

Abstract
BACKGROUND AND PURPOSE
Angiotensin II has been implicated in the development of various cardiovascular ailments, including cardiac hypertrophy and heart failure. The fact that inhibiting its signalling reduced the incidences of both sudden cardiac death and heart failure in several large-scale clinical trials suggests that angiotensin II is involved in increased cardiac arrhythmogenicity during the development of heart failure. However, because angiotensin II also promotes structural remodelling, including cardiomyocyte hypertrophy and cardiac fibrosis, it has been difficult to assess its direct contribution to cardiac arrhythmogenicity independently of the structural effects.
EXPERIMENTAL APPROACH
We induced cardiac hypertrophy in wild-type (WT) and angiotensin II type 1a receptor knockout (AT1aR-KO) mice by transverse aortic constriction (TAC). The susceptibility to ventricular tachycardia (VT) assessed in an in vivo electrophysiological study was compared in the two genotypes. The effect of acute pharmacological blockade of AT1R on the incidences of arrhythmias was also assessed.
KEY RESULTS
As described previously, WT and AT1aR-KO mice with TAC developed cardiac hypertrophy to the same degree, but the incidence of VT was much lower in the latter. Moreover, although TAC induced an increase in tyrosine phosphorylation of connexin 43, a critical component of gap junctional channels, and a reduction in ventricular levels of connexin 43 protein in both genotypes, the effect was significantly ameliorated in AT1aR-KO mice. Acute pharmacological blockade of AT1R also reduced the incidence of arrhythmias.
CONCLUSIONS AND IMPLICATIONS
Our findings demonstrate that AT1aR-mediated signalling makes a direct contribution to the increase in arrhythmogenicity in hypertrophied hearts independently of structural remodelling.
Keywords: angiotensin, arrhythmia, hypertrophy, connexin
Introduction
Heart failure is a leading cause of mortality and morbidity in the Western world (McKinsey and Olson, 2005). Despite the recent progress in both medical and non-medical treatment, the prognosis for patients with heart failure remains poor, with a 5-year survival rate of only about 50% (Zannad et al., 1999; Levy et al., 2002). Furthermore, up to 50% of the deaths among heart failure patients are sudden and unexpected, and are presumed to be the result of lethal arrhythmias (Tomaselli and Marban, 1999). For that reason, a fuller understanding of the molecular mechanisms underlying the increased arrhythmogenicity seen in failing hearts would be highly desirable.
Heart failure is often preceded by pathological enlargement of the heart due to cardiac muscle cell hypertrophy (Frey and Olson, 2003). Indeed, cardiac hypertrophy is a major risk factor for heart failure (Lauer et al., 1999; Kannel, 2000; Vakili et al., 2001; Devereux et al., 2004; Gardin and Lauer, 2004; Okin et al., 2004) and is also a risk factor for ventricular arrhythmias and sudden cardiac death (Levy et al., 1987; Haider et al., 1998). This suggests that there are shared pathways linking cardiac hypertrophy, heart failure and arrhythmias. Recent data showing a close association between ventricular hypertrophy and increased susceptibility to arrhythmia in a genetically engineered mouse model of hypertrophic cardiomyopathy support this notion (Wolf et al., 2005). This increased susceptibility to arrhythmia reflects electrical remodelling that includes changes in the function and expression of membrane ion channels, gap junction proteins and Ca2+-cycling proteins, and structural remodelling that includes alterations in composition of the extracellular matrix, that predisposes the hypertrophied ventricle to arrhythmogenic events such as early and delayed afterdepolarization and re-entry (Tomaselli and Marban, 1999).
Angiotensin II has been implicated in the development and progression of various cardiovascular diseases, including cardiac hypertrophy and heart failure. A number of large-scale randomized clinical trials have shown that reducing angiotensin II signalling by inhibiting ACE or blocking angiotensin type 1 receptor improves the prognosis and reduces disease severity in patients with heart failure (Howard et al., 2006). Inhibiting angiotensin II signalling also reportedly reduces the incidences of both heart failure and sudden cardiac death (Kober et al., 1995; Cleland et al., 1997). Moreover, several studies of genetically modified animal models have clearly demonstrated the arrhythmic potential of angiotensin II signalling (Xiao et al., 2004; Domenighetti et al., 2007; Fischer et al., 2007). However, because angiotensin II signalling also plays an important role in the structural remodelling (e.g. cardiomyocyte hypertrophy and cardiac fibrosis) seen during the development and progression of cardiac hypertrophy or cardiomyopathy, it has been difficult to obtain clear evidence of a direct contribution by angiotensin II signalling to the increase in arrhythmogenicity independently of structural remodelling.
