

Abstract
The ghrelin receptor is a 7 transmembrane (7TM) receptor involved in a variety of physiological functions including growth hormone secretion, increased food intake and fat accumulation as well as modulation of reward and cognitive functions. Because of its important role in metabolism and energy expenditure, the ghrelin receptor has become an important therapeutic target for drug design and the development of anti-obesity compounds. However, none of the compounds developed so far have been approved for commercial use. Interestingly, the ghrelin receptor is able to signal through several different signalling pathways including Gαq, Gαi/o, Gα12/13 and arrestin recruitment. These multiple signalling pathways allow for functionally biased signalling, where one signalling pathway may be favoured over another either by selective ligands or through mutations in the receptor. In the present review, we have described how ligands and mutations in the 7TM receptor may bias the receptors to favour either one G-protein over another or to promote G-protein independent signalling pathways rather than G–protein-dependent pathways. For the ghrelin receptor, both agonist and inverse agonists have been demonstrated to signal more strongly through the Gαq-coupled pathway than the Gα12/13-coupled pathway. Similarly a ligand that promotes Gαq coupling over Gαi coupling has been described and it has been suggested that several different active conformations of the receptor may exist dependent on the properties of the agonist. Importantly, ligands with such biased signalling properties may allow the development of drugs that selectively modulate only the therapeutically relevant physiological functions, thereby decreasing the risk of side effects.
LINKED ARTICLES
This article is part of a themed section on Neuropeptides. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.170.issue-7
Keywords: ghrelin receptor, biased agonism, biased signalling, GPCR, 7TM receptor
Introduction to ghrelin receptor physiology
The ghrelin receptor, previously known as the growth hormone secretagogue (GHS) receptor 1a (for receptor nomenclature see Alexander et al., 2011), is the receptor for the anabolic hormone ghrelin. This receptor is involved in GH secretion, appetite regulation, fat accumulation and energy expenditure. In addition it can also modulate behaviour and mood (van der Lely et al., 2004; Dickson et al., 2011; Figure 1).
Figure 1.
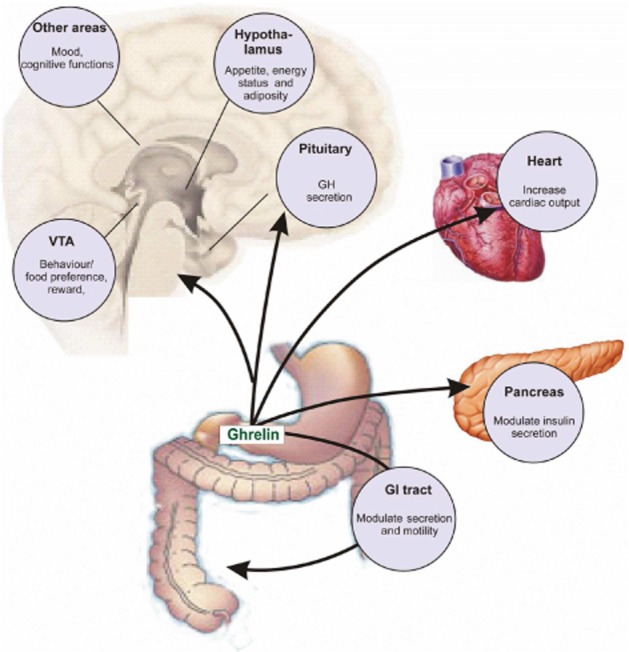
Cartoon of the most well-known ghrelin-mediated physiological functions.
GH
The ghrelin receptor was initially cloned as a receptor important for GH secretion. In the mid-1970s, it was found that enkephalin-like peptides had the ability to release GH from pituitary cells (Bowers, 2001), acting through a receptor different from the previously discovered GH-releasing hormone receptor, but did not affect the release of other pituitary hormones (Bowers et al., 1990). The pharmaceutical industry was encouraged by this observation and established large drug discovery programmes in order to develop GH-releasing substances (Smith et al., 1997). Highly potent peptide and non-peptide compounds, collectively named GHS, were developed and were important for the cloning of the receptor for these synthetic compounds (Howard et al., 1996). Some of the GHS compounds were tested for their ability to induce GH secretion in a large clinical trial. However, the results were not good enough to support further clinical development with GH secretion as primary focus (Murphy et al., 2001; Bach et al., 2004).
Appetite and adipose tissue regulation
The ghrelin receptor is mostly associated with its expression in the neuropeptide Y (NPY)/agouti-related peptide (AgRP) neurons of the arcuate nucleus of the hypothalamus, from where it exerts its orexigenic effect (Howard et al., 1996; Kojima et al., 1999). Acute administration of ghrelin induces a rapid increase in food intake, while continuous ghrelin administration augments both food intake and weight gain, primarily by increasing the mass of adipose tissue (Tschop et al., 2000). Plasma levels of ghrelin increase before a meal and subsequently decrease after energy intake in human subjects, suggesting ghrelin is a meal initiator or meal preparator (Cummings et al., 2001). Additionally, the plasma levels of ghrelin are regulated in response to the general energy balance, as obese individuals have decreased plasma ghrelin levels compared with lean individuals (Tschop et al., 2001). The orexigenic effect of ghrelin has been shown to be dependent on various other neuropeptides and signalling molecules, in addition to NPY and AgRP, acting in the hypothalamic nuclei, such as cannabinoids, melanocortin and orexin.
The intracellular signalling leading to activation of the orexigenic pathways by ghrelin has been studied in the hypothalamus; primarily in the ventromedial hypothalamus and the arcuate nucleus. Part of this process involves activation of sirtuin 1, which deacetylates p53 and forkhead box protein O1 (FoxO1) and thereby makes these two transcription factors available for regulating gene transcription. This leads to modulation of the activity of AMP-activated PK (AMPK; Velasquez et al., 2011). The phosphorylation of AMPK leads to the inactivation of acetyl-CoA carboxylase (Munday et al., 1988), preventing malonyl-CoA formation, the substrate of fatty acid synthase (FAS). In addition, phosphoAMPK decreases mRNA expression levels of FAS, resulting in a decreased level of free fatty acids in the brain (Zhou et al., 2001; Lopez et al., 2008). The decreased malonyl-CoA levels then lead to increased activity of carnitine palmitoyl transferase 1 and changes in the mitochondrial respiration and production of ROS, a process that is dependent on the activation of uncoupling protein 2 (UCP2) (Andrews et al., 2008; Lopez et al., 2008). UCP2 has been shown to be highly important for not only ghrelin-induced mitochondrial proliferation but also activation of NPY/AgRP neurons and ghrelin-induced food intake (Andrews et al., 2008). Ghrelin also increases the activation of the transcription factors cAMP response element binding protein (CREB), FoxO1 and brain-specific homobox transcription factor that are important for the transcription of NPY and AgRP genes (Sakkou et al., 2007; Lage et al., 2010). In addition, results from a recent study indicate that the mammalian target of rapamycin and its substrate S6K are also involved in the orexigenic effect of ghrelin (Martins et al., 2012; Stevanovic et al., 2012).
Extra-hypothalamic regions are involved in the hedonic aspects of ghrelin-induced appetite regulation. For example, activation of the ghrelin receptor in dopaminergic neurons within the ventral tegmental area (VTA) increases their firing rates and food intake. This effect was reversed by infusion of a ghrelin receptor antagonist into the VTA, which reduced ghrelin-induced feeding behaviour (Abizaid et al., 2006).
Other functions
Extra-hypothalamic expression of the ghrelin receptor has been shown to be important for a number of other functions. For example administration of ghrelin into the VTA augmented alcohol intake by increasing the alcohol reward signal, whereas a ghrelin receptor antagonist attenuated alcohol intake in mice (Jerlhag et al., 2009). Additionally the ghrelin receptor appears to be important for other reward related behaviours such as sucrose preference, and the responses to cocaine and amphetamine (Jerlhag et al., 2012; Figure 1).
As mRNA for the ghrelin receptor is present in many areas of the brain, several groups have investigated the importance of the ghrelin/ghrelin receptor-axis in cognitive function. In early experiments it was shown that ghrelin improves memory retention and anxiety behaviour (Carlini et al., 2002) and synthetic ghrelin receptor agonists have been shown to enhance learning and exploratory behaviour (Atcha et al., 2009). It was also shown that ghrelin promotes dendritic spine formation and contributes to long-term potentiation, consistent with its involvement in memory retention (Diano et al., 2006).
In addition to the ubiquitious expression of ghrelin receptors in the brain, ghrelin receptors are also present in peripheral tissues, but at much lower expression levels (Guan et al., 1997; Gnanapavan et al., 2002). Their pattern of expression in the heart has led to the speculation that the ghrelin receptor system influences cardiovascular function, both in the presence and absence of growth hormone (Papotti et al., 2000). For example, it was demonstrated that ghrelin has a beneficial effect on blood pressure, can prevent ischaemia, heart failure and even angiogenesis in vascular endothelial tissue. However, these effects on the CVS may, to a large extent, result from central ghrelin-mediated regulation of the sympathetic nervous system (Nagaya et al., 2001; Chang et al., 2004; Li et al., 2007; Sato et al., 2011; Yang et al., 2012).
In the pancreas, ghrelin receptors have been shown to be expressed both on the endocrine α and β cells as well as potentially on a novel cell type, the ε cells. Conflicting data have been presented with respect to the functional effect of ghrelin on the pancreas (Broglio et al., 2003a; Briggs and Andrews, 2011). However, in most studies it was demonstrated that acute administration of ghrelin induces a transient decrease in spontaneous insulin secretion and that antagonists are able block this effect (Broglio et al., 2003b; Esler et al., 2007). Interestingly, non-acylated ghrelin has been shown to counteract the ghrelin-induced modulation of insulin and glucose levels without affecting the neuroendocrine actions of ghrelin (Broglio et al., 2004).
