | (Lang et al., 2006) |
| Cys2, Cys7 | cyclo [Asp2, Lys7]-CGRP | Twofold decrease in affinity | Rat spleen | (Dennis et al., 1989) |
| Cys2, Cys7 | cyclo [Asp2, Lys7]-CGRP | No agonist activity | Guinea pig atria | (Dennis et al., 1989) |
| Cys2, Cys7 | [Cys(Acm)]2,7-CGRP | Kd 40 nM ∼400-fold decrease in affinity. Partial agonist, ∼300-fold decrease in potency where measurable | CGRP receptor Cos 7 cells | (Bailey and Hay, 2006) |
| Cys2, Cys7 | [Cys(Et)2,7]-CGRP | 10-fold decrease in potency | CGRP receptor Cos 7 cells | (Bailey and Hay, 2006) |
| Residues 1–5 | [Pro7,8,Cys31,36]-CGRP6–37 (SH992) | Twofold reduction in affinity, threefold reduction in potency, 80% reduction in Emax | Porcine iris ciliary body | (Heino et al., 1998) |
All differences are reported with respect to human α-CGRP.
Table 4.
Mutations of residues 27–37
| Residue | Mutation | Effect | Comment | Reference |
|---|---|---|---|---|
| Phe27 | [Phe27]-CGRP27–37 | Eightfold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Val28 | [Ala28]-Tyr0CGRP27–37 | Threefold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Pro29 | [Ala29]-Tyr0CGRP27–37 | No binding | SK-N-MC cells | (Rist et al., 1998) |
| Pro29 | [Ala29Ala34Phe35]-CGRP27–37 | Sevenfold decrease in affinity | SK-N-MC cells | (Carpenter et al., 2001) |
| Pro29 | [▴29D31P34F35]-CGRP27−37 | No change in affinity compared with CGRP8–37 | SK-N-MC cells | (Lang et al., 2006) |
| Thr30 | [Ala30]-Tyr0CGRP27–37 | No binding | SK-N-MC cells | (Rist et al., 1998) |
| Thr30 | [Ser30]-Tyr0CGRP27–37 | >30-fold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Thr30 | [Ala30Ala34Phe35]-CGRP27–37 | 100-fold less potent than [Ala34Phe35]-CGRP27–37 | SK-N-MC cells | (Carpenter et al., 2001) |
| Asn31 | [Ala31]-Tyr0CGRP27–37 | >30-fold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Asn31 | [Leu31]-Tyr0CGRP27–37 | Twofold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Asn31 | [Gln31]-Tyr0CGRP27–37 | 30-fold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Asn31 | [Asp31]-Tyr0CGRP27–37 | Twofold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Asn31 | [Ala31Ala34Phe35]-CGRP27–37 | Fivefold less potent than [Ala34Phe35]-CGRP27–37 | SK-N-MC cells | (Carpenter et al., 2001) |
| Val32 | [Ala32]-Tyr0CGRP27–37 | No binding | SK-N-MC cells | (Rist et al., 1998) |
| Val32 | [Ala32Ala34Phe35]-CGRP27–37 | 240-fold decrease in affinity compared with [Ala34Phe35]-CGRP27–37 | SK-N-MC cells | (Carpenter et al., 2001) |
| Gly33 | [Ala33]-Tyr0CGRP27–37 | No binding | SK-N-MC cells | (Rist et al., 1998) |
| Gly33 | [Ala33Ala34Phe35]-CGRP27–37 | 23-fold decrease in affinity | SK-N-MC cells | (Carpenter et al., 2001) |
| Gly33 | [Asp31,azaGly33,Pro34,Phe35]-CGRP29–37 | No binding | CGRP receptor, HEK293 cells | (Boeglin et al., 2007) |
| Gly33 | [Asp31,azaGly33,Pro34,Phe35]-CGRP27–37 | 10-fold increase in affinity compared with [Asp31,Pro34,Phe35]-CGRP27–37 | CGRP receptor, HEK293 cells | (Boeglin et al., 2007) |
| Ser34 | [Ala34]-Tyr0CGRP27–37 | No change in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Ser34 | [D31▾34F35]- CGRP8−37 | Threefold decrease in affinity compared with CGRP8–37 | SK-N-MC cells | (Lang et al., 2006) |
| Glu35 | [Ala35]-Tyr0CGRP27–37 | Threefold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Glu35 | [His35]-Tyr0CGRP27–37 | No change in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Glu35 | [Gln35]-Tyr0CGRP27–37 | Twofold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Glu35 | [Leu35]-Tyr0CGRP27–37 | Threefold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Glu35 | [Ala35Ala34Phe35]-CGRP27–37 | 14-fold less potent than [Ala34Phe35]-CGRP27–37 | SK-N-MC cells | (Carpenter et al., 2001) |
| Ala36 | [Phe36]-Tyr0CGRP27–37 | No binding | SK-N-MC cells | (Rist et al., 1998) |
| Ala36 | [Gly36]-Tyr0CGRP27–37 | 16-fold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Phe37 | [Ala37]-Tyr0CGRP27–37 | No binding | SK-N-MC cells | (Rist et al., 1998) |
| Phe37 | [Tyr37]-Tyr0CGRP27–37 | 11-fold decrease in affinity | SK-N-MC cells | (Rist et al., 1998) |
| Phe37 | [Ala37Ala34Phe35]-CGRP27–37 | >340-fold decrease in affinity compared with [Ala34Phe35]-CGRP27–37 | SK-N-MC cells | (Carpenter et al., 2001) |
| Phe37 | [Gly37]-CGRP8–37 | 2050-fold decrease in affinity of CGRP8–37 | Porcine coronary artery | (Smith et al., 2003) |
| Phe37 | [Tyr37]-CGRP8–37 | No decrease in affinity of CGRP8–37 | Porcine coronary artery | (Smith et al., 2003) |
| Phe37 | Replacement by rotational restricted Phe analogues | 40–60-fold decrease in affinity of CGRP8–37 | Porcine coronary artery | (Smith et al., 2003) |
| Phe37 | [Ala37]-CGRP8–37 | >100-fold decrease in affinity of CGRP8–37 | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Asn31Ser34Glu35 | [Asp31Ala34Phe35]-CGRP27–37 | 10-fold decrease in affinity compared with CGRP | SK-N-MC cells | (Carpenter et al., 2001) |
| Asn31Ser34Glu35 | [Asp31Phe34Phe35]-CGRP27–37 | Twofold decrease in affinity compared with CGRP | SK-N-MC cells | (Carpenter et al., 2001) |
| Ser19Gly20 and Gly33Ser34 | Both replaced by BTD | No decrease in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Residues 1–27 | CGRP27–37 | 16000-fold decrease in affinity compared with CGRP8–37 | SK-N-MC cells | (Ladram et al., 2008) |
| Residues 1–27 | Tyr0-CGRP19–37 | 900-fold decrease in affinity to CGRP8-37 | SK-N-MC cells | (Heino et al., 1998) |
| Phyllomedusa CGRP27–37 | Residues 1–28 | Ki 95 nM (fivefold less potent than human CGRP) | SK-N-MC cells | (Ladram et al., 2008) |
Unless otherwise stated, all differences are with respect to the parent compound used to create the analogue: human α-CGRP27–37, Tyr°CGRP27–37 or CGRP8–37. ▾, ▴: the ▾ and ▴ conformers of β-aminocyclopropane carboxylic acid.
BTD, β-turn dipeptide.
The structure of CGRP
A number of NMR and circular dichroism studies have been conducted on CGRP and its derivatives, although no structure of CGRP has been deposited in the protein structure database. The data collectively are consistent with residues 8–18 of CGRP in aqueous solution forming an α-helix; there is some evidence for this for CGRP8–37 but the helix is less well defined. For both peptides, the helix is stabilized by increasing the hydrophobicity of the solution (Breeze et al., 1991; Hubbard et al., 1991; Sagoo et al., 1991; Matsuura and Manning, 1993; O'Connell et al., 1993; Boulanger et al., 1995; 1996; Robinson et al., 2009). There is some evidence for a β or γ turnaround residues 18–21 (Sagoo et al., 1991; Boulanger et al., 1995). Thereafter, it becomes difficult to detect any well-defined structure until the last 11 amino acids. A hydrogen bond between the carbonyl of Val28 and the amide of Thr30 has been detected in one study of CGRP (Boulanger et al., 1996). In another study, a β-1 turn was seen between Gly33 and Val34 (Sagoo et al., 1991). As will be discussed below, these observations of a set of turns at the extreme C-terminus of CGRP fit well with structure activity work on CGRP27–37.