Our aim in the present study was therefore to clarify the direct contribution of angiotensin II type1a receptor (AT1aR)-mediated signalling to the increase in arrhythmogenicity seen during cardiac remodelling. To accomplish this, we first induced cardiac hypertrophy in wild-type (WT) and angiotensin II type 1a receptor knockout (AT1aR-KO) mice by subjecting their hearts to chronic pressure overload caused by transverse aortic constriction (TAC) (Harada et al., 1998b). We then carried out an in vivo electrophysiological study to compare arrhythmogenicity in the two genotypes. We also assessed the inhibitory effect of acute pharmacological blockade of AT1R on inducible arrhythmias in mice with TAC and the molecular mechanism by which AT1aR-mediated signalling makes a direct contribution to the electrical remodelling. Our results demonstrate that AT1aR-mediated signalling can make a direct contribution to the increase in arrhythmogenicity in hypertrophied hearts independently of structural remodelling.
Methods
Animal preparations
The animal care and all experimental protocols were reviewed and approved by the Animal Research Committee at the Kyoto University Graduate School of Medicine. AT1aR-KO (AT1aR−/−) mice (C57BL/6 background) were kindly provided by Dr Fukamizu (The University of Tsukuba) (Sugaya et al., 1995). WT C57BL/6 littermates served as controls.
Transverse aortic constriction
Eight- to ten-week-old mice were anaesthetized, intubated and artificially ventilated as previously described (Izumi et al., 2001). Pressure overload was then induced by TAC (Hill et al., 2000; Nakagawa et al., 2006). Briefly, the chest cavity was opened in the second intercostal space, at the left sternal border, to expose the aortic arch. The segment of aortic arch between the right innominate and left carotid arteries was then constricted using a 7-0 silk suture tied firmly against a 27-gauge needle. The needle was subsequently removed, leaded to approximately 70% constriction. Sham-operated mice underwent the identical surgical procedure, except the aortic arch was not constricted. Four weeks after the sham or TAC operation, mice were killed by cervical dislocation and excised hears were used for further analyses.
Echocardiography
Echocardiography was carried out before (baseline) and 4 weeks after surgery using an echocardiographic system (Toshiba Power Vision 8000) equipped with a 12 MHz imaging transducer as described previously (Adachi et al., 2003). Mice were anaesthetized with spontaneous respiration by i.p. injection of tribromoethanol/amylene hydrate (avertin) 2.5% w/v solution (8 L·g−1). We measured left ventricular (LV) end-diastolic dimension (LVDd), LV posterior wall (LVPW) thickness and percent fractional shortening (%FS).
Histological examination
Hearts were fixed in 10% formalin and prepared for histological analysis as described previously (Li et al., 2002). To quantitate the myocardial fibrosis, we determined the ratios of the Sirius red-stained area to the total area. The data were then shown as % fibrosis, as previously reported (Li et al., 2002).
Quantitative RT-PCR analysis
Using 50 ng of total RNA prepared from ventricles, levels of mouse ANP, BNP, Acta1, SERCA2, TGF-β1, collagen 1, fibronectin, CTGF, SCN5A, CACNA1c, KCND2, Kchip2, KCNJ2, KCNH2, KCNJ11 and GAPDH mRNA were determined using quantitative real-time PCR according to the manufacture's protocol (Applied Biosystems, Zaventam, Belgium). The sequences of the forward (F) and reverse (R) primers and of the probes (P) with fluorescent dye (FAM) and quencher (TAMRA) for ANP, BNP, Acta1, SERCA2, TGF-β1 and collagen 1 were reported previously (Li et al., 2002; Kuwahara et al., 2003). The primers and probes for fibronectin, CTGF, SCN5A, CACNA1c, KCND2, Kchip2, KCNJ2, KCNH2, KCNJ11 and GAPDH were purchased from Applied Biosystems.