Given the wide range of effects of the ghrelin receptor system on physiology, a key challenge for ghrelin receptor drug discovery is to develop drugs that selectively modulate the desired aspects of ghrelin receptor function – for example, hypothalamic feeding behaviour. In potential treatments for obesity or cachexia, the effects of ghrelin receptor ligands on various metabolic parameters may be beneficial. However, the risks of side effects related to pituitary growth hormone secretion, mood and memory, and peripheral cardiovascular events, must be minimized. Modulation of the ghrelin/ghrelin receptor axis by exogenous ligands is clearly a delicate process that requires control of the full array of physiological responses induced by ghrelin. Therefore, we should pursue opportunities for functionally biased ligands towards the relevant signalling pathways, if we want to create a new generation of superior drugs for disease treatment.
7TM receptor signalling and activation
7TM receptor signalling
The ghrelin receptor belongs to the large family of 7 transmembrane segment receptors (7TM receptors), which are localized in the plasma membrane and transduce extracellular signals to intracellular responses. In addition to peptide hormones such as ghrelin, these extracellular stimuli also include large proteins, lipids, small organic molecules, ions and photons. Importantly a large percentage of drugs on the market targets the 7TM receptor superfamily, including some anti-histamines and β-blockers (Rask-Andersen et al., 2011). To differentiate the extracellular stimuli from this large array of 7TM receptors, cells have developed a sophisticated intracellular signalling network, governed by G–protein-dependent and G–protein-independent mechanisms. The G-protein is a heterotrimer composed of Gα and Gβγ subunits, which when inactive is assembled and bound to GDP. Upon receptor activation the GDP is exchanged with GTP, thereby allowing the Gα and Gβγ subunits to dissociate and initiate their respective signalling cascades. There are four highly studied Gα-mediated signalling pathways: (i) Gαs activates adenylate cyclase, leading to cAMP accumulation and cAMP-mediated responses whereas (ii) Gαi/o inhibits adenylate cyclase and thereby the cAMP accumulation; (iii) Gαq/11 activates PLC leading to inositol tris-1,4,5-phosphate [IP3; in the rest of this review article we will refer to the inositol phosphates (IP3, IP2, IP) as IP, IP is the most predominant form of the IPs measured in experiments of Gαq and subsequently PLC activation] and DAG production; and (iv) Gα12/13 activates Rho guanine exchange factors, resulting in activation of Rho associated kinases, and cytoskeletal rearrangement. The Gβγ subunits have also been shown to regulate signalling pathways such as calcium release (Stehno-Bittel et al., 1995), PLB, adenylate cyclase, but also MAPK, nucleic histone deacetylase 5 and adipocyte enhancer-binding protein 1 among others (Tang and Gilman, 1991; Crespo et al., 1994; Zhang et al., 1996; Park et al., 1999; Spiegelberg and Hamm, 2005).
The most commonly described G–protein-independent signalling pathway involves phosphorylation of the 7TM receptor by GPCR kinases (GRKs) followed by β-arrestin adaptor recruitment. β-Arrestins are able to activate various kinases such as Src, Akt and MAPK (e.g. ERK 1/2 and p38; Violin and Lefkowitz, 2007). β-Arrestin recruitment is also one of the possible pathways for internalization of most 7TM receptors, initiating processes of intracellular trafficking, which determine whether receptors are recycled or degraded during chronic agonist exposure (Zhang et al., 1998; Hanyaloglu and von Zastrow, 2008).
Ghrelin receptor mediated signalling
The ghrelin receptor was originally discovered to induce calcium release during an investigation into its growth hormone-releasing properties (Howard et al., 1996). When its endogenous ligand, ghrelin, was eventually detected (Kojima et al., 1999) more thorough studies of its signal transduction properties demonstrated a ghrelin receptor-dependent elevation of the PLC endproduct IP. Ghrelin has also been reported to activate other downstream signalling pathways, such as CREB-mediated transcription, in a dose-dependent manner – presumably through Gαq, as CREB can be activated by calcium calmodulin kinase (Dash et al., 1991; Kojima et al., 1999; Holst et al., 2003; 2004). In addition to the Gαq-coupled signalling the ghrelin receptor couples to Gα12/13 and thereby activates RhoA kinase. The combined actions of Gαq and Gα12/13 are responsible for the majority of the ghrelin-induced activation of serum response element (SRE), whereas Gαi coupling is not relevant for this pathway (Sivertsen et al., 2011), as the signal is unaffected by pertussis toxin. However Gαi/o coupling has been demonstrated in GTPγS assays in model systems (Bennett et al., 2009) as well as in isolated lipid discs (Damian et al., 2012; Mary et al., 2012). Furthermore, activation of the ghrelin receptor leads to the recruitment of the clathrin adaptor-related protein complex 2 (AP2), or β-arrestins, in a manner that is independent of G-protein coupling (Damian et al., 2012; Mokrosinski et al., 2012). Stimulation of the ghrelin receptor also induces ERK1/2 phosphorylation in a dose-dependent manner. This process has been shown to be dependent on PKC stimulation and phosphatidylcholine accumulation in a G–protein-dependent manner. In contrast β-arrestin does not play a role in this signalling, because a dominant negative mutant of β-arrestin failed to decrease ERK1/2 phosphorylation (Mousseaux et al., 2006; Chu et al., 2007). This wide range of signalling possibilities has allowed the development of ligands with functionally biased signalling properties (Figure 2).
Figure 2.
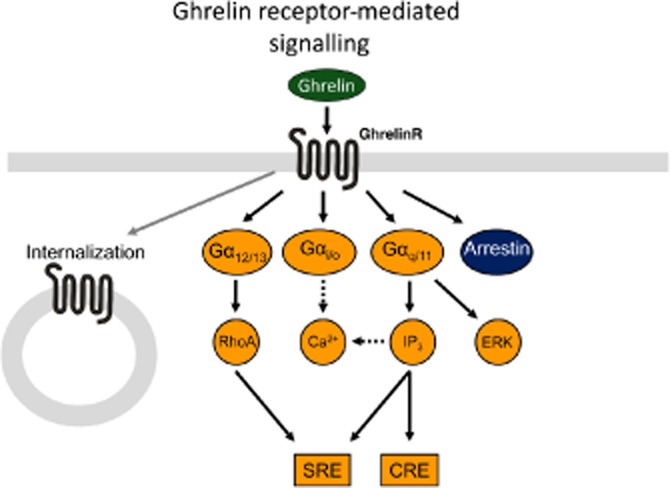
Multiple signalling pathways of the ghrelin receptor. Dotted arrows represent not fully verified signalling pathways, and black full arrows indicate pathways that have been described or suggested for the ghrelin receptor. The ghrelin receptor is able to signal through three different G-proteins, for example Gαq, Gαi/o, Gα12/13 in addition to G–protein-independent arrestin coupling and internalization. Gαq, that activates PLC and leads to increased IP and DAG formation can induce an increase in Ca2+ signalling. However, the pharmacological profiling of different ghrelin receptor agonists indicates that the Ca2+ signalling and IP accumulation originate from separate signalling pathways (Holst et al., 2005), which explains the dotted line. In addition, Gαq coupling may also lead to CRE-mediated transcription activity and probably contributes to the SRE-mediated transcriptional activity. Finally, ligand-mediated Gαq coupling may also stimulate ERK1/2 phosphorylation. Gα12/13 activates RhoA and ROCK resulting in SRE transcription. Gαi/o, generally inhibits adenylate cylase to decrease cAMP accumulation; however, this has not been shown for the ghrelin receptor and it is possible that it can couple to Gαi/o and induce Ca2+ release. Ligand activation of the ghrelin receptor induces recruitment of β-arrestin, which might lead to receptor internalization. Both the constitutive and ligand activation of the receptor induce internalization of the receptor. β-Arrestins might lead to ERK phosphorylation but this is still uncertain.
Unusually, ligand binding is not required for significant ghrelin receptor activation. In the absence of any ligand present, the receptor signals with almost 50% constitutive activity as measured in IP accumulation assays. This property has been demonstrated for several different signalling pathways including SRE- and CREB-mediated transcriptional activity (Holst et al., 2004). Recruitment of arrestin has also been shown to occur in a ligand-independent manner in heterologous expression systems, whereas in isolated lipid discs the arrestin recruitment is ligand dependent (Mary et al., 2012; Mokrosinski et al., 2012). Internalization of the ghrelin receptor can occur in a ligand-independent manner, but is dependent on receptor constitutive activity and domains within the C-terminal tail (Holliday et al., 2007). The recent demonstration of ligand-independent AP2 recruitment to the ghrelin receptor may therefore contribute to these basal receptor internalization properties (Damian et al., 2012). Interestingly, however, some signalling pathways, such as ERK phosphorylation and Gαi coupling, do require receptor activation by exogenous agonists (Holst et al., 2004; Damian et al., 2012). Recent studies have shown that the constitutive activity of the ghrelin receptor is an intrinsic property of the receptor; when the receptor is embedded in lipid discs it can induce IP accumulation and GTPγS binding and these signals can be reduced by addition of the inverse agonist or increased by the agonist (Damian et al., 2012). Thus, the scope for functionally selective ghrelin receptor ligands may also include those that might differentially modulate constitutively active receptor signalling rather than ghrelin-stimulated responses, known as ‘protean’ agonism (Ganguli et al., 1998; Gbahou et al., 2003).
The ghrelin receptor has been shown to dimerize both with other 7TM receptors as heterodimers and with itself as a homodimer. The interface responsible for the dimerization has not been studied for this receptor family. However, it has been demonstrated that the dopamine D1 receptor (Jiang et al., 2006), dopamine D2 receptor (Kern et al., 2012), melanocortin MC3 receptor (Rediger et al., 2012) and the 5-HT2C receptor (Schellekens et al., 2013) are co-expressed with the ghrelin receptor under physiological conditions. Importantly it has been shown that co-expression of the ghrelin receptor with either the MC3, the D1 or the 5HT2C receptor leads to decreased ghrelin-mediated signalling in heterologous expression systems. In addition α-melanocyte stimulating hormone (α-MSH) signalling is enhanced by co-expression of the ghrelin receptor with the MC3 receptor (Rediger et al., 2012).