No structure is available for CGRP bound to lipid or detergent micelles; these provide a hydrophobic environment that is often considered to provide a better mimic of the environment that the peptide encounters in association with a receptor. Such structures are available for other peptides that bind to family B GPCRs. They show that the majority has at least some stretch of α-helix (see Watkins et al., 2012, for a recent review). The assumption that micelle-bound peptides resemble receptor-bound peptides has been most rigorously tested for pituitary adenylate cyclase-activating polypeptide (PACAP), where NMR structures are available for the micelle-bound PACAP27 and receptor-bound PACAP21. In this case, the C-terminus of the peptide forms very similar α-helices, but the receptor-bound N-terminus of PACAP21 forms a β-coil not seen in the micelle-bound PACAP27 (Inooka et al., 2001). This suggests that the micelle approach gives a reasonable approximation to the likely structure of parts of the peptide (probably at the C-terminus) but there may be localized changes induced by the receptor. It is not unreasonable to assume that these are most likely to be at either end of the peptide, where secondary structural elements have seldom been observed and the peptide forms an extended conformation. For CGRP, the most relevant structures that can be used for comparison are those of amylin, which has 37% sequence identity with CGRP (55% over the first 20 amino acids). In SDS micelles at pH 7.4, there is a clear helical region in amylin, extending over residues 7–17, immediately beyond the disulphide-bonded loop. At the end of this helix, there is a sharp turn then another helical section involving residues 21–28 or 23–29. There is yet another bend and then a final section of helix from 33 to 35 (Nanga et al., 2011). This is broadly similar to the pattern of turns and helices found in CGRP. A second set of structures for micelle-bound amylin conducted at pH 4.4 showed amylin to be predominantly helical from residues 5–17 and 18–28, with a bend of around 30° between these segments (Patil et al., 2009). The theme of a helix followed by an area of more flexibility is also seen in structures of micelle-bound AM (Perez-Castells et al., 2012) and CT (Hashimoto et al., 1999).
Overall, if micelle-bound structures provide any guide to the receptor-bound structure, it seems reasonable to assume that CGRP exists as an α-helix from approximately residues 8–18, with the disulphide bond at its N-terminus. It is possible to align the various micelle-bound structures of amylin (2KB8), AM (2L7S) and salmon calcitonin (2GLH), so that the helices and disulphide-bonded loops overlap. Given the sequence overlap in the disulphide-bonded loop (discussed below), it is an attractive notion that all members of this family have this region bound to the receptor in a common orientation. Figure 7A gives a schematic illustration of CGRP showing the main structural features and also a speculative view as to where on the receptor they may interact.
Figure 7.
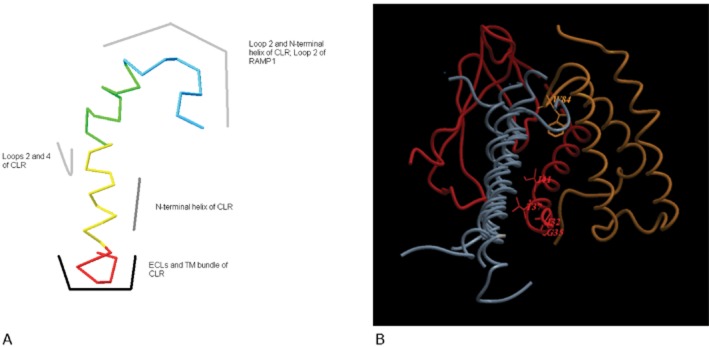
(A) Schematic model of the possible interaction of CGRP with its receptor. Red, residues 1–7 of CGRP; yellow, residues 8–18; green, residues 19–27; blue, residues 28–37. The structure shown of CGRP is simply for illustrative purposes. (B) Superimposition of the bound structures of PTH, GIP, GLP-1 and exendin (all grey) on the extracellular domains of CLR (red) and RAMP1 (orange). Residues on CLR and RAMP1 known to be important for CGRP binding are shown.
Stabilizing the N-terminus
There has been some discussion about the roles of N-caps in stabilizing helical regions of family B peptide ligands. The N-cap is a structure formed by the amino acids at the N-terminal end of the helix, stabilizing it, and it relies on a combination of hydrogen bonds and hydrophobic interactions. It has been suggested that the disulphide-bonded loop serves this function in the CGRP/CT family (Neumann et al., 2008). On the other hand, a classic N-cap motif is also present (Watkins et al., 2012). Potentially the side chain of Thr9 in CGRP could form a hydrogen bond with the carbonyl backbone of the disulphide-bonded ring at Ala5; Thr9 has been shown to be involved in a hydrogen bond in CGRP in an NMR study, although the authors were not able to identify its partner (Breeze et al., 1991). Thr9 is conserved in amylin, although only 10% of the structures of the micelle-bound form show this hydrogen bond (Patil et al., 2009; Nanga et al., 2011). The carbonyl of Ala5 could also hydrogen bond to the backbone amides of Thr9 and/or Val8 and these interactions will also help stabilize the 8–18 helix; in CGRP8–37, N-cap motifs are likely to stabilize this helix (Watkins et al., 2012). Beyond the helical region, the structure of bound CGRP is less certain. By analogy with the known structures of peptides bound to family B GPCRs, it might be expected to adopt a predominantly linear form (Archbold et al., 2011), but as will be argued below, there is good evidence that bound CGRP incorporates a number of bends.
The role of residues 1–7: the disulphide-bonded loop
Removal of residues 1–7 creates CGRP8–37, the prototypical CGRP antagonist. This binds approximately 10-fold less potently than CGRP and has lost all efficacy (Chiba et al., 1989; Dennis et al., 1990). Thus, residues 1–7 have a dual role in activating the receptor, contributing to the overall affinity of bound CGRP, and stabilizing the helix. It seems likely that these residues interact predominantly with the ECLs and associated TM regions of CLR, based on the two domain models for family B GPCRs (Hoare, 2005). A variety of effects have been reported for N-terminal extension of CGRP (Dennis et al., 1989; van Valen et al., 1989; Poyner et al., 1992). Although as Tyr0-CGRP can be used successfully as a radioligand, it seems that any reduction in affinity is modest ( Table 1). This is consistent with the fact that AM and AM2, both of which bind to CLR, have substantial N-terminal extensions beyond the disulphide-bonded ring, although these can be removed without substantially decreasing potency (Eguchi et al., 1994; Bailey and Hay, 2006). The first residue in CGRP is conserved as a small, uncharged amino acid: serine/glycine/alanine (Figure 1). The conservation suggests that the side chain of this residue is subject to some steric constraints, perhaps adjacent to a hydrophobic surface.
Residue 2 is a cysteine which forms the disulphide bond; the role of this will be considered below. There is extensive conservation of the next four amino acids: Asp/Asn-Thr-Ala-Thr. They are conserved in both CGRP and amylin; in addition, Thr6 is conserved in almost every member of the CT family (Figures 1, 2). It is possible to extend Asp3 by coupling it to 4-azidoaniline to produce a photoaffinity derivative of CGRP that can be used to label the receptor and which shows little change in affinity (Stangl et al., 1993). This derivative could potentially retain any hydrogen bonds formed by the carboxyl or carbonyl amide moieties of aspartic acid and asparagine. However, it does suggest that the residue is not sitting in a particularly constrained pocket. It can also be acylated by a long-chain fatty acid with only a modest change in affinity (Patent WO/2011/051312). The γ-carbonyl of Asp3/Asn3 can potentially hydrogen bond to the backbone amide of Ala5, but whether this is of any relevance to bound CGRP is unknown.
Figure 2.

Sequence alignment from different members of the calcitonin peptide family generated using ClustalW and visualized using EsPript using the %Equivalence algorithm. Conserved regions are shown in red boxes with regions with similar physicochemical parameters in yellow.
The high conservation of the Thr-Ala-Thr motif in positions 4–6 suggests that Thr4 and Thr6 take part in hydrogen bonds while residue 5 sits in a sterically constrained environment. Substitution of Thr4 by valine (Heino et al., 1998) or acylation by a long-chain substituent (Patent WO/2011/051312) causes modest decreases in affinity. [Val6]-CGRP is inactive at stimulating cAMP production in porcine iris ciliary body (Heino et al., 1998), suggesting that this position is of especial importance. Interestingly, an AM derivative with substitution of the equivalent of Thr6 (Thr20) was reported to have a reduced potency on blood pressure (Kuwasako et al., 2011). The residue may play an important role in the activation of CLR and CTR by all members of the peptide family.