Intracardiac electrophysiology
The mice underwent intracardiac electrophysiological examination 4 weeks after TAC or sham operation. They were initially anaesthetized with ether and placed on a warm pad maintained at 37°C. The trachea of each mouse was then cannulated with a polyethylene tube, and the mice were respirated using a rodent respirator (Shinano Co., Tokyo, Japan) with the tidal volume set at 0.9 mL and the respiration rate set at 110 min−1. The mice were anaesthetized with 0.5–1.5% isoflurane for the remainder of the surgical procedure. A surface frontal plane 6-lead ECG was obtained using clips placed on each limb, and a midline cervical incision was made to expose the right jugular vein. Using a jugular vein cutdown approach, a catheter (2.0F octapolar catheter with inter-electrode spacing of 0.5 mm, CIBer mouse EP; NuMed, Hopkinton, NY, USA) was blindly placed into the right ventricle (RV). We confirmed the proper positioning of the catheter at the end of the experiment. A standard electrophysiology protocol was performed as described previously (Gehrmann and Berul, 2000; Kuwahara et al., 2003). Rapid RV pacing using the extra stimulation (S1S2) technique with 1–3 extra stimuli was performed to determine the RV refractory period and to attempt induction of ventricular arrhythmias: 10 pacing stimuli (S1; a coupling interval is 80–90 ms) followed by 2–3 extra stimulations (S2-4) were used to induce VT in experiments to see effects of TAC on WT and AT1aR-KO, and 10 pacing stimuli (S1; a coupling interval is 80 ms) followed by 2 extra stimulations (S2, S3) were used to induce VT in experiments to see effects of EXP-3174. Between these two slightly different protocols, VT induction rates in WT mice subjected to TAC were not significantly different (Tables 1 and 2). Note that the similar protocol is commonly used in electrophysiological studies with humans. The stimulation was applied at twice the ventricular diastolic capture threshold. VT was defined as an induction of three or more consecutive premature ventricular contractions. The operator who performed electrophysiological study was blinded to the genotype and procedure status of the experimental animals. In some experiments, 0.3 mg·kg−1 EXP-3174, an AT1R-blocking losartan metabolite, or vehicle was i.v. administered 60 min before the examination. We used EXP-3174 because it is the active carboxylic acid metabolite of losartan, exerts prompt pharmacological action, and has more potent and selective AT1R blocking activity than losartan (Chang et al., 1995).
Table 1.
VT inducibility in WT and AT1aR-KO mice subjected to TAC or sham operation
| Total number of VT(+) mice | Induction rate of VT | |
|---|---|---|
| WT-Sham (n = 15) | 0 | 0% |
| KO-Sham (n = 13) | 0 | 0% |
| WT-TAC (n = 15) | 11 | 73.3% |
| KO-TAC (n = 13) | 4 | 30.8%* |
VT, ventricular tachyarrhythmia; WT-Sham, wild-type mice subjected to sham operation; KO-Sham, AT1aR-KO mice subjected to sham operation; WT-TAC, wild-type mice subjected to TAC; KO-TAC, AT1aR-KO mice subjected to TAC.
P < 0.05 versus WT-TAC.
Table 2.
Heart weight-to-body weight ratio, lung weight-to-body weight ratio and VT inducibility in WT mice subjected to TAC for 4 weeks then treated with either vehicle or EXP-3174
| Vehicle | EXP-3174 | |
|---|---|---|
| Body weight (g) | 27.7 ± 0.1 | 27.4 ± 0.2 |
| Heart weight (mg) | 211.1 ± 5.6 | 213.4 ± 6.7 |
| Lung weight (mg) | 226.1 ± 3.9 | 241.9 ± 3.9 |
| HW/BW (mg·g−1) | 7.62 ± 0.23 | 7.82 ± 0.26 |
| LW/BW (mg·g−1) | 8.18 ± 1.43 | 8.87 ± 1.43 |
| Total number of VT (+) mice | 8 | 3 |
| Induction rate of VT (%) | 73 | 25* |
HW/BW, heart weight-to-body weight ratio; LW/BW, lung weight-to-body weight ratio; vehicle, WT mice treated acutely with vehicle 4 weeks after TAC; EXP-3174, WT mice treated acutely with EXP-3174 4 weeks after TAC; VT, ventricular tachyarrhythmia; TAC, transverse aortic constriction. EXP-3174 (0.3 mg·kg−1) was given intravenously 1 h prior to the EPS study.
n = 11 in the vehicle group and 12 in the EXP-3174 group.
P < 0.05 versus control vehicle.