Ligand development for the ghrelin receptor was initially focused on agonists to increase growth hormone secretion. Several high-potency efficacious agonists – based on either peptide or non-peptide scaffolds – were studied in clinical trials, when the appetite promoting effects of the ghrelin system was revealed and ligands to block the function of ghrelin subsequently became the major focus. The substance P analogue ([D-Arg1, D-Phe5, D-Trp7,9, Leu11]-substance P), was the first antagonist and inverse agonist to be identified for the ghrelin receptor. Since then, several other ligands for the ghrelin receptor have been discovered. Based on a conserved motif wFwLL (where wFwLL denotes one letter abbreviations of amino acids, using the small letters to denote the D-form and capital letters to denote the L-form.) of the substance P analogue, a series of inverse agonists has been developed (Holst et al., 2003). The penta-peptide wFwLL in itself binds to the ghrelin receptor with submicromolar affinity. However, it has the potential to act both as an agonist with a high potency and as an inverse agonist with a slightly lower potency. Modification by the N-terminal elongation of this motif with an alanine (AwFwLL) produced a more effective partial agonist whereas addition of lysine (KwFwLL) produced an inverse agonist (Holst et al., 2007). Thus, even subtle changes in the structure of this motif can give rise to large variations ranging from negative to positive efficacy. This family of peptides interacts with the ghrelin receptor through a different subset of receptor interaction points compared with the endogenous ligand ghrelin, as shown by mutational analysis and computational docking simulations (Holst et al., 2006). More recently, in an attempt to develop a treatment for obesity, small-molecule antagonists have been discovered for the ghrelin receptor. However, the molecular pharmacological properties of these compounds have yet to be fully characterized (Xin et al., 2006; Rudolph et al., 2007; Palus et al., 2011).
Using mutation studies in combination with computational chemistry, the interaction of ligands with the ghrelin receptor has also been studied for several agonist compounds and for the previously mentioned peptide-based inverse agonists. The most important ligand interaction site described in the ghrelin receptor is a glutamic acid in the extracellular part of TM III. This is pivotal for the binding and function of both ghrelin and almost all ghrelin receptor ligands. Furthermore, mutations of aromatic and positively charged residues in TM VI affect the potency of ghrelin significantly (Feighner et al., 1998; Holst et al., 2006; 2009). Apart from these residues, no other substitutions in the binding pocket affect ghrelin-induced activation, indicating that ghrelin only makes a few key interactions within the centre of the binding pocket of the receptor in addition to potential interactions in the extracellular loops (ECLs; Holst et al., 2006; 2009). However, the constitutive activity of the ghrelin receptor is diminished by substitutions in several other receptor domains, including substitutions of aromatic and charged residues in the extracellular part of TM III, TM VI and VII (Holst et al., 2004; Goze et al., 2010). These studies demonstrate the importance of a hydrophobic cluster connecting the extracellular parts of TM VI and VII for the activation of the receptor. Finally, inverse agonists generally require interactions deep in the transmembrane-binding pocket compared with the interactions made by agonists, which occur more superficially towards the extracellular ends of the TM domains (Holst et al., 2007; 2009).
Biased 7TM receptor signalling
Traditionally, the same agonist–receptor complex has been thought to signal with the same intrinsic efficacies to all downstream signalling pathways. This classical paradigm is sufficient to account for variations in agonist potency and maximum response, which depend on the extent of signal amplification and receptor reserve. However, multiple lines of evidence challenge this classical view, suggesting that that some ligands may activate 7TM receptors differently through pathway-dependent intrinsic efficacies (Kenakin and Miller, 2010; Kahsai et al., 2011; Kenakin, 2012). From a cellular signalling perspective, this phenomenon predicts ligands with the capacity to bias the functional response and favour a selection of some signalling pathways over others of the receptor's signalling repertoire. This allows the receptor to activate only a subset of specific effectors and thereby fine tune cellular responses and associated physiological responses. In addition to the ligand-induced signalling bias, structural changes in the receptor by amino acid substitutions may also allow the receptor to selectively favour some signal transduction pathways over others. This concept, known as ‘ligand-directed trafficking’, ‘functional selectivity’ or ‘biased agonism’, indicates that receptors are capable of generating a more complex response than classical pharmacological agonism or antagonism would predict.
Quantifying the bias component of a ligand by application of an operational model may facilitate prediction of the biased signalling properties in all systems without actually quantifying biased signalling in all systems (Ehlert, 2008; Kenakin, 2009). In the model defined by Ehlert, the relative intrinsic response of a biased agonist compared with the standard agonist can be estimated based on data from simple binding and dose–response curves, for example affinity and efficacy. This method has been applied successfully to the muscarinic M2 receptor system where agonists with variable efficacy through Gαi/o, Gαs and Gα15 signalling have been calculated and observed (Griffin et al., 2007).
G-protein coupling versus G–protein-independent signalling
The most thoroughly investigated biased ligands are those that selectively differentiate between G-protein and G–protein-independent mediated signalling (Berg et al., 1998). The clinical and pharmaceutical perspective for this kind of ligand is that it may allow for activation of only the therapeutic or clinically relevant pathways, not activating pathways associated with adverse effects. An example of such a biased ligand was discovered for the niacin receptor, GPR109A, where novel nicotinic acid derivatives only induce the anti-lipolytic response mediated through inhibition of cAMP accumulation. This is in contrast to nicotinic acid itself, which also activated an unwanted flushing response mediated through the G–protein-independent pathway of ERK1/2 activation (McKenney et al., 1994; Richman et al., 2007). This kind of success story has encouraged research within the field of functionally biased ligands targeting the 7TM GPCRs.
Examples of receptors
Several different 7TM receptors have the ability to couple to more than one signalling pathway and within the last decade an increasing number of receptor ligands have been developed or characterized for biased signalling properties. (Kenakin, 1995a; Berg et al., 1998)
For the angiotensin II (AT1A) receptor, the ligand TRV120027 is an excellent example of how biased ligands can provide advantages over classical unbiased drugs. TRV120027 stimulates ERK1/2 phosphorylation in a β–arrestin-dependent manner while antagonizing G-protein coupling such as Gαq-mediated IP production or GTPγS recruitment (Wei et al., 2003). This signalling profile allows the ligand to inhibit angiotensin II-mediated vasoconstriction while increasing cardiomyocyte contractility via β-arrestin coupling (Violin et al., 2010; Boerrigter et al., 2011). In vivo, TRV120027 reduces mean arterial pressure in a manner similar to that of unbiased AT1A receptor antagonists. However, unlike the unbiased antagonists, which tend to decrease cardiac performance, TRV120027 increases cardiac performance and preserves cardiac stroke volume. Thus, this ligand alleviates the symptoms of acute heart failure, and has now reached phase 2 clinical trials. The striking differences in vivo between unbiased and arrestin-biased ligands illustrate the potential of biased ligands for optimized targeting of therapeutic effects while minimizing adverse events.
For the chemokine CCR5 receptor synthetic ligands have been developed in an attempt to prevent HIV entry. Importantly, to decrease the risk of side effects, these ligands have been selected for biased signalling properties that favour internalization and do not affect G-protein coupling (O'Hayre et al., 2010; Steen et al., 2013).
Mutation-induced signalling bias
In a large number of other class A 7TM receptors, functionally biased ligands have also been found to distinguish between G-protein coupled and arrestin-mediated signalling (Stallaert et al., 2011). Interestingly, such bias can also be generated by mutations located in various areas of the 7TM receptor, which selectively shift the balance for activating one pathway rather than another. Substitutions of residues located in the ligand binding pocket can in some cases be responsible for biasing the signalling towards either the G-protein or G–protein-independent pathways. For example, in the muscarinic M2 cholinoceptor, mutations in both the orthosteric and the allosteric pockets have been identified that selectively affect coupling to the ERK1/2 pathway. Substitution of a tyrosine in the orthosteric site selectively abolished signalling to the ERK1/2 pathway while retaining the ability to activate G-proteins and mediate intracellular calcium mobilization. In contrast, mutation of a second tyrosine in the allosteric site selectively enhanced signalling through the ERK1/2 pathway (Gregory et al., 2010).
More common are substitutions in the intracellular part of the receptor that selectively uncouple either G-protein or β-arrestin coupling. For example, mutations in the conserved TM III DRY motif of the AT1A receptor result in the loss of G-protein coupling, measured by GTPγS binding or IP production. However, this receptor mutant retains the ability to induce ERK1/2 phosphorylation via β-arrestin recruitment (Wei et al., 2003). Another AT1A mutation with a substitution in the second intracellular loop displayed impaired IP accumulation compared with wild type (WT) receptors, but still retained WT-like receptor internalization properties (Gaborik et al., 2003). Several other mutations in intracellular loop 2 preserved receptor activation of Src and ERK1/2, but could not induce Gαq-mediated activation of PLC or Gαi-mediated inhibition of forskolin-induced cAMP accumulation (Seta et al., 2002). These studies suggest that mutations of the intracellular surface of the AT1A receptor are important for discrimination between the intracellular signalling proteins, e.g. β-arrestins versus G proteins. Thus some types of signalling bias, normally invoked by substitutions in the intracellular domains, induce selective manipulation of the interaction sites between an active receptor conformation and its G-protein or arrestin effectors. However other biasing mutations, which can be located in almost any region of the 7TM receptor, clearly indicate the possibility of pathway-specific receptor conformations.