The disulphide bond is of course a defining feature of the CGRP/CT family of peptides. Within CGRP, breakage of the disulphide bond usually reduces potency (Lang et al., 2006). However, the effect depends on the nature of the modification. Much of the older literature of necessity relied on in vivo or ex vivo assays where CGRP and its derivatives could interact with a range of receptors. Using a recombinant CGRP receptor expressed in Cos 7 cells, it has been shown that [Cys(Acm)2,7]-CGRP is a partial agonist with an apparent pKB of 7.34, suggesting it bound 100–1000 times less well than human α-CGRP. However, [Cys(Et)2,7]-CGRP was a full agonist which only had a 10-fold reduction in potency (Bailey and Hay, 2006). In older studies, analogues such as cyclo2,7[Asp2,Lys7] human α-CGRP indicated that modification of the disulphide bridge can give modest effects on affinity, although they are typically more deleterious to efficacy (Dennis et al., 1989). Thus, while the disulphide bridge enhances binding and biological activity in the endogenous peptide, at least some derivatives retain activity without it. It has been reported that a derivative SH992, where the first five amino acids of CGRP were deleted, and Cys7-Val8 was replaced by diproline, had weak partial agonist activity at stimulating cAMP production on the porcine iris-ciliary body and rabbit lung (Hakala et al., 1994; Heino et al., 1998). It would be of interest to examine this peptide on a recombinant CGRP receptor to confirm its retention of agonist activity, but it suggests that some form of receptor activation can be achieved with just Thr6.
Taken as a whole, some degree of modification is possible for most residues in 1–7, but it is likely that each contributes a modest amount to the binding or efficacy for CGRP. However, Thr6 is probably a key residue for activation for all members of the CT family of peptides and may interact with one of the ECLs or TM helices (Figure 3).
Figure 3.
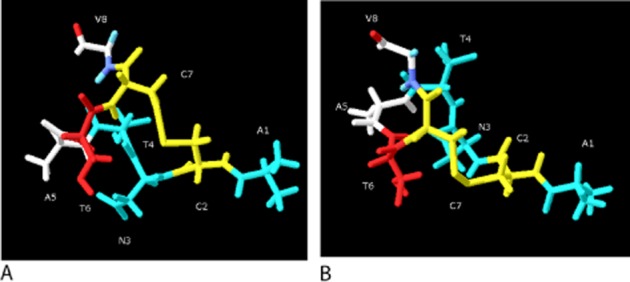
Residues 1–8 of CGRP. (A) Side view. (B) View looking down on Val8. Blue: residues where modification makes a 2–10-fold change in potency; red, modification makes >10-fold change in potency; white, no information; yellow, Cys2-Cys7 disulphide bond. The structure is derived from a homology model of SDS-bound amylin (2KB8) and which was then minimized in water for 50 ns using CHARMM, as described previously (Bailey et al., 2010). The orientation of the side chains is intended only to be illustrative.
The role of residues 8–18: the α-helix
The sequence of the presumed α-helix is highly conserved in CGRP, although in placental mammals residues 14–15 are Gly-Leu, rather than Asp-Phe in almost all other species. Across all members of the family, the general consensus sequence is x-x-polar-polar-Leu-Ala/Ser-x-x-Leu/Ile-polar-polar. This pattern does not create a particularly strongly conserved hydrophobic face but does give a series of hydrophobic and hydrophilic patches, a pattern that is clear in CGRP itself (Figure 4).
Figure 4.
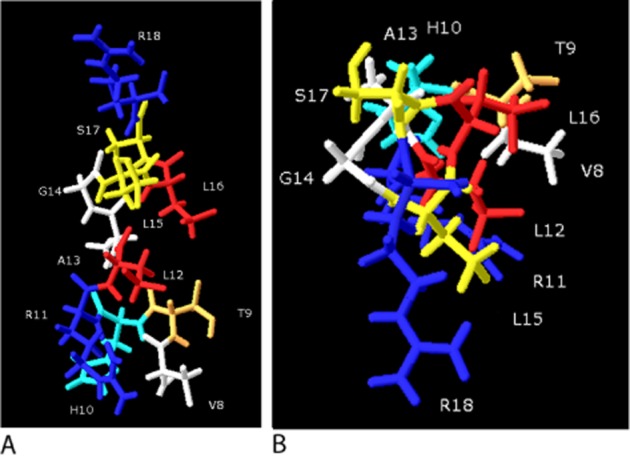
CGRP 8–18. (A) Side view. (B) View looking down on Arg18. Val8, Ala13 and Gly14 (white) have only minor effects on binding of CGRP or CGRP8–37 when mutated. His10 (light blue), Arg11 and Arg18 (dark blue) potentially form distant coulombic interactions with positively charged residues. Thr9 (orange), Leu15 and Ser17 make minor contributions to the binding of this region; Ala13 and Gly14 appear of little importance. However, Leu12 and Leu16 (red) lie on a sterically constrained face. The structure is derived from a homology model of SDS-bound amylin (2KB8) and which was then minimized in water for 50 ns using CHARMM, as described previously (Bailey et al., 2010). The orientation of the side chains is intended only to be illustrative.
CGRP8–37 has a pKB of around 8; a study of deletion analogues of CGRP8–37 indicates that by the time the peptide has been cut back to CGRP11–37 the pKB is close to 6 (Mimeault et al., 1991), although this probably reflects a destabilizing effect on the α-helix (Watkins et al., 2012). CGRP19–37 generally binds with an affinity two orders of magnitude lower than CGRP8–37 (Rovero et al., 1992; Howitt and Poyner, 1997; Poyner et al., 1998). Thus, the 8–18 helix plays an important part in conferring high affinity to native CGRP. A graphic demonstration of its importance is that when it is substituted into AM22–52, it increases its pA2 at CGRP receptors from 6 to over 8 (Robinson et al., 2009). Generally, substitutions of individual amino acids cause only modest changes in affinity ( Table 2). Sometimes, the results are best interpreted as due to changes in peptide secondary structure of either the helix (by introducing prolines) or elsewhere in the peptide [perturbation of residues 28–37 caused by replacement of Ser17 by alanine (Boulanger et al., 1996)]. The substitution of the leucines can produce larger effects. Leu15 can be replaced by a large benzoyl-phenylalanine moiety with only small changes in affinity; however, replacement of Leu12 causes a 30-fold decrease and replacement of Leu16 well over a 100-fold decrease in affinity, suggesting both residues are in sterically confined pockets when bound to the receptor (Howitt et al., 2003). The introduction of two lactam bridges between residues 9 and 13, and 15 and 19 resulted in a peptide that bound with around fivefold lower affinity then the parent compound (Miranda et al., 2008). This suggests reasonable toleration of the modification; the bridges would lie away from the face of the helix defined by Leu12 and Leu16, where it is suggested that there are strict steric constraints. Replacement of Arg18 by alanine has virtually no effect but the double alanine mutant with Arg11 shows 100-fold reduction in affinity. Replacing either of the arginines with glutamic acid caused over a 10-fold reduction in affinity; however, it was possible to substitute with glutamine with retention of high-affinity binding (Poyner et al., 1998; Howitt et al., 2003; Miranda et al., 2008). The arginines may interact with an area of negative charge on the receptor. Potentially, His10 could also have a role in this interaction, although evidence is lacking.
Table 2.
Mutation of residues 8–18
| Residue | Mutation | Effect | Comment | Reference |
|---|---|---|---|---|
| Val8 | Deletion (CGRP9–37) | No change in affinity | Guinea pig atria | (Mimeault et al., 1991) |
| Val8 | [Pro8]-CGRP8–37 | No change in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Val8 | [Gly8]-CGRP8–37 | No change in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Thr9 | Deletion (CGRP10–37) | Fivefold decrease in affinity | Guinea pig atria | (Mimeault et al., 1991) |
| Thr9 | [Ala8]-CGRP8–37 | Threefold decrease in affinity | Guinea pig atria | (Mimeault et al., 1992) |
| Thr9His10 | Deletion (CGRP11–37) | Further twofold decrease in affinity compared with CGRP9–37 | Guinea pig atria | (Mimeault et al., 1991) |
| His10 | [Ala10]-CGRP8–37 | Threefold decrease in affinity | Guinea pig atria | (Mimeault et al., 1992) |
| His10 | Benzoylation of His | 50-fold increase in affinity of CGRP8–37 | Porcine coronary artery | (Smith et al., 2003) |
| Val8His10Gly14 | [Pro8Glu10Glu14]-CGRP8–37 | >100-fold decrease in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Thr9His10Arg11 | Deletion (CGRP12–37) | Further twofold decrease in affinity compared with CGRP11–37 | Guinea pig atria | (Mimeault et al., 1991) |
| Arg11 | [Ala11]-CGRP8–37 | Fivefold decrease in affinity | Guinea pig atria | (Mimeault et al., 1992) |
| Arg11 | [Ala11]-CGRP8–37 | Sixfold decrease in affinity | L6 myocytes | (Howitt and Poyner, 1997) |
| Arg11 | [Glu11]-CGRP8–37 | 60-fold decrease in affinity | SK-N-MC cells | (Howitt et al., 2003) |
| Arg11 | Long-chain acylation | Threefold decrease in affinity | CGRP receptor-expressing cell line | Patent WO/2011/051312 |
| Leu12 | Replacement by Bpa [Bpa12]-CGRP8–37 | 30-fold decrease in affinity | SK-N-MC cells | (Howitt et al., 2003) |
| Gly14 | [Ala14]-CGRP8–37 | No decrease in affinity | Porcine coronary artery | (Li et al., 1997) |
| Gly14 | Replacement by Aib [Aib14]-CGRP8–37 | Twofold decrease in affinity | Porcine coronary artery | (Li et al., 1997) |
| Gly14 | [Asp14]-CGRP8–37 | Twofold decrease in affinity | Porcine coronary artery | (Li et al., 1997) |
| Gly14 | [Asn14]-CGRP8–37 | Threefold decrease in affinity | Porcine coronary artery | (Li et al., 1997) |
| Gly14 | [Pro14]-CGRP8–37 | 100-fold decrease in affinity | Porcine coronary artery | (Li et al., 1997) |
| Leu15 | Replacement by Bpa [Bpa15]-CGRP8–37 | Fivefold decrease in affinity | SK-N-MC cells | (Howitt et al., 2003) |
| Leu16 | Replacement by Bpa [Bpa16]-CGRP8–37 | 700-fold decrease in affinity | SK-N-MC cells | (Howitt et al., 2003) |
| Leu16 | [Pro16]-CGRP8–37 | >1000-fold decrease in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Leu16 | [Ala16]-CGRP8–37 | Fivefold decrease in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Ser17 | [Ala17]-CGRP8–37 | Twofold increase in affinity | Guinea pig atrium | (Boulanger et al., 1996) |
| Arg18 | [Ala18]-CGRP8–37 | No decrease in affinity | L6 myocytes | (Howitt and Poyner, 1997) |
| Arg18 | [Glu18]-CGRP8–37 | 60-fold decrease in affinity | SK-N-MC cells | (Howitt et al., 2003) |
| Arg11,18 | [Ala11,18]-CGRP8–37 | 300-fold decrease in affinity | L6 myocytes | (Howitt and Poyner, 1997) |
| Arg11,18 | [Ser11,18]-CGRP8–37 | 1000-fold decrease in affinity | SK-N-MC cells | (Howitt et al., 2003) |
All differences are with respect to human α-CGRP8–37.
Bpa, 3-(4-benzoylphenyl)alanine.
The affinity of CGRP8–37 can be increased by hydrophobic substituents at the N-terminus; for example, by the introduction of a benzoyl group at N1 and a benzene at C4 of the imidazole ring of His10. Presumably, these are able to interact with a hydrophobic residue on the receptor which is normally too distant to interact with the bound native ligand (Smith et al., 2003). This may be part of the pocket that normally accommodates residues 1–7. His10 is often exploited in radio-iodinated forms of CGRP; the introduction of an iodine atom here may have some influence on the properties of the resulting derivative, given the effects of benzoylation noted above on CGRP8–37.
In summary, the segment from residues 8–18 potentially has two roles: to orientate CGRP to allow efficient binding and to interact directly with the receptor itself [either the upper reaches of the ECLs or the ECD (Figure 7A)]. The segment can orientate both the N-terminus of CGRP and may also stabilize the C-terminus in a high-affinity conformation. Individual residues make only small contributions to the overall binding energy but, collectively these may make a substantial contribution to the binding affinity. Leu12 and Leu16, which lie on the same face of the helix, (Figure 4), may be in close proximity to the receptor. Arg11, Arg18 and possibly His10 may interact with an area of negative charge, probably arising from one or more residues some distance from the bound ligand.
As CGRP8–37 is an antagonist, it might be expected that it contributes little to the process of receptor activation, as opposed to ligand binding. However, at the guinea pig atrium, CGRP1–7 acts as an antagonist at high concentrations (apparent pKB of 6.6), not an agonist (Dennis et al., 1989). CGRP1–12, CGRP1–15 and CGRP1–22 all mimicked the hypotensive effects of CGRP when injected into anaesthetized rats, albeit with considerable loss of potency (Maggi et al., 1990). This suggests that residues 8–12 (and possibly beyond) are needed to promote the efficacy of any activator switches in 1–7 of CGRP. There is of course no proof that these effects were mediated by CGRP receptors, but given the data that N-terminal fragments of other family B GPCRs can show agonist activity, it would not be surprising if this was the case. If it is accepted that CGRP1–7 is an antagonist but CGRP1–12 is an agonist, then this suggests that residues in the regions 8–12 are able to promote receptor activation, perhaps by allowing the correct orientation of 1–7, although the mechanism for this is unclear.
Residues 19–26
The key structural feature of this part of CGRP is the evidence for a bend covering residues 19–21. The sequence of Ser-Gly-Gly is almost completely conserved in all known CGRP sequences. Not only is there evidence for this from the structural studies both on CGRP and other members of the peptide family reviewed above, but introduction of a β-turn dipeptide at residues 19–20 maintained the normal affinity of CGRP8–37 at the CGRP receptor of rat pulmonary artery ( Table 3) (Wisskirchen et al., 2000). This raises questions about whether residues 8–37 of CGRP bind to its receptor predominantly as a helix as presumed or observed for the equivalent regions of many other peptide ligands for family B GPCRs. The bend at this position is likely to be important for its local effects on the structure of CGRP, but there is also NMR evidence that it helps maintain structure at the extreme C-terminus of the peptide (Boulanger et al., 1996).
Table 3.
Mutations of residues 19–26
| Residue | Mutation | Effect | Comment | Reference |
|---|---|---|---|---|
| Ser19Gly20 | Replaced by BTD [BTD19–20]-CGRP8–37 | No decrease in affinity | Rat pulmonary artery | (Wisskirchen et al., 2000) |
| Gly20 | [Gly20]-CGRP8–37 | Twofold decrease in affinity | Guinea pig atrium | (Boulanger et al., 1996) |
| Lys24 | Long-chain acylation | Threefold decrease in affinity | CGRP receptor-expressing cell line | Patent WO/2011/051312 |
| Asn25 | Long-chain acylation | Threefold decrease in affinity | CGRP receptor-expressing cell line | Patent WO/2011/051312 |
| Asn26 | Long-chain acylation | Threefold decrease in affinity | CGRP receptor-expressing cell line | Patent WO/2011/051312 |
| Residues 1–18 | CGRP19–37 | 40-fold decrease in affinity | SK-N-MC cells | (Poyner et al., 1998) |
| Residues 1–7 and 19–27 | CGRP8–18, 28–37 | 18-fold decrease in affinity | SK-N-MC cells | (Poyner et al., 1998) |
| Residues 1–27 | Tyr0-CGRP28–37 | 170-fold decrease in affinity (fourfold decrease with respect to CGRP19–37) | SK-N-MC cells | (Poyner et al., 1998) |
All differences are with respect to human α-CGRP8–37.
BTD, β-turn dipeptide.
Residues 19–26 are important for CGRP binding, as demonstrated by their deletion in a CGRP8–37 analogue; this bound with around 10-fold lower affinity than CGRP8–37 itself (Poyner et al., 1998). There is however little detailed structure–activity information for this part of CGRP. Replacement of the glycines at positions 20 and 21 reduces affinity, probably by disrupting the α-turn (Boulanger et al., 1996). Introduction of a long-chain acylated lysine at positions 24, 25 or 26 is tolerated with a three- to fourfold loss in potency (Patent WO/2011/051312), suggesting that the residues are subject to a rather loose steric constraint. Substitution of Lys24Asn25 by Arg-Lys gives a small increase in potency, possibly suggesting that the region is influenced by a negative charge on the receptor (Miranda et al., 2008). Substitution of Asn25 by leucine gives only a small change in affinity but the corresponding replacement of Asn26 is reported to give almost a 100-fold reduction in affinity, by far the largest change seen for any substitution in this part of the molecule (Heino et al., 1998). This residue aside, the main function of this part of the molecule may be to orientate CGRP in a hinge-like manner. There may well be contacts between individual amino acids and the receptor, but as with 8–18, singly these may be relatively weak.