Biased coupling to one G-protein versus another
The majority of the biased ligands described in the literature distinguish between G-protein coupled and G–protein-independent signalling (Violin and Lefkowitz, 2007). However, in some cases signal transduction of one G-protein is favoured over another G-protein (Berg et al., 1998). Both mutations and ligands can bias receptors to distinguish between the different G-proteins as described later.
The melanocortin MC4 receptor represents an example of a receptor where ligands have been shown to favour one G-protein coupling over another. Activation of the MC4 receptor stimulates both cAMP accumulation and Ca2+-release, which involve both Gαs and Gαi proteins (Buch et al., 2009). These G-protein coupled pathways can be separated by different agonists. For example, the non-peptide ligand tetrahydroisoquinoline (Van der Ploeg et al., 2002) displays a 100-fold higher potency for Gαs coupling, as measured by cAMP accumulation, compared to Ca2+ release. In contrast, other non-peptide agonists such as NBI 58704 show the opposite signalling bias with an 11-fold impaired potency for cAMP accumulation compared with Ca2+-release (Nickolls et al., 2005). Even the well-described MC4 receptor antagonist AgRP displays biased signalling properties as it is able to activate Gαi/o coupling by stimulating the MC4 receptor (Buch et al., 2009).
The thyrotropin (TSH) receptor represents an example of a receptor where substitutions of residues in the transmembrane region alter G-protein signalling preferences. In the constitutively active TSH receptor, subtle substitutions of aromatic and hydrophobic residues in TM V and VI resulted in bias of the functional response with respect to Gαs coupled cAMP accumulation compared with Gαq coupled IP accumulation. Substitutions in TM VI resulted in strongly decreased Gαs coupled constitutive efficacy as measured by cAMP, whereas the constitutive Gαq coupled IP accumulation was only slightly decreased. The same phenomenon was observed for ligand-induced activation, as the same substitutions showed almost WT-like ligand-induced stimulation of cAMP accumulation, but strongly decreased efficacy for IP accumulation. This suggests that specific residues in these helices are involved in G–protein-specific coupling (Kleinau et al., 2011).
Biased signalling of the ghrelin receptor
The ghrelin receptor is coupled to at least three different G-proteins, Gαq, Gαi and Gα12/13, and in addition it is also able to recruit β-arrestin and AP2 upon activation. Most of these pathways may be activated ligand-independently, whereas ERK phosphorylation is only activated by agonists (Holst et al., 2004). The wide spectrum of signalling pathways available for the ghrelin receptor provide many possibilities for ligand- and mutation-induced selectivity of the functional response.
A classical biased agonist, wFw-isonipecotic acid (Isn), has been discovered for the ghrelin receptor, which favours signalling through one G-protein rather than another (Sivertsen et al., 2011). This development was built on the knowledge of the wFw peptide binding modalities and the transition from inverse to positive agonism that this peptide series displays at the ghrelin receptor (described earlier). Thus, the biased agonist contains the core binding wFw motif, and is linked to a synthetic amino acid Isn at the C-terminus. Mutational studies and computational docking studies revealed that the interaction pattern of wFw-Isn is different from that of all the other peptide and non-peptide ghrelin receptor ligands described. Importantly, the activity of this agonist is not dependent on the highly conserved negatively charged glutamic acid in TM III that serves as the receptor anchoring point for all the other ligands described for the ghrelin receptor. This ligand induces IP accumulation and ERK1/2 phosphorylation, but it is unable to activate the Gα12/13 coupled SRE-induced transcriptional activity. These data and calculations suggest that wFw-Isn is a biased ligand that favours pathways such as Gαq and ERK1/2 phosphorylation over Gα12/13 coupling. In support of this notion, wFw-Isn does not activate RhoA in a pituitary cell line naturally expressing the ghrelin receptor, unlike ghrelin itself (Sivertsen et al., 2011; Figures 3B and 4B). Interestingly, wFw-Isn administered i.c.v. to rats was unable to stimulate food intake, which is one of the most solid and reproducible effects of ghrelin receptor stimulation. These data suggest that based on the elucidation of the ligand receptor interaction it is possible to rationally develop biased agonists.
Figure 3.
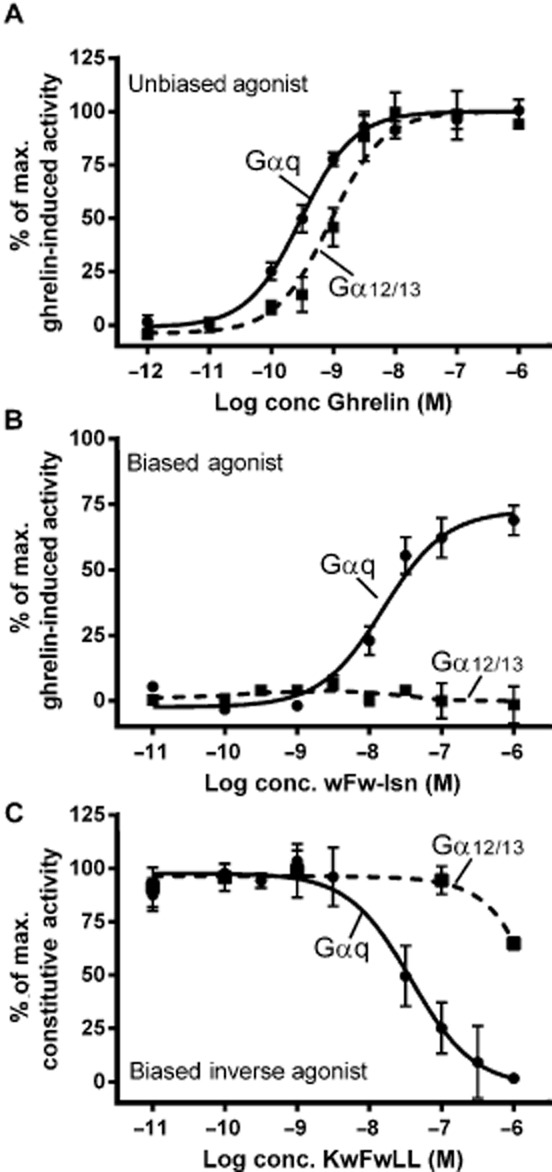
Dose–response curves of an example of an unbiased agonist, a biased agonist and a biased inverse agonist. The dose–response curve was performed in HEK293 cells transiently transfected with the ghrelin receptor where the response of the ligands is measured by the accumulation of IP (Gαq coupled) and by activation of SRE-mediated transcriptional control (Gα12/13 mediated; Holst et al., 2006). Dose–response curves for (A) the un-biased ligand, ghrelin, (B) the biased agonist, wFw-Isn and (C) the biased inverse agonist are shown.
Figure 4.
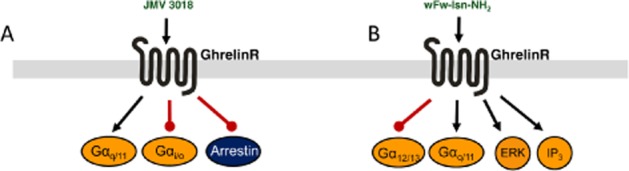
The effects of two different biased agonists. (A) The biased agonist JMV3018 is able to stimulate Gαq recruitment in purified ghrelin receptors in lipid discs, but no recruitment of Gαq or arrestin was observed for this compound. (B) The biased agonist wFw-Isn is able to couple to Gαq and the downstream signalling pathways IP and ERK phophorylation, but it is unable to activate the Gα12/13 coupled SRE pathway when compared with ghrelin.
In addition the hexapeptide ligand, KwFwLL, has been identified, unusually, as a biased inverse agonist for the ghrelin receptor. It is a potent inverse agonist, when measuring IP accumulation, with an EC50 value of 32 nM. However, when measuring SRE transcriptional activity it is a very low-potent inverse agonist that only induces a weak decrease in the signalling at 1 μM (Holst et al., 2007). This observation is most likely due to biased inverse agonist signalling favouring inhibition of the Gαq coupled pathway compared to that coupled to Gα12/13 (Figure 3C).
The examples mentioned earlier of biased ligands for the ghrelin receptor were characterized in vitro, predominantly in heterologous expression systems. However, recently the ghrelin receptor was reconstituted in lipid discs, a phospholipid bilayer lined by amphipathic helical membrane scaffold proteins to form a disc like structure (Bayburt et al., 2002). In this reconstitution method, the receptor is expressed in bacteria, extracted via inclusion body formation, which requires solubilization before purification, but yields functional receptors (Mary et al., 2012). Receptor activation was measured by monitoring conformational changes via fluorescent probes in the intracellular and in the extracellular domains. Under these conditions the ghrelin receptor was able to couple to Gαi in addition to the previously mentioned signalling pathways. In this system, a biased agonist, JMV3018, was demonstrated to couple to Gαq to the same extent as ghrelin, but was unable to couple to Gαi or recruit β-arrestin (Mary et al., 2012; Figure 4B).
The phenomenon of signalling bias in the ghrelin receptor system has also been demonstrated by the introduction of mutations in the receptor. For example, we observed that substitutions of an alanine residue in one of the ECLs (position 204) resulted in clear differences in relative receptor signalling through IP production, SRE-induced transcriptional activity or β-arrestin mobilization. Substitution of the alanine with either a charged residue or an aromatic residue only decreased the basal constitutive Gαq-mediated signalling, measured as IP accumulation. However, these mutations had a greater effect on both SRE-mediated transcription activity and arrestin recruitment, where decreased ghrelin-induced efficacy was also observed. In particular arrestin recruitment was almost eliminated (Mokrosinski et al., 2012). The potencies of ghrelin-induced SRE-induced transcription activity and IP accumulation were only affected when the alanine was substituted with a small negatively charged aspartic acid. However, the potency of ghrelin in the β-arrestin mobilization assay was not affected by any substitution. The molecular background for this substitution-induced signalling bias is probably due to changes in the secondary structure in ECL2 resulting in an extended constrained α-helix that then affects the intracellular coupling repertoire of the receptor. The findings of this study also indicate that the less constrained ECL2 is important for the constitutive activity of the ghrelin receptor (Mokrosinski et al., 2012).
7TM receptor conformational changes during biased signalling
Biased signalling properties may, from a molecular perspective, refer to the fact that several different distinct ‘active’ as well as ‘inactive’ conformations of the receptor exist. These ‘active’ conformations may interact differently with the intracellular signalling partners and generate different signalling patterns (Kenakin, 1995b). It has recently been shown that multiple active conformations are stabilized during ligand binding in the β2-adrenoceptor (Kahsai et al., 2011).
The active conformation of 7TM receptors
Transition to the active conformation of 7TM receptors, stabilized by ligands of highly variable chemical nature, is believed to involve similar global changes. These conformational changes between the active and the inactive conformation have been intensively studied over the last two decades by use of various biochemical and biophysical methods, and more recently, complementary X-ray crystallographic data have been obtained. Using spin-labelling studies in rhodopsin, Farrens et al. (1996) showed that the intracellular parts of the TM segments moved relative to each other upon activation and that constraining the intracellular part of the transmembrane segments by covalent bonds impaired signal transduction. The most dramatic changes were an outward movement and rotation of the TM VI relative to TM III (Farrens et al., 1996). These data were confirmed both by environment-sensitive probes in the β2-adrenoceptor, by NMR studies and by introduction of zinc sites (Sheikh et al., 1996; Gether and Kobilka, 1998; Ye et al., 2010). More recently crystal structures of the β2-adrenoceptor bound to either an agonist or an inverse agonist, as well as the receptor in complex with the G-protein have been resolved (Rasmussen et al., 2007; 2011). The difference between these structures is primarily that the intracellular part of TM VI is rotated and moved away from the helix bundle in the structure of the agonist-bound receptor compared with that of the inverse agonist (Schwartz and Sakmar, 2011). This allows for an interaction with the C-terminal part of Gαs protein that initiates the intracellular activation cascade (Rasmussen et al., 2011). The crystal structure of the A2A adenosine receptor shows a similar outwards rotation of the intracellular part of TM VI upon binding to its endogenous ligand, adenosine (Jaakola et al., 2008; Lebon et al., 2011). This mechanism of activation is therefore believed to be a general characteristic of 7TM receptors of the rhodopsin family. The conformation of the extracellular part of the receptor has only been shown to differ to a very small degree between the active and the inactive state (Bokoch et al., 2010). This change involves weakening of a salt bridge between ECL II and III in the active conformation (Bokoch et al., 2010) and in the extracellular located transmembrane part the most significant changes involved minor movements of TM V (Rasmussen et al., 2011).
Several active conformations
Interestingly, biophysical data suggest that full agonists and partial agonists targeting the most well studied receptor, the β2-adrenoceptor, apparently bind to and activate the receptor through different mechanisms and with different kinetics – irrespective of the fact that they have very similar chemical structures (Swaminath et al., 2005). For example fluorescent probes in the intracellular part of the receptor revealed different conformations induced by the full agonists, adrenaline and isoprenaline, and the partial agonist noradrenaline (Swaminath et al., 2005; Reiner et al., 2010).
Distinct receptor conformations could also lead to differential recruitment of intracellular regulatory proteins, such as G-proteins, β-arrestins and GRKs, biasing the receptor towards a given pattern of response (Breit et al., 2011; Ulloa-Aguirre et al., 2011). This is supported by data showing that a β–arrestin-biased ligand of the β2-adrenoceptor, carvedilol, selectively mediates a specific phosphorylation pattern, thus leading to stimulation of ERK (Nobles et al., 2011). The specific phosphorylation pattern has been shown to be due to selective recruitment of GRK6, suggesting a specific active conformation stabilized by the biased agonist compared with the un-biased agonists. The distinct active conformation induced by β–arrestin-biased agonists has also been studied by site-specific 19F-NMR label studies of the β2 adrenoceptor in complex with ligands. These data suggest that the biased agonist stabilizes a conformation in the intracellular part of the receptor, slightly different from un-biased ligands. Specifically for the β2-adrenoceptor, β–arrestin-biased ligands that do not, or only to a small extent, activate G–protein-dependent signalling, induced a characteristic pattern of TM VII movements. This effect on TM VII was different from the changes observed for unbiased ligands that induced major changes in TM VI and VII (Bokoch et al., 2010; Liu et al., 2012). A similar observation has been made in a prototypical peptide activated 7TM receptor, the V2 arginine-vasopressin, in which the receptor conformations stabilized, either by an un-biased agonist or by two different agonists biased either towards β-arrestin or Gαs coupling, were studied. In this receptor system, ligand-induced conformational changes were studied by fluorescent probes incorporated strategically into the purified V2 receptor. Gαs biased agonists induced changes in the conformation between intracellular loop 3 and TMVI, whereas the β-arrestin biased agonist required movements in the interphase between TMVII and helix VIII (Rahmeh et al., 2012).
Given that several 7TM receptors can exist as dimers or oligomers (Pin et al., 2005; Milligan, 2008), it seems feasible that signalling would be modulated in these states compared with signalling of a monomeric receptor. Dimerization both as homo-dimerization and as heterodimerization has been demonstrated to modulate signalling bias. For example, when co-expressing the cannabinoid with the dopamine D2 receptor a switch in CB1-mediated Gs coupling was observed, even in the absence of a D2 agonist (Jarrahian et al., 2004), supposedly because the D2 receptor stabilizes the CB1 receptor in a conformation more favourable for Gs coupling (Ellis et al., 2006; Hudson et al., 2010). A recent publication showed that homodimerization may affect the signalling bias in the family B 7TM receptor glucagon like peptide-1 (GLP-1) receptor; intracellular Ca2+ release was completely abolished when dimerization was disrupted, whereas ERK and cAMP signalling was unchanged (Harikumar et al., 2012). The association of a 7TM receptor with other membrane proteins could therefore affect signalling bias of 7TM receptors.
Active and inactive conformations of the ghrelin receptor
The ghrelin receptor has also been studied in terms of conformational changes during activation and inactivation. The observation that inverse agonists interact differently with the receptor compared with agonists and thereby may stabilize a different conformation, has been observed in mutational studies. The KwFwLL, an inverse agonist, and the structurally similar AwFwLL, an agonist, interact in a different way with the receptor. They are both dependent on key residues in TM II, III, IV, V and VII; however, mutations in TM V strongly decreased the potency of AwFwLL, whereas the inhibitory potency of KwFwLL was unchanged. This indicates that the two structurally-related molecules interact differently with the receptor to generate different pharmacological effects by stabilizing the inactive and active receptor conformations respectively (Holst et al., 2007). In agreement with the crystal structure data, the active state requires conformational changes in TM V and agonism was dependent on residues in TM V. In contrast, the inverse agonist did not share these interaction sites. Biophysical data from fluorescence spectroscopy studies using reconstituted monomeric ghrelin receptors in lipid discs support the occurrence of distinct active and inactive ghrelin receptor conformations differentially stabilized by agonists and inverse agonists. Thus, the ghrelin receptor occupied by the inverse agonist [D-Arg1, D-Phe5, D-Trp7,9, Leu11]-substance P analogue adopts a confirmation that is different from the conformation stabilized by full agonists, MK-677 and ghrelin. Notably this conformation is also different from that of the unoccupied receptor, which may suggest multiple inactive conformations. The conformation induced by the inverse agonist leads to decreased binding of β-arrestins and G-proteins (Damian et al., 2012). Importantly, the arrestin-biased ligand JMV3018 also induced changes in receptor conformation that differed from those induced by an unbiased agonist. Interestingly, conformational selection was also observed when the ghrelin-receptor complex was exposed to either β-arrestin or Gαq. In the presence of β-arrestin, ghrelin induced an active conformation that was different from the conformation stabilized in the presence of Gαq, supporting the hypothesis that minor variations in the active conformation can determine the coupling pattern of 7TM receptors.
From the present data, it is postulated that 7TM receptors may adopt many different active and inactive conformations, stabilized by agonists or inverse agonists respectively. Several different active conformations may exist depending on the properties of the agonist. Biased agonists stabilize receptor conformations that couple with different intrinsic efficacies to the different possible signalling pathways (Schellekens et al., 2013). Finally dimerization and intracellular cross-talk between the ghrelin receptor and other 7TM receptors might also contribute to the biased signalling properties, as the ghrelin receptor has been demonstrated to dimerize with MC3, D1, D2 and other receptors (Jiang et al., 2006; Kern et al., 2012; Rediger et al., 2012; Schellekens et al., 2013).
Biased signalling in a physiological setting
Several studies performed in animal models suggest that it is possible to dissociate the physiological effects of the ghrelin receptor system dependent on biased signalling properties of the applied ligand. In particular, the actions of ligands developed in the search for either anti-obesity treatment through inhibition of the ghrelin receptor system, or for anti-cachexia treatment through activation of the system, can be dissociated on the basis of their effects on GH secretion, food intake and body weight.
The small molecule compound GSK161443 represents one example of a physiologically biased agonist. In vitro, this compound was shown to act as a potent competitive antagonist in calcium screening assays, although in IP accumulation assays, weak partial agonism was observed. In the initial in vivo studies monitoring GH release, GSK161443 behaved as an antagonist. However, this compound turned out to behave as an agonist with regard to food intake and body weight regulation both in acute and chronic studies (Costantini et al., 2011). Importantly, this effect was found to be mediated entirely by the ghrelin receptor as the compound only increased body weight in WT mice and not in ghrelin receptor knockout mice (Sabbatini et al., 2010; Costantini et al., 2011). A similar dissociation between GH secretion and food intake effects was observed for another class of small-molecule ligands – but in the opposite manner (Moulin et al., 2007). These ligands were also described as antagonists in calcium mobilization assays, but they were only able to antagonize the agonist effects induced by ghrelin receptors in food intake studies, not in the GH secretion studies (Moulin et al., 2007). Peptide ligands based on ghrelin itself have also been shown to display a strong degree of selection between the modulation of GH secretion and food intake. The ghrelin analogue BIM-28163 was shown to act as a ghrelinR antagonist in the in vitro calcium mobilization assay (Halem et al., 2004). However, in in vivo assays, BIM-28163 blocked ghrelin-induced GH secretion but acted as a full agonist on food intake with comparable efficacy to ghrelin (Halem et al., 2004).