Residues 27–37; the C-terminus
Human α-CGRP27–37 and Tyr0-CGRP28–37 have only low affinity for the CGRP receptor (pKB∼6–7) (Howitt and Poyner, 1997; Heino et al., 1998). However, analogues comprising this region have been produced which have nanomolar-binding affinity; CGRP27–37 derived from the frog Phyllomedusa bicolor also has a high affinity (Ladram et al., 2008), but generally substitution is necessary to achieve this. P. bicolor CGRP naturally contains many of the substitutions that are used to increase the affinity of human α-CGRP27–37 (Figure 1). In some studies, constraints have been introduced such as with a disulphide bond, or others have used amino acid substitutions to promote bends, either in fragments or full-length CGRP (Hakala et al., 1994; Heino et al., 1998; Rist et al., 1998; 1999; Wisskirchen et al., 2000; Carpenter et al., 2001; Lang et al., 2006; Boeglin et al., 2007; Yan et al., 2011) ( Table 4, Figure 5). The most significant residues seem to be in the 30–37 segment of peptide, but with a turn, around residue 29 being important to orientate Thr30 (Carpenter et al., 2001; Lang et al., 2006). Within residues 30–37, a β-turn between amino acids 33 and 34 is extremely important for high-affinity interactions (Wisskirchen et al., 2000; Carpenter et al., 2001; Lang et al., 2006; Boeglin et al., 2007). As noted above, these two elements of secondary structure can be detected among the assemblages of NMR structures obtained for CGRP or CGRP8–37.
Figure 5.
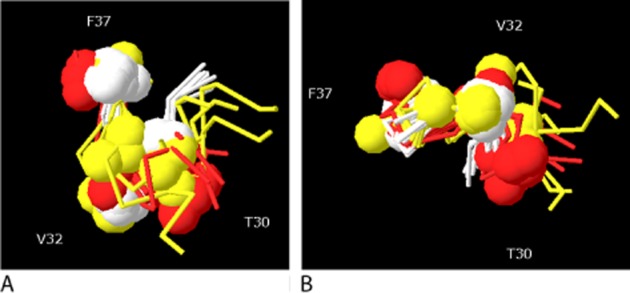
Overlay of residues 27–37 for CGRP derivatives. (A) Side view. (B) View looking up on Val32. White, cyclo-[Cys29, Cys36]CGRP27–37; yellow, cyclo-[Cys31, Cys36]CGRP27–37; red, [D31,A34,F35]CGRP27−37. Homology models were created from SDS-bound amylin (2KB8) and this was then minimized in water using CHARMM, as described previously (Bailey et al., 2010). For cyclo-[Cys29, Cys36]CGRP27–37, structures at 50 s intervals to 350 ns are superimposed. For cyclo-[Cys31, Cys36]CGRP27–37, structures after 10, 15, 20, 22, 27. 32, 37, 41 and 89 ns are superimposed. For [D31,A34,F35]CGRP27−37, structures at 50 s intervals to 300 ns are superimposed. Spheres indicate the position of the α-carbons of Thr30, Val32 and Phe37.
Two key short peptide antagonists have been identified: [D31,P34,F35]CGRP27−37 and [D31,A34,F35]CGRP27−37. Structure–activity studies have shown that Thr30, Val32, Gly33 and Phe37 are key residues (Rist et al., 1998; 1999). While the Phe27-Val28-Pro29 sequence is highly conserved in all forms of CGRP, its role is probably chiefly to orientate the subsequent C-terminal residues. Gly33 is likely to be involved in the β-turn; the side chains of Thr30, Val32 and Phe37 are plausibly involved in receptor contacts. Thr30 cannot be replaced by serine without some loss of affinity, and Val32 cannot be replaced by alanine or phenylalanine, indicating extremely strict steric constraints at these positions. Asn31 can tolerate replacement with Asp or Leu but not Glu, suggesting steric constraints; substitution with Ala reduces affinity, but there is no agreement on the magnitude of the effect (Rist et al., 1998; Carpenter et al., 2001) depending on whether A34F35CGRP28–37 or Tyr°CGRP28–37 is used. The effect of substitutions at Gly33 also differs depending on the peptide analogue being studied (Carpenter et al., 2001; Boeglin et al., 2007). Position 35 tolerates not only phenylalanine but numerous bulky synthetic analogues such as β-(2-naphthyl)alanine (Miranda et al., 2008); in native CGRP this position can either be positively or negatively charged or uncharged (Figure 1), suggesting that the side chain is probably not normally in contact with the receptor. Phe37 is another residue where the structure–activity relationship depends on the nature of the peptide used. It is possible to substitute this with Tyr or Leu in CGRP8–37 with little loss of affinity, but attempts to constrain the rotational flexibility of the aromatic ring are not tolerated (Heino et al., 1998; Smith et al., 2003). By contrast, Tyr substitution is not tolerated in Tyr0-CGRP28–37 (Rist et al., 1999). In a disulphide-constrained analogue, Phe37 can be replaced by pyridylalanine (Yan et al., 2011). There is agreement that the C-terminal amide is essential for all analogues (Rist et al., 1999; Smith et al., 2003).
Constraining the conformation of the C-terminus by a disulphide bond or other bridge unsurprisingly can change affinity. However, as with amino acid substitutions, the details of the effects depend on the analogue. A bridge between residues 31 and 36 is well tolerated for CGRP8–37 but not in a CGRP27–37 derivative, where only a 29–36 bridge was effective (Heino et al., 1998; Yan et al., 2011).
A number of conclusions can be drawn about the role of the C-terminus. It is of major significance for the binding affinity of CGRP; there is good evidence for a number of key contacts between residue side chains and the receptor, particularly involving Thr30, Val32, Phe37 and the C-terminal amide. There are strong steric constraints to ensure these residues are orientated correctly, and other parts of the C-terminus such as Gly33Ser34 help orientate the contact residues. It is possible to overlay the C-termini of the two highest affinity disulphide-constrained CGRP analogues (cyclo-[Cys29, Cys36]CGRP27–37 and [Pro7,8,Cys31,36]-CGRP6–37) with CGRP27–37 incorporating a backbone hydrogen bond between Val28 and Thr30, and β-turn between Gly33 and Ser34 (Figure 5); in all of these structures, there is good correlation between the positions of individual amino acids and these are probably a fair approximation of the structure of the bound peptide. There are differences in detail as to the structure activity relationships between different peptides, indicating they bind in different ways. This can be seen in Figure 6, where comparison is made between the effects of equivalent substitutions at either Tyr0-CGRP27–37 and [Ala34Phe35]-CGRP27–37 or [Asp31Pro34Phe35]-CGRP27–37 and [Asp31Pro34Phe35]-CGRP29–37. The most striking example is with the latter pair of peptides, where substitution of Gly33 with its aza equivalent either abolishes high-affinity binding or increases it 10-fold (Boeglin et al., 2007). For these short analogues, it is possible that the principle-binding interactions seen with CGRP are conserved, but that the substitutions act to strengthen weak interactions by providing extra or better contacts which are of little significance for the longer peptides.
Figure 6.
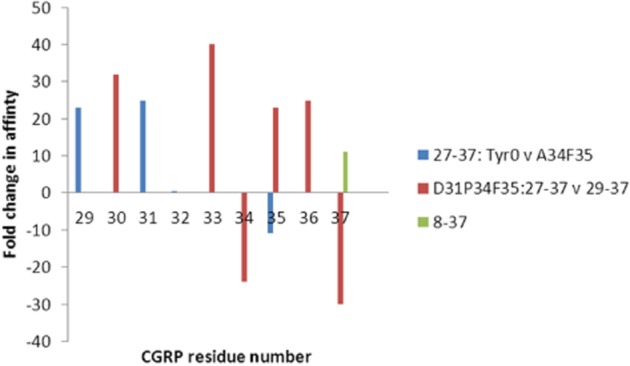
Effect of analogue on the C-terminal CGRP structure activity relationship. For each set of comparisons, the change in affinity between pairs of analogues at individual residues has been plotted. Blue, comparison between Tyr0-CGRP27–37 and [Ala34Phe35]-CGRP27–37 replacing residues with alanine (Rist et al., 1998; Carpenter et al., 2001). Blue dots indicate no difference, no comparison was made at positions 34 and 35. Red, comparison between [Asp31Pro34Phe35]-CGRP27–37 and [Asp31Pro34Phe35]-CGRP29–37 replacing residues with aza Gly (Boeglin et al., 2007). No comparisons were done at positions 29 and 32. Green, comparison between [Tyr0,37]-CGRP27–37 and [Tyr37]-CGRP8–37 (Smith et al., 2003). There are no data for CGRP8–37 at other positions.
CGRP-receptor contacts
It is very difficult to predict the contact points on the receptor for CGRP with any level of useful detail. This is especially the case as a number of binding modes have been either identified or proposed for other family B GPCRs for both the N- and C-termini of the peptides. (Archbold et al., 2011; Barwell et al., 2011; Couvineau and Laburthe, 2011). We have previously suggested that the N-terminus of CGRP may contact the CGRP receptor in a pocket formed chiefly by ECLs 1 and 2 (Barwell et al., 2012). Residues 8–18 and 19–27 are most likely to contact the ECD of CLR; by analogy with bound glucagon-like peptide 1 (GLP-1), exendin, glucose-dependent insulinotropic polypeptide, parathyroid hormone (PTH) and PTHrP (Parthier et al., 2007; Pioszak and Xu, 2008; Runge et al., 2008; Pioszak et al., 2009; Underwood et al., 2010). The main interactions are likely to be with the N-terminal helix and the second and fourth loops. For most of the above structures, the C-termini of the ligands make contacts in the vicinity of loop 4 of their receptors. However, in the CGRP receptor, we suggest that the β-bend around residues 20–21 and the turns at the extreme C-terminus of the peptide act to direct the latter part of the molecule towards a cavity formed from helices 2 and 3 of RAMP1 and the interconnecting loop 2 and the adjacent portions of the ECD of CLR, particularly the N-terminal helix and the second loop (Barwell et al., 2010; Moore et al., 2010). It is interesting that the C-terminus of exendin, which forms a pronounced loop stabilizing a Trp-cage, also points away from loops 2 and 4 of the GLP-1 receptor into the space where it is suggested that the C-terminal loop of CGRP may sit (Figure 7). These predictions are of course speculative, although the model is consistent with the structures of the bound non-peptide CGRP antagonists, which also occupy the space between loop 2 of RAMP1 and the N-terminus and loops 2 and 4 of CLR (ter Haar et al., 2010).
Conclusions
There is enough evidence in the literature to identify both areas of CGRP that make key contacts with the receptor and those that are more distant. A case can be made that there are relatively few individual high-affinity or efficacy contacts. These might include Thr6, important for receptor activation, a hydrophobic patch involving Leu12 and Leu16, Asn26, Thr30, Val32, Phe37 and the C-terminal amide. There are likely to be a more extensive range of minor contacts, for example, His10 and Arg11, possibly responding to negatively charged amino acids on the ECD of CLR some distance (>5 Å) from the bound ligand. Collectively, these probably make a substantial contribution to the binding affinity. A third set of residues are those that are required for stabilizing an element of secondary structure: Cys2, Cys7, residues 9–12 for the 8–18 α-helix, 19–21, 28–30 and 33–34 for bends.
The importance of bends for high-affinity binding of CGRP raises questions as to the nature of the bound peptide. The conventional model for ligand binding of family B GPCRs to their receptors envisages either helical or slightly bent helices binding to the N-terminus. It is not clear how far this can be applied to CGRP; the β-bend that seems likely to exist over residues 19–21 and the potential sharp turns at the C-terminus may imply a more complicated structure for the bound peptide. Of course, the alternative is that in the bound receptor, the bends can be accommodated within a broadly linear binding mode.
Clearly, a crystal structure of CGRP bound to its receptor will clarify many of the points raised in this review. Given progress with GPCR crystallization, this is not unrealistic, although it is disappointing that it has not so far been possible to co-crystallize any part of the peptide with the ECD of the receptor; an alternative would be an NMR structure of bound CGRP, as has been reported for other family B GPCR peptide ligands (Inooka et al., 2001; Watkins et al., 2011). Even if crystals are obtained, these are unlikely to solve all problems. A crystal structure is simply a single picture of what is likely to be a dynamic process; the predictions need to be verified by mutagenesis and extended by NMR, molecular modelling and other allied techniques. In terms of therapeutics, there remains a need for additional CGRP antagonists and especially, novel agonists. Work to understand how the N-terminus of CGRP interacts and activates its receptor is particularly needed, especially if molecules can be produced, which retain selectivity for CGRP receptors (or other receptors for which CGRP is a physiological agonist).
Glossary
- AM
adrenomedullin
- CGRP
calcitonin gene-related peptide
- CLR
calcitonin receptor-like receptor
- CT
calcitonin
- ECD
extracellular domain
- ECL
extracellular loop
- GLP-1
glucagon-like peptide 1
- PACAP
pituitary adenylate cyclase-activating polypeptide
- PTH
parathyroid hormone
- RAMP
receptor activity-modifying protein
- TM
transmembrane domain
Conflict of interest
None.
References
- Archbold JK, Flanagan JU, Watkins HA, Gingell JJ, Hay DL. Structural insights into RAMP modification of secretin family G protein-coupled receptors: implications for drug development. Trends Pharmacol Sci. 2011;32:591–600. doi: 10.1016/j.tips.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Bailey RJ, Hay DL. Pharmacology of the human CGRP1 receptor in Cos 7 cells. Peptides. 2006;27:1367–1375. doi: 10.1016/j.peptides.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Bailey RJ, Bradley JW, Poyner DR, Rathbone DL, Hay DL. Functional characterization of two human receptor activity-modifying protein 3 variants. Peptides. 2010;31:579–584. doi: 10.1016/j.peptides.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Barwell J, Miller PS, Donnelly D, Poyner DR. Mapping interaction sites within the N-terminus of the calcitonin gene-related peptide receptor; the role of residues 23-60 of the calcitonin receptor-like receptor. Peptides. 2010;31:170–176. doi: 10.1016/j.peptides.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwell J, Gingell JJ, Watkins HA, Archbold JK, Poyner DR, Hay DL. Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br J Pharmacol. 2011;166:51–65. doi: 10.1111/j.1476-5381.2011.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwell J, Woolley MJ, Wheatley M, Conner AC, Poyner DR. The role of the extracellular loops of the CGRP receptor, a family B GPCR. Biochem Soc Trans. 2012;40:433–437. doi: 10.1042/BST20110726. [DOI] [PubMed] [Google Scholar]
- Boeglin D, Hamdan FF, Melendez RE, Cluzeau J, Laperriere A, Heroux M, et al. Calcitonin gene-related peptide analogues with aza and indolizidinone amino acid residues reveal conformational requirements for antagonist activity at the human calcitonin gene-related peptide 1 receptor. J Med Chem. 2007;50:1401–1408. doi: 10.1021/jm061343w. [DOI] [PubMed] [Google Scholar]
- Boulanger Y, Khiat A, Chen Y, Senecal L, Tu YS, Pierre S, et al. Structure of human calcitonin gene-related peptide (hCGRP) and of its antagonist hCGRP 8-37 as determined by NMR and molecular modeling. Pept Res. 1995;8:206–213. [PubMed] [Google Scholar]
- Boulanger Y, Khiat A, Larocque A, Fournier AS, Pierre S. Structural comparison of alanine-substituted analogues of the calcitonin gene-related peptide 8-37. Importance of the C-terminal segment for antagonistic activity. Int J Pept Protein Res. 1996;47:477–483. doi: 10.1111/j.1399-3011.1996.tb01098.x. [DOI] [PubMed] [Google Scholar]
- Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84:903–934. doi: 10.1152/physrev.00037.2003. [DOI] [PubMed] [Google Scholar]
- Breeze AL, Harvey TS, Bazzo R, Campbell ID. Solution structure of human calcitonin gene-related peptide by 1H NMR and distance geometry with restrained molecular dynamics. Biochemistry. 1991;30:575–582. doi: 10.1021/bi00216a036. [DOI] [PubMed] [Google Scholar]