These in vivo studies show that activation or inhibition of the ghrelin receptor is not a simple one-way road to modulation of all the downstream physiological functions. In particular, it is obvious that modulation of GH secretion and food intake is dependent on distinct subsets of the complex intracellular signal transduction pathways initiated by this receptor. Thus compounds simply screened for antagonism of ghrelin-induced calcium release (via Gαq) cannot be predicted to have the same physiological effects in vivo, as illustrated in the examples mentioned previously (Halem et al., 2004; Moulin et al., 2007; Sabbatini et al., 2010; Costantini et al., 2011). Even with calcium release, multiple mechanisms may be involved, for example both Gαq coupling and Gαi coupling may also induce an increase in the calcium level through Gβγ-subunit potentiation of PLC (Lee et al., 1993). The dissociation between the pathways modulating GH secretion and food intake has also been shown in in vivo studies where a specific inhibitor of the intracellular enzyme SIRT1 selectively blocked the ghrelin-induced food intake, but did not affect GH secretion (Velasquez et al., 2011). In order to predict the physiological effect of a given ligand based on results from in vitro studies, it is therefore important to dissect the signalling repertoire of the ghrelin receptor carefully and take into consideration the many possible coupling pathways.
Conclusion
The ghrelin receptor constitutes an interesting therapeutic target because of its important effect on food intake and energy expenditure. However, the drug development process is complicated by the fact that the ghrelin receptor system also modulates neuropsychiatric functions such as mood, reward behaviour and cognition, and GH secretion. The development of functionally biased ghrelin receptor ligands, illustrated in principle by current in vitro and in vivo studies, demonstrates that ligand modulation of a selected subset of the possible ghrelin receptor signalling pathways is achievable, and a route that could be used to develop drugs with minimal potential side effects.
Acknowledgments
This work was supported by the Danish Health Science Research Council (B. H.), the Novo Nordic Foundation Center for Basic Metabolic Research (B. H., A. N. M., B. S.), Faculty of Health Sciences at the University of Copenhagen (B.S.) and the UK Medical Research Council Grant G0700049 (N. H.).
Glossary
- 7TM
7 transmembrane segment
- AgRP
agouti-related peptide
- AMPK
AMP-activated PK
- AP2
adaptor-related protein complex 2
- AT1A receptor
angiotensin 1A receptor
- AVP
arginine-vasopressin
- CREB
cAMP response element binding protein
- ECL
extracellular loop
- FAS
fatty acid synthase
- GH
growth hormone
- GHS
growth hormone secretagogues
- GRK
GPCR kinase
- IP
inositol phosphate
- IP3
inositol tris-1,4,5-phosphate
- Isn
isonipecotic acid
- MC4
melanocortin 4
- NPY
neuropeptide Y
- SRE
serum response element
- UCP2
uncoupling protein 2
- VTA
ventral tegmental area
- WT
wild type
Conflict of interest
None.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454:846–851. doi: 10.1038/nature07181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atcha Z, Chen WS, Ong AB, Wong FK, Neo A, Browne ER, et al. Cognitive enhancing effects of ghrelin receptor agonists. Psychopharmacology (Berl) 2009;206:415–427. doi: 10.1007/s00213-009-1620-6. [DOI] [PubMed] [Google Scholar]
- Bach MA, Rockwood K, Zetterberg C, Thamsborg G, Hebert R, Devogelaer JP, et al. The effects of MK-0677, an oral growth hormone secretagogue, in patients with hip fracture. J Am Geriatr Soc. 2004;52:516–523. doi: 10.1111/j.1532-5415.2004.52156.x. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Grinkova YG, Sligar SG. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002;8:853–856. [Google Scholar]
- Bennett KA, Langmead CJ, Wise A, Milligan G. Growth hormone secretagogues and growth hormone releasing peptides act as orthosteric super-agonists but not allosteric regulators for activation of the G protein Galpha(o1) by the ghrelin receptor. Mol Pharmacol. 2009;76:802–811. doi: 10.1124/mol.109.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC., Jr Cardiorenal actions of TRV120027, a novel ss-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail. 2011;4:770–778. doi: 10.1161/CIRCHEARTFAILURE.111.962571. [DOI] [PubMed] [Google Scholar]
- Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers CY. Unnatural growth hormone-releasing peptide begets natural ghrelin. J Clin Endocrinol Metab. 2001;86:1464–1469. doi: 10.1210/jcem.86.4.7431. [DOI] [PubMed] [Google Scholar]
- Bowers CY, Reynolds GA, Durham D, Barrera CM, Pezzoli SS, Thorner MO. Growth hormone (GH)-releasing peptide stimulates GH release in normal men and acts synergistically with GH-releasing hormone. J Clin Endocrinol Metab. 1990;70:975–982. doi: 10.1210/jcem-70-4-975. [DOI] [PubMed] [Google Scholar]
- Breit A, Buch TR, Boekhoff I, Solinski HJ, Damm E, Gudermann T. Alternative G protein coupling and biased agonism: new insights into melanocortin-4 receptor signalling. Mol Cell Endocrinol. 2011;331:232–240. doi: 10.1016/j.mce.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Briggs DI, Andrews ZB. A recent update on the role of ghrelin in glucose homeostasis. Curr Diabetes Rev. 2011;7:201–207. doi: 10.2174/157339911795843140. [DOI] [PubMed] [Google Scholar]
- Broglio F, Gottero C, Arvat E, Ghigo E. Endocrine and non-endocrine actions of ghrelin. Horm Res. 2003a;59:109–117. doi: 10.1159/000069065. [DOI] [PubMed] [Google Scholar]
- Broglio F, Gottero C, Benso A, Prodam F, Destefanis S, Gauna C, et al. Effects of ghrelin on the insulin and glycemic responses to glucose, arginine, or free fatty acids load in humans. J Clin Endocrinol Metab. 2003b;88:4268–4272. doi: 10.1210/jc.2002-021940. [DOI] [PubMed] [Google Scholar]
- Broglio F, Gottero C, Prodam F, Gauna C, Muccioli G, Papotti M, et al. Non-acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J Clin Endocrinol Metab. 2004;89:3062–3065. doi: 10.1210/jc.2003-031964. [DOI] [PubMed] [Google Scholar]
- Buch TR, Heling D, Damm E, Gudermann T, Breit A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J Biol Chem. 2009;284:26411–26420. doi: 10.1074/jbc.M109.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlini VP, Monzon ME, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, et al. Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun. 2002;299:739–743. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- Chang L, Ren Y, Liu X, Li WG, Yang J, Geng B, et al. Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J Cardiovasc Pharmacol. 2004;43:165–170. doi: 10.1097/00005344-200402000-00001. [DOI] [PubMed] [Google Scholar]
- Chu KM, Chow KB, Leung PK, Lau PN, Chan CB, Cheng CH, et al. Over-expression of the truncated ghrelin receptor polypeptide attenuates the constitutive activation of phosphatidylinositol-specific phospholipase C by ghrelin receptors but has no effect on ghrelin-stimulated extracellular signal-regulated kinase 1/2 activity. Int J Biochem Cell Biol. 2007;39:752–764. doi: 10.1016/j.biocel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Costantini VJ, Vicentini E, Sabbatini FM, Valerio E, Lepore S, Tessari M, et al. GSK1614343, a novel ghrelin receptor antagonist, produces an unexpected increase of food intake and body weight in rodents and dogs. Neuroendocrinology. 2011;94:158–168. doi: 10.1159/000328968. [DOI] [PubMed] [Google Scholar]
- Crespo P, Xu N, Simonds WF, Gutkind JS. Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719. doi: 10.2337/diabetes.50.8.1714. [DOI] [PubMed] [Google Scholar]
- Damian M, Marie J, Leyris JP, Fehrentz JA, Verdie P, Martinez J, et al. High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J Biol Chem. 2012;287:3630–3641. doi: 10.1074/jbc.M111.288324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- Dickson SL, Egecioglu E, Landgren S, Skibicka KP, Engel JA, Jerlhag E. The role of the central ghrelin system in reward from food and chemical drugs. Mol Cell Endocrinol. 2011;340:80–87. doi: 10.1016/j.mce.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:549–577. doi: 10.1007/s00210-008-0260-4. [DOI] [PubMed] [Google Scholar]
- Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem. 2006;281:38812–38824. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- Esler WP, Rudolph J, Claus TH, Tang W, Barucci N, Brown SE, et al. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology. 2007;148:5175–5185. doi: 10.1210/en.2007-0239. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Feighner SD, Howard AD, Prendergast K, Palyha OC, Hreniuk DL, Nargund R, et al. Structural requirements for the activation of the human growth hormone secretagogue receptor by peptide and nonpeptide secretagogues. Mol Endocrinol. 1998;12:137–145. doi: 10.1210/mend.12.1.0051. [DOI] [PubMed] [Google Scholar]
- Gaborik Z, Jagadeesh G, Zhang M, Spat A, Catt KJ, Hunyady L. The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. Endocrinology. 2003;144:2220–2228. doi: 10.1210/en.2002-0135. [DOI] [PubMed] [Google Scholar]
- Ganguli SC, Park CG, Holtmann MH, Hadac EM, Kenakin TP, Miller LJ. Protean effects of a natural peptide agonist of the G protein-coupled secretin receptor demonstrated by receptor mutagenesis. J Pharmacol Exp Ther. 1998;286:593–598. [PubMed] [Google Scholar]
- Gbahou F, Rouleau A, Morisset S, Parmentier R, Crochet S, Lin JS, et al. Protean agonism at histamine H3 receptors in vitro and in vivo. Proc Natl Acad Sci U S A. 2003;100:11086–11091. doi: 10.1073/pnas.1932276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gether U, Kobilka BK. G protein-coupled receptors. II. Mechanism of agonist activation. J Biol Chem. 1998;273:17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, et al. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J Clin Endocrinol Metab. 2002;87:2988. doi: 10.1210/jcem.87.6.8739. [DOI] [PubMed] [Google Scholar]
- Goze C, Berge G, M'Kadmi C, Floquet N, Gagne D, Galleyrand JC, et al. Involvement of tryptophan W276 and of two surrounding amino acid residues in the high constitutive activity of the ghrelin receptor GHS-R1a. Eur J Pharmacol. 2010;643:153–161. doi: 10.1016/j.ejphar.2010.06.018. [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem. 2010;285:7459–7474. doi: 10.1074/jbc.M109.094011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin MT, Figueroa KW, Liller S, Ehlert FJ. Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J Pharmacol Exp Ther. 2007;321:1193–1207. doi: 10.1124/jpet.107.120857. [DOI] [PubMed] [Google Scholar]
- Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48:23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- Halem HA, Taylor JE, Dong JZ, Shen Y, Datta R, Abizaid A, et al. Novel analogs of ghrelin: physiological and clinical implications. Eur J Endocrinol. 2004;151(Suppl. 1):S71–S75. doi: 10.1530/eje.0.151s071. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Harikumar KG, Wootten D, Pinon DI, Koole C, Ball AM, Furness SG, et al. Glucagon-like peptide-1 receptor dimerization differentially regulates agonist signaling but does not affect small molecule allostery. Proc Natl Acad Sci U S A. 2012;109:18607–18612. doi: 10.1073/pnas.1205227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday ND, Holst B, Rodionova EA, Schwartz TW, Cox HM. Importance of constitutive activity and arrestin-independent mechanisms for intracellular trafficking of the ghrelin receptor. Mol Endocrinol. 2007;21:3100–3112. doi: 10.1210/me.2007-0254. [DOI] [PubMed] [Google Scholar]
- Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. High constitutive signaling of the ghrelin receptor – identification of a potent inverse agonist. Mol Endocrinol. 2003;17:2201–2210. doi: 10.1210/me.2003-0069. [DOI] [PubMed] [Google Scholar]
- Holst B, Holliday ND, Bach A, Elling CE, Cox HM, Schwartz TW. Common structural basis for constitutive activity of the ghrelin receptor family. J Biol Chem. 2004;279:53806–53817. doi: 10.1074/jbc.M407676200. [DOI] [PubMed] [Google Scholar]
- Holst B, Brandt E, Bach A, Heding A, Schwartz TW. Nonpeptide and peptide growth hormone secretagogues act both as ghrelin receptor agonist and as positive or negative allosteric modulators of ghrelin signaling. Mol Endocrinol. 2005;19:2400–2411. doi: 10.1210/me.2005-0059. [DOI] [PubMed] [Google Scholar]
- Holst B, Lang M, Brandt E, Bach A, Howard A, Frimurer TM, et al. Ghrelin receptor inverse agonists: identification of an active peptide core and its interaction epitopes on the receptor. Mol Pharmacol. 2006;70:936–946. doi: 10.1124/mol.106.024422. [DOI] [PubMed] [Google Scholar]
- Holst B, Mokrosinski J, Lang M, Brandt E, Nygaard R, Frimurer TM, et al. Identification of an efficacy switch region in the ghrelin receptor responsible for interchange between agonism and inverse agonism. J Biol Chem. 2007;282:15799–15811. doi: 10.1074/jbc.M609796200. [DOI] [PubMed] [Google Scholar]
- Holst B, Frimurer TM, Mokrosinski J, Halkjaer T, Cullberg KB, Underwood CR, et al. Overlapping binding site for the endogenous agonist, small-molecule agonists, and ago-allosteric modulators on the ghrelin receptor. Mol Pharmacol. 2009;75:44–59. doi: 10.1124/mol.108.049189. [DOI] [PubMed] [Google Scholar]
- Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974–977. doi: 10.1126/science.273.5277.974. [DOI] [PubMed] [Google Scholar]
- Hudson BD, Hebert TE, Kelly ME. Ligand- and heterodimer-directed signaling of the CB(1) cannabinoid receptor. Mol Pharmacol. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahian A, Watts VJ, Barker EL. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J Pharmacol Exp Ther. 2004;308:880–886. doi: 10.1124/jpet.103.057620. [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, et al. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A. 2009;106:11318–11323. doi: 10.1073/pnas.0812809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Janson AC, Waters S, Engel JA. Concomitant release of ventral tegmental acetylcholine and accumbal dopamine by ghrelin in rats. PLoS ONE. 2012;7:e49557. doi: 10.1371/journal.pone.0049557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Betancourt L, Smith RG. Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol Endocrinol. 2006;20:1772–1785. doi: 10.1210/me.2005-0084. [DOI] [PubMed] [Google Scholar]
- Kahsai AW, Xiao K, Rajagopal S, Ahn S, Shukla AK, Sun J, et al. Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol. 2011;7:692–700. doi: 10.1038/nchembio.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. I: mechanisms of efficacy and receptor promiscuity. Trends Pharmacol Sci. 1995a;16:188–192. doi: 10.1016/s0165-6147(00)89020-3. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. II. agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995b;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin TP. '7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci. 2009;30:460–469. doi: 10.1016/j.tips.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron. 2012;73:317–332. doi: 10.1016/j.neuron.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinau G, Mueller S, Jaeschke H, Grzesik P, Neumann S, Diehl A, et al. Defining structural and functional dimensions of the extracellular thyrotropin receptor region. J Biol Chem. 2011;286:22622–22631. doi: 10.1074/jbc.M110.211193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Lage R, Vazquez MJ, Varela L, Saha AK, Vidal-Puig A, Nogueiras R, et al. Ghrelin effects on neuropeptides in the rat hypothalamus depend on fatty acid metabolism actions on BSX but not on gender. FASEB J. 2010;24:2670–2679. doi: 10.1096/fj.09-150672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Shin SH, Hepler JR, Gilman AG, Rhee SG. Activation of phospholipase C-beta 2 mutants by G protein alpha q and beta gamma subunits. J Biol Chem. 1993;268:25952–25957. [PubMed] [Google Scholar]
- van der Lely AJ, Tschop M, Heiman ML, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004;25:426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- Li A, Cheng G, Zhu GH, Tarnawski AS. Ghrelin stimulates angiogenesis in human microvascular endothelial cells: implications beyond GH release. Biochem Biophys Res Commun. 2007;353:238–243. doi: 10.1016/j.bbrc.2006.11.144. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M, Lage R, Saha AK, Perez-Tilve D, Vazquez MJ, Varela L, et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab. 2008;7:389–399. doi: 10.1016/j.cmet.2008.03.006. [DOI] [PubMed] [Google Scholar]
- McKenney JM, Proctor JD, Harris S, Chinchili VM. A comparison of the efficacy and toxic effects of sustained- vs immediate-release niacin in hypercholesterolemic patients. JAMA. 1994;271:672–677. [PubMed] [Google Scholar]
- Martins L, Fernandez-Mallo D, Novelle MG, Vazquez MJ, Tena-Sempere M, Nogueiras R, et al. Hypothalamic mTOR signaling mediates the orexigenic action of ghrelin. PLoS ONE. 2012;7:e46923. doi: 10.1371/journal.pone.0046923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary S, Damian M, Louet M, Floquet N, Fehrentz JA, Marie J, et al. Ligands and signaling proteins govern the conformational landscape explored by a G protein-coupled receptor. Proc Natl Acad Sci U S A. 2012;109:8304–8309. doi: 10.1073/pnas.1119881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol. 2008;153(Suppl. 1):S216–S229. doi: 10.1038/sj.bjp.0707490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrosinski J, Frimurer TM, Sivertsen B, Schwartz TW, Holst B. Modulation of constitutive activity and signaling bias of the ghrelin receptor by conformational constraint in the second extracellular loop. J Biol Chem. 2012;287:33488–33502. doi: 10.1074/jbc.M112.383240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulin A, Demange L, Berge G, Gagne D, Ryan J, Mousseaux D, et al. Toward potent ghrelin receptor ligands based on trisubstituted 1,2,4-triazole structure. 2. Synthesis and pharmacological in vitro and in vivo evaluations. J Med Chem. 2007;50:5790–5806. doi: 10.1021/jm0704550. [DOI] [PubMed] [Google Scholar]
- Mousseaux D, Le GL, Ryan J, Oiry C, Gagne D, Fehrentz JA, et al. Regulation of ERK1/2 activity by ghrelin-activated growth hormone secretagogue receptor 1A involves a PLC/PKCvarepsilon pathway. Br J Pharmacol. 2006;148:350–365. doi: 10.1038/sj.bjp.0706727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 1988;175:331–338. doi: 10.1111/j.1432-1033.1988.tb14201.x. [DOI] [PubMed] [Google Scholar]