- Carpenter KA, Schmidt R, von Mentzer B, Haglund U, Roberts E, Walpole C. Turn structures in CGRP C-terminal analogues promote stable arrangements of key residue side chains. Biochemistry. 2001;40:8317–8325. doi: 10.1021/bi0102860. [DOI] [PubMed] [Google Scholar]
- Chiba T, Yamaguchi A, Yamatani T, Nakamura A, Morishita T, Inui T, et al. Calcitonin gene-related peptide receptor antagonist human CGRP-(8-37) Am J Physiol. 1989;256(2 Pt 1):E331–E335. doi: 10.1152/ajpendo.1989.256.2.E331. [DOI] [PubMed] [Google Scholar]
- Christopoulos G, Perry KJ, Morfis M, Tilakaratne N, Gao Y, Fraser NJ, et al. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol. 1999;56:235–242. doi: 10.1124/mol.56.1.235. [DOI] [PubMed] [Google Scholar]
- Couvineau A, Laburthe M. VPAC receptors: structure, molecular pharmacology and interaction with accessory proteins. Br J Pharmacol. 2011;166:42–50. doi: 10.1111/j.1476-5381.2011.01676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis T, Fournier A, St Pierre S, Quirion R. Structure-activity profile of calcitonin gene-related peptide in peripheral and brain tissues. Evidence for receptor multiplicity. J Pharmacol Exp Ther. 1989;251:718–725. [PubMed] [Google Scholar]
- Dennis T, Fournier A, Cadieux A, Pomerleau F, Jolicoeur FB, St Pierre S, et al. hCGRP8-37, a calcitonin gene-related peptide antagonist revealing calcitonin gene-related peptide receptor heterogeneity in brain and periphery. J Pharmacol Exp Ther. 1990;254:123–128. [PubMed] [Google Scholar]
- Devigny C, Perez-Balderas F, Hoogeland B, Cuboni S, Wachtel R, Mauch CP, et al. Biomimetic screening of class-B G protein-coupled receptors. J Am Chem Soc. 2011;133:8927–8933. doi: 10.1021/ja200160s. [DOI] [PubMed] [Google Scholar]
- Durham PL, Vause CV. Calcitonin gene-related peptide (CGRP) receptor antagonists in the treatment of migraine. CNS Drugs. 2010;24:539–548. doi: 10.2165/11534920-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi S, Hirata Y, Iwasaki H, Sato K, Watanabe TX, Inui T, et al. Structure-activity relationship of adrenomedullin, a novel vasodilatory peptide, in cultured rat vascular smooth muscle cells. Endocrinology. 1994;135:2454–2458. doi: 10.1210/endo.135.6.7988431. [DOI] [PubMed] [Google Scholar]
- Garber AJ. Novel GLP-1 receptor agonists for diabetes. Expert Opin Investig Drugs. 2012;21:45–57. doi: 10.1517/13543784.2012.638282. [DOI] [PubMed] [Google Scholar]
- ter Haar E, Koth CM, Abdul-Manan N, Swenson L, Coll JT, Lippke JA, et al. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Hakala JM, Valo T, Vihavainen S, Hermonen J, Heino P, Halme M, et al. Constrained analogues of the calcitonin gene-related peptide. Biochem Biophys Res Commun. 1994;202:497–503. doi: 10.1006/bbrc.1994.1956. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Toma K, Nishikido J, Yamamoto K, Haneda K, Inazu T, et al. Effects of glycosylation on the structure and dynamics of eel calcitonin in micelles and lipid bilayers determined by nuclear magnetic resonance spectroscopy. Biochemistry. 1999;38:8377–8384. doi: 10.1021/bi983018j. [DOI] [PubMed] [Google Scholar]
- Hay DL, Howitt SG, Conner AC, Doods H, Schindler M, Poyner DR. A comparison of the actions of BIBN4096BS and CGRP(8-37) on CGRP and adrenomedullin receptors expressed on SK-N-MC, L6, Col 29 and Rat 2 cells. Br J Pharmacol. 2002;137:80–86. doi: 10.1038/sj.bjp.0704844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DL, Poyner DR, Quirion R. International Union of Pharmacology. LXIX. Status of the calcitonin gene-related peptide subtype 2 receptor. Pharmacol Rev. 2008;60:143–145. doi: 10.1124/pr.108.00372. [DOI] [PubMed] [Google Scholar]
- Heino P, Oksala O, Palkama A, Valo T, Vihavainen S, Koskinen A, et al. Binding of CGRP analogs and their effect on adenylate cyclase activity in porcine iris-ciliary body. J Ocul Pharmacol Ther. 1998;14:543–554. doi: 10.1089/jop.1998.14.543. [DOI] [PubMed] [Google Scholar]
- Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Howitt SG, Poyner DR. The selectivity and structural determinants of peptide antagonists at the CGRP receptor of rat, L6 myocytes. Br J Pharmacol. 1997;121:1000–1004. doi: 10.1038/sj.bjp.0701212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitt SG, Kilk K, Wang Y, Smith DM, Langel U, Poyner DR. The role of the 8-18 helix of CGRP8-37 in mediating high affinity binding to CGRP receptors; coulombic and steric interactions. Br J Pharmacol. 2003;138:325–332. doi: 10.1038/sj.bjp.0705040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JA, Martin SR, Chaplin LC, Bose C, Kelly SM, Price NC. Solution structures of calcitonin-gene-related-peptide analogues of calcitonin-gene-related peptide and amylin. Biochem J. 1991;275(Pt 3):785–788. doi: 10.1042/bj2750785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inooka H, Ohtaki T, Kitahara O, Ikegami T, Endo S, Kitada C, et al. Conformation of a peptide ligand bound to its G-protein coupled receptor. Nat Struct Biol. 2001;8:161–165. doi: 10.1038/84159. [DOI] [PubMed] [Google Scholar]
- Kuwasako K, Kitamura K, Nagata S, Hikosaka T, Takei Y, Kato J. Shared and separate functions of the RAMP-based adrenomedullin receptors. Peptides. 2011;32:1540–1550. doi: 10.1016/j.peptides.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Ladram A, Besne I, Breton L, de Lacharriere O, Nicolas P, Amiche M. Pharmacologic study of C-terminal fragments of frog skin calcitonin gene-related peptide. Peptides. 2008;29:1150–1156. doi: 10.1016/j.peptides.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Lang M, De Pol S, Baldauf C, Hofmann HJ, Reiser O, Beck-Sickinger AG. Identification of the key residue of calcitonin gene related peptide (CGRP) 27-37 to obtain antagonists with picomolar affinity at the CGRP receptor. J Med Chem. 2006;49:616–624. doi: 10.1021/jm050613s. [DOI] [PubMed] [Google Scholar]
- Li J, Matsuura JE, Waugh DJ, Adrian TE, Abel PW, Manning MC, et al. Structure-activity studies on position 14 of human alpha-calcitonin gene-related peptide. J Med Chem. 1997;40:3071–3076. doi: 10.1021/jm9608164. [DOI] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- Maggi CA, Rovero P, Giuliani S, Evangelista S, Regoli D, Meli A. Biological activity of N-terminal fragments of calcitonin gene-related peptide. Eur J Pharmacol. 1990;179:217–219. doi: 10.1016/0014-2999(90)90422-3. [DOI] [PubMed] [Google Scholar]
- Mallee JJ, Salvatore CA, LeBourdelles B, Oliver KR, Longmore J, Koblan KS, et al. Receptor activity-modifying protein 1 determines the species selectivity of non-peptide CGRP receptor antagonists. J Biol Chem. 2002;277:14294–14298. doi: 10.1074/jbc.M109661200. [DOI] [PubMed] [Google Scholar]
- Matsuura J, Manning MC. Conformation of human calcitonin gene-related peptide (8-37) in aqueous solution as determined by circular dichroism spectroscopy. J Pharm Biomed Anal. 1993;11:89–93. doi: 10.1016/0731-7085(93)80128-n. [DOI] [PubMed] [Google Scholar]
- Mimeault M, Fournier A, Dumont Y, St-Pierre S, Quirion R. Comparative affinities and antagonistic potencies of various human calcitonin gene-related peptide fragments on calcitonin gene-related peptide receptors in brain and periphery. J Pharmacol Exp Ther. 1991;258:1084–1090. [PubMed] [Google Scholar]
- Mimeault M, Quirion R, Dumont Y, St-Pierre S, Fournier A. Structure-activity study of hCGRP8-37, a calcitonin gene-related peptide receptor antagonist. J Med Chem. 1992;35:2163–2168. doi: 10.1021/jm00090a003. [DOI] [PubMed] [Google Scholar]
- Miranda LP, Holder JR, Shi L, Bennett B, Aral J, Gegg CV, et al. Identification of potent, selective, and metabolically stable peptide antagonists to the calcitonin gene-related peptide (CGRP) receptor. J Med Chem. 2008;51:7889–7897. doi: 10.1021/jm8009298. [DOI] [PubMed] [Google Scholar]
- Moore EL, Gingell JJ, Kane SA, Hay DL, Salvatore CA. Mapping the CGRP receptor ligand binding domain: Tryptophan-84 of RAMP1 is critical for agonist and antagonist binding. Biochem Biophys Res Commun. 2010;394:141–145. doi: 10.1016/j.bbrc.2010.02.131. [DOI] [PubMed] [Google Scholar]
- Nanga RP, Brender JR, Vivekanandan S, Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim Biophys Acta. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro A, Lionetto L, Simmaco M, Martelletti P. CGRP receptor antagonists: an expanding drug class for acute migraine? Expert Opin Investig Drugs. 2012;21:807–818. doi: 10.1517/13543784.2012.681044. [DOI] [PubMed] [Google Scholar]
- Neumann JM, Couvineau A, Murail S, Lacapere JJ, Jamin N, Laburthe M. Class-B GPCR activation: is ligand helix-capping the key? Trends Biochem Sci. 2008;33:314–319. doi: 10.1016/j.tibs.2008.05.001. [DOI] [PubMed] [Google Scholar]
- O'Connell JP, Kelly SM, Raleigh DP, Hubbard JA, Price NC, Dobson CM, et al. On the role of the C-terminus of alpha-calcitonin-gene-related peptide (alpha CGRP). The structure of des-phenylalaninamide37-alpha CGRP and its interaction with the CGRP receptor. Biochem J. 1993;291(Pt 1):205–210. doi: 10.1042/bj2910205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoshi M, Inoue K, Naruse K, Takei Y. Evolutionary history of the calcitonin gene-related peptide family in vertebrates revealed by comparative genomic analyses. Peptides. 2006;27:3154–3164. doi: 10.1016/j.peptides.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D, et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci U S A. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C, Reedtz-Runge S, Rudolph R, Stubbs MT. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Patil SM, Xu S, Sheftic SR, Alexandrescu AT. Dynamic alpha-helix structure of micelle-bound human amylin. J Biol Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Castells J, Martin-Santamaria S, Nieto L, Ramos A, Martinez A, Pascual-Teresa B, et al. Structure of micelle-bound adrenomedullin: a first step toward the analysis of its interactions with receptors and small molecules. Biopolymers. 2012;97:45–53. doi: 10.1002/bip.21700. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci U S A. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Gardella TJ, Xu HE. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J Biol Chem. 2009;284:28382–28391. doi: 10.1074/jbc.M109.022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR. Calcitonin gene-related peptide: multiple actions, multiple receptors. Pharmacol Ther. 1992;56:23–51. doi: 10.1016/0163-7258(92)90036-y. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Andrew DP, Brown D, Bose C, Hanley MR. Pharmacological characterization of a receptor for calcitonin gene-related peptide on rat, L6 myocytes. Br J Pharmacol. 1992;105:441–447. doi: 10.1111/j.1476-5381.1992.tb14272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR, Soomets U, Howitt SG, Langel U. Structural determinants for binding to CGRP receptors expressed by human SK-N-MC and Col 29 cells: studies with chimeric and other peptides. Br J Pharmacol. 1998;124:1659–1666. doi: 10.1038/sj.bjp.0702032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- Prado MA, Evans-Bain B, Dickerson IM. Receptor component protein (RCP): a member of a multi-protein complex required for G-protein-coupled signal transduction. Biochem Soc Trans. 2002;30:460–464. doi: 10.1042/bst0300460. [DOI] [PubMed] [Google Scholar]
- Qi T, Dong M, Watkins HA, Wootten D, Miller LJ, Hay DL. Receptor activity-modifying protein dependent impairment of calcitonin receptor splice variant Delta(1-47)hCT((a)) function. Br J Pharmacol. 2012;168:644–657. doi: 10.1111/j.1476-5381.2012.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rist B, Entzeroth M, Beck-Sickinger AG. From micromolar to nanomolar affinity: a systematic approach to identify the binding site of CGRP at the human calcitonin gene-related peptide 1 receptor. J Med Chem. 1998;41:117–123. doi: 10.1021/jm970533r. [DOI] [PubMed] [Google Scholar]
- Rist B, Lacroix JS, Entzeroth M, Doods HN, Beck-Sickinger AG. CGRP 27-37 analogues with high affinity to the CGRP1 receptor show antagonistic properties in a rat blood flow assay. Regul Pept. 1999;79:153–158. doi: 10.1016/s0167-0115(98)00159-1. [DOI] [PubMed] [Google Scholar]
- Robinson SD, Aitken JF, Bailey RJ, Poyner DR, Hay DL. Novel peptide antagonists of adrenomedullin and calcitonin gene-related peptide receptors: identification, pharmacological characterization, and interactions with position 74 in receptor activity-modifying protein 1/3. J Pharmacol Exp Ther. 2009;331:513–521. doi: 10.1124/jpet.109.156448. [DOI] [PubMed] [Google Scholar]
- Rovero P, Giuliani S, Maggi CA. CGRP antagonist activity of short C-terminal fragments of human alpha CGRP, CGRP(23-37) and CGRP(19-37) Peptides. 1992;13:1025–1027. doi: 10.1016/0196-9781(92)90067-d. [DOI] [PubMed] [Google Scholar]
- Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Sagoo JK, Bose C, Beeley NR, Tendler SJ. Structural studies on the [Bu(t)-Cys18](19-37)-fragment of human beta-calcitonin-gene-related peptide. Biochem J. 1991;280(Pt 1):147–150. doi: 10.1042/bj2800147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Potts JT, Jr, Gardella TJ. Minimization of parathyroid hormone. Novel amino-terminal parathyroid hormone fragments with enhanced potency in activating the type-1 parathyroid hormone receptor. J Biol Chem. 2000;275:21836–21843. doi: 10.1074/jbc.M909861199. [DOI] [PubMed] [Google Scholar]
- Smith DD, Saha S, Fang G, Schaffert C, Waugh DJ, Zeng W, et al. Modifications to the N-terminus but not the C-terminus of calcitonin gene-related peptide(8-37) produce antagonists with increased affinity. J Med Chem. 2003;46:2427–2435. doi: 10.1021/jm020507f. [DOI] [PubMed] [Google Scholar]
- Stangl D, Muff R, Schmolck C, Fischer JA. Photoaffinity labeling of rat calcitonin gene-related peptide receptors and adenylate cyclase activation: identification of receptor subtypes. Endocrinology. 1993;132:744–750. doi: 10.1210/endo.132.2.8381072. [DOI] [PubMed] [Google Scholar]
- Tippins JR, Di Marzo V, Panico M, Morris HR, MacIntyre I. Investigation of the structure/activity relationship of human calcitonin gene-related peptide (CGRP) Biochem Biophys Res Commun. 1986;134:1306–1311. doi: 10.1016/0006-291x(86)90392-x. [DOI] [PubMed] [Google Scholar]
- Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Valen F, Keck E, Jurgens H. Functional characteristics of calcitonin gene-related peptide receptors in human Ewing's sarcoma WE-68 cells. FEBS Lett. 1989;256:170–174. doi: 10.1016/0014-5793(89)81742-9. [DOI] [PubMed] [Google Scholar]
- Watkins HA, Au M, Bobby R, Williams G, Abdul Manan N, Archbold J, et al. 2011. Adrenomedullin: a structural paradigm shift in class B G-protein coupled receptor peptide hormones? Proceedings of the British Pharmacological Society. Available at: http://www.pA2online.org/abstracts/Vol9Issue3abst070P.pdf (accessed 2/25/2013) [DOI] [PMC free article] [PubMed]
- Watkins HA, Au M, Hay DL. The structure of secretin family GPCR peptide ligands: implications for receptor pharmacology and drug development. Drug Discov Today. 2012;17:1006–1014. doi: 10.1016/j.drudis.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Wisskirchen FM, Doyle PM, Gough SL, Harris CJ, Marshall I. Bioactive beta-bend structures for the antagonist halpha CGRP(8–37) at the CGRP(1) receptor of the rat pulmonary artery. Br J Pharmacol. 2000;129:1049–1055. doi: 10.1038/sj.bjp.0703152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LZ, Johnson KW, Rothstein E, Flora D, Edwards P, Li B, et al. Discovery of potent, cyclic calcitonin gene-related peptide receptor antagonists. J Pept Sci. 2011;17:383–386. doi: 10.1002/psc.1358. [DOI] [PubMed] [Google Scholar]