- Murphy MG, Weiss S, McClung M, Schnitzer T, Cerchio K, Connor J, et al. Effect of alendronate and MK-677 (a growth hormone secretagogue), individually and in combination, on markers of bone turnover and bone mineral density in postmenopausal osteoporotic women. J Clin Endocrinol Metab. 2001;86:1116–1125. doi: 10.1210/jcem.86.3.7294. [DOI] [PubMed] [Google Scholar]
- Nagaya N, Uematsu M, Kojima M, Date Y, Nakazato M, Okumura H, et al. Elevated circulating level of ghrelin in cachexia associated with chronic heart failure: relationships between ghrelin and anabolic/catabolic factors. Circulation. 2001;104:2034–2038. doi: 10.1161/hc4201.097836. [DOI] [PubMed] [Google Scholar]
- Nickolls SA, Fleck B, Hoare SR, Maki RA. Functional selectivity of melanocortin 4 receptor peptide and nonpeptide agonists: evidence for ligand-specific conformational states. J Pharmacol Exp Ther. 2005;313:1281–1288. doi: 10.1124/jpet.105.083337. [DOI] [PubMed] [Google Scholar]
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hayre M, Salanga CL, Handel TM, Hamel DJ. Emerging concepts and approaches for chemokine-receptor drug discovery. Expert Opin Drug Discov. 2010;5:1109–1122. doi: 10.1517/17460441.2010.525633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palus S, Schur R, Akashi YJ, Bockmeyer B, Datta R, Halem H, et al. Ghrelin and its analogues, BIM-28131 and BIM-28125, improve body weight and regulate the expression of MuRF-1 and MAFbx in a rat heart failure model. PLoS ONE. 2011;6:e26865. doi: 10.1371/journal.pone.0026865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papotti M, Ghe C, Cassoni P, Catapano F, Deghenghi R, Ghigo E, et al. Growth hormone secretagogue binding sites in peripheral human tissues. J Clin Endocrinol Metab. 2000;85:3803–3807. doi: 10.1210/jcem.85.10.6846. [DOI] [PubMed] [Google Scholar]
- Park JG, Muise A, He GP, Kim SW, Ro HS. Transcriptional regulation by the gamma5 subunit of a heterotrimeric G protein during adipogenesis. EMBO J. 1999;18:4004–4012. doi: 10.1093/emboj/18.14.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Kniazeff J, Liu J, Binet V, Goudet C, Rondard P, et al. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005;272:2947–2955. doi: 10.1111/j.1742-4658.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A. 2012;109:6733–6738. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rediger A, Piechowski CL, Habegger K, Gruters A, Krude H, Tschop MH, et al. MC4R dimerization in the paraventricular nucleus and GHSR/MC3R heterodimerization in the arcuate nucleus: is there relevance for body weight regulation? Neuroendocrinology. 2012;95:277–288. doi: 10.1159/000334903. [DOI] [PubMed] [Google Scholar]
- Reiner S, Ambrosio M, Hoffmann C, Lohse MJ. Differential signaling of the endogenous agonists at the beta2-adrenergic receptor. J Biol Chem. 2010;285:36188–36198. doi: 10.1074/jbc.M110.175604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman JG, Kanemitsu-Parks M, Gaidarov I, Cameron JS, Griffin P, Zheng H, et al. Nicotinic acid receptor agonists differentially activate downstream effectors. J Biol Chem. 2007;282:18028–18036. doi: 10.1074/jbc.M701866200. [DOI] [PubMed] [Google Scholar]
- Rudolph J, Esler WP, O'Connor S, Coish PD, Wickens PL, Brands M, et al. Quinazolinone derivatives as orally available ghrelin receptor antagonists for the treatment of diabetes and obesity. J Med Chem. 2007;50:5202–5216. doi: 10.1021/jm070071+. [DOI] [PubMed] [Google Scholar]
- Sabbatini FM, Di FR, Corsi M, Cavanni P, Bromidge SM, St-Denis Y, et al. Discovery process and characterization of novel carbohydrazide derivatives as potent and selective GHSR1a antagonists. ChemMedChem. 2010;5:1450–1455. doi: 10.1002/cmdc.201000185. [DOI] [PubMed] [Google Scholar]
- Sakkou M, Wiedmer P, Anlag K, Hamm A, Seuntjens E, Ettwiller L, et al. A role for brain-specific homeobox factor Bsx in the control of hyperphagia and locomotory behavior. Cell Metab. 2007;5:450–463. doi: 10.1016/j.cmet.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Sato T, Nakashima Y, Nakamura Y, Ida T, Kojima M. Continuous antagonism of the ghrelin receptor results in early induction of salt-sensitive hypertension. J Mol Neurosci. 2011;43:193–199. doi: 10.1007/s12031-010-9414-1. [DOI] [PubMed] [Google Scholar]
- Schellekens H, van Oeffelen WE, Dinan TG, Cryan JF. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. J Biol Chem. 2013;288:181–191. doi: 10.1074/jbc.M112.382473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TW, Sakmar TP. Structural biology: snapshot of a signalling complex. Nature. 2011;477:540–541. doi: 10.1038/477540a. [DOI] [PubMed] [Google Scholar]
- Seta K, Nanamori M, Modrall JG, Neubig RR, Sadoshima J. AT1 receptor mutant lacking heterotrimeric G protein coupling activates the Src-Ras-ERK pathway without nuclear translocation of ERKs. J Biol Chem. 2002;277:9268–9277. doi: 10.1074/jbc.M109221200. [DOI] [PubMed] [Google Scholar]
- Sheikh SP, Zvyaga TA, Lichtarge O, Sakmar TP, Bourne HR. Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature. 1996;383:347–350. doi: 10.1038/383347a0. [DOI] [PubMed] [Google Scholar]
- Sivertsen B, Lang M, Frimurer TM, Holliday ND, Bach A, Els S, et al. Unique interaction pattern for a functionally biased ghrelin receptor agonist. J Biol Chem. 2011;286:20845–20860. doi: 10.1074/jbc.M110.173237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RG, Van der Ploeg LH, Howard AD, Feighner SD, Cheng K, Hickey GJ, et al. Peptidomimetic regulation of growth hormone secretion. Endocr Rev. 1997;18:621–645. doi: 10.1210/edrv.18.5.0316. [DOI] [PubMed] [Google Scholar]
- Spiegelberg BD, Hamm HE. G betagamma binds histone deacetylase 5 (HDAC5) and inhibits its transcriptional co-repression activity. J Biol Chem. 2005;280:41769–41776. doi: 10.1074/jbc.M504066200. [DOI] [PubMed] [Google Scholar]
- Stallaert W, Christopoulos A, Bouvier M. Ligand functional selectivity and quantitative pharmacology at G protein-coupled receptors. Expert Opin Drug Discov. 2011;6:811–825. doi: 10.1517/17460441.2011.586691. [DOI] [PubMed] [Google Scholar]
- Steen A, Thiele S, Guo D, Hansen LS, Frimurer TM, Rosenkilde MM. Biased and constitutive signaling in the CC-chemokine receptor CCR5 by manipulating the interface between transmembrane helices 6 and 7. J Biol Chem. 2013;288:12511–12521. doi: 10.1074/jbc.M112.449587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehno-Bittel L, Krapivinsky G, Krapivinsky L, Perez-Terzic C, Clapham DE. The G protein beta gamma subunit transduces the muscarinic receptor signal for Ca2+ release in Xenopus oocytes. J Biol Chem. 1995;270:30068–30074. doi: 10.1074/jbc.270.50.30068. [DOI] [PubMed] [Google Scholar]
- Stevanovic D, Janjetovic K, Misirkic M, Vucicevic L, Sumarac-Dumanovic M, Micic D, et al. Intracerebroventricular administration of metformin inhibits ghrelin-induced Hypothalamic AMP-kinase signalling and food intake. Neuroendocrinology. 2012;96:24–31. doi: 10.1159/000333963. [DOI] [PubMed] [Google Scholar]
- Swaminath G, Deupi X, Lee TW, Zhu W, Thian FS, Kobilka TS, et al. Probing the beta2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- Tang WJ, Gilman AG. Type-specific regulation of adenylyl cyclase by G protein beta gamma subunits. Science. 1991;254:1500–1503. doi: 10.1126/science.1962211. [DOI] [PubMed] [Google Scholar]
- Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50:707–709. doi: 10.2337/diabetes.50.4.707. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Crepieux P, Poupon A, Maurel MC, Reiter E. Novel pathways in gonadotropin receptor signaling and biased agonism. Rev Endocr Metab Disord. 2011;12:259–274. doi: 10.1007/s11154-011-9176-2. [DOI] [PubMed] [Google Scholar]
- Van der Ploeg LH, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, et al. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci U S A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez DA, Martinez G, Romero A, Vazquez MJ, Boit KD, Dopeso-Reyes IG, et al. The central Sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes. 2011;60:1177–1185. doi: 10.2337/db10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, et al. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579. doi: 10.1124/jpet.110.173005. [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, et al. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z, Serby MD, Zhao H, Kosogof C, Szczepankiewicz BG, Liu M, et al. Discovery and pharmacological evaluation of growth hormone secretagogue receptor antagonists. J Med Chem. 2006;49:4459–4469. doi: 10.1021/jm060461g. [DOI] [PubMed] [Google Scholar]
- Yang C, Wang Y, Liu H, Li N, Sun Y, Liu Z, et al. Ghrelin protects H9c2 cardiomyocytes from angiotensin II-induced apoptosis through the endoplasmic reticulum stress pathway. J Cardiovasc Pharmacol. 2012;59:465–471. doi: 10.1097/FJC.0b013e31824a7b60. [DOI] [PubMed] [Google Scholar]
- Ye S, Zaitseva E, Caltabiano G, Schertler GF, Sakmar TP, Deupi X, et al. Tracking G-protein-coupled receptor activation using genetically encoded infrared probes. Nature. 2010;464:1386–1389. doi: 10.1038/nature08948. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, et al. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci U S A. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Coso OA, Lee C, Gutkind JS, Simonds WF. Selective activation of effector pathways by brain-specific G protein beta5. J Biol Chem. 1996;271:33575–33579. doi: 10.1074/jbc.271.52.33575. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]