Immunoprecipitation and Western analyses
Details of the methods used to prepare lysates from the ventricles of sham- and TAC-operated mice, as well as those used to carry out co-immunoprecipitation assays, were described previously (Nakagawa et al., 2006). Briefly, frozen samples of the ventricle were homogenized and sonicated in lysis buffer containing 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg·mL−1 leupeptin and 1 mM phenylmethylsulfonyl fluoride (Cell Signaling Technology, Inc., Danvers, MA, USA). After the debris was cleared by centrifugation, the protein concentration in the supernatant was quantified as the crude extract. For immunoprecipitation, 1 mg of extract was mixed with anti-connexin43 (Cx43) antibodies (1:50 dilution; Santa Cruz Biotechnology, Dallas, TX, USA) and protein A/G-PLUS-agarose (Santa Cruz) and then incubated overnight at 4°C. Precipitated proteins were separated by SDS-PAGE using 10% polyacrylamide gels and then analysed by Western blotting using anti-phosphotyrosine (1:200; Santa Cruz) and anti-Cx43 (1:1000; Santa Cruz) as probes. In addition, extract containing 50 μg of protein was used for Western blotting with anti-Cx43 (1:1000; Santa Cruz) or anti-β-actin (1:1000; Sigma-Aldrich, St. Louis, MO, USA) antibody as Input to control for variation in levels of protein expression. In another experiment, extracts containing 50 μg of protein were separated by SDS-PAGE using 15% polyacrylamide gels and then analysed by Western blotting using anti-Tyr265-phosphorylated Cx43 (1:1000; Santa Cruz), anti-Cx43 (1:1000; Santa Cruz) or anti-GAPDH (1:1000; Merck Millipore, Billerica, MA, USA) antibody.
Statistical Analysis
Data are presented as means ± SEM. We compared continuous variables using Student's t-test. Frequency analysis was performed using chi square test. anova with post hoc Fisher's protected least significant difference test was used for comparison among groups. Values of P < 0.05 were considered significant.
Results
TAC induced comparable cardiac hypertrophy in AT1aR-KO and WT mice
An earlier report showed that, in response to chronic pressure overload created by abdominal aortic banding, AT1aR-KO mice developed cardiac hypertrophy just as WT mice did (Harada et al., 1998b). With that in mind, we aimed to use these mice to examine the role of AT1aR in the electrical remodelling seen during cardiac hypertrophy induced by chronic pressure overload caused by TAC. Echocardiographic examination revealed that before TAC or the sham operation, there were no significant differences in LVDd, LVPW thickness or %FS among the four groups studied (Figure 1A–C). However, LVPW thickness was significantly higher in both AT1aR-KO and WT mice 4 weeks after TAC than before it (0.61 ± 0.01 mm → 0.82 ± 0.02 mm and 0.63 ± 0.01 mm → 0.86 ± 0.02 mm, respectively, P < 0.01). As previously reported, there was no difference between the heart weight-to-body weight ratios in AT1aR-KO and WT mice following either TAC or sham operation (AT1aR-KO with sham, 4.3 ± 0.05 mg·g−1; AT1aR-KO with TAC, 5.7 ± 0.22 mg·g−1; WT with sham, 4.6 ± 0.06 mg·g−1; WT with TAC, 6.1 ± 0.25 mg·g−1) (Figure 1D). Likewise, expression of hypertrophic and fibrotic marker genes induced in response to the chronic pressure overload was quite similar in AT1aR-KO and WT mice (Figures 2A–D and 3A–D), and histological analysis showed that pressure overload induced similar degrees of cardiac fibrosis in AT1aR-KO and WT mice (Figure 3E and F), which is consistent with the notion that AT1aR-mediated signalling is dispensable for the structural remodelling induced by TAC. Angiotensin type 2 receptor (AT2R) expression did not significantly differ between AT1aR-KO and WT mice, with or without TAC, as reported previously (data not shown here) (Harada et al., 1998b).
Figure 1.
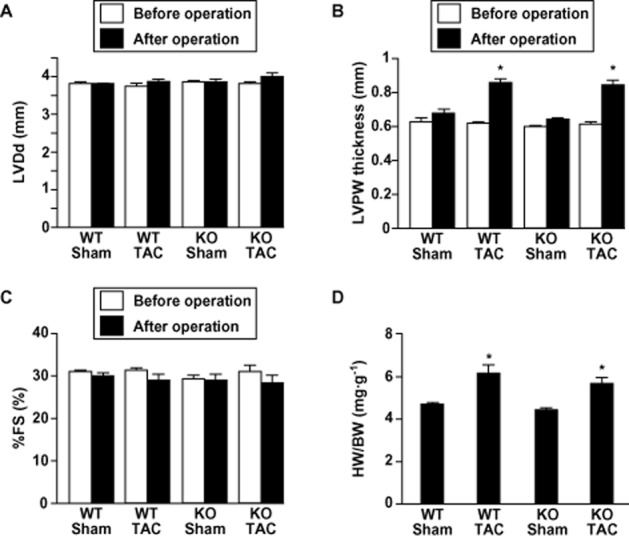
Angiotensin II type 1a receptor knockout (AT1a-KO) and wild-type (WT) mice develop left ventricular hypertrophy to a similar degree after transverse aortic constriction (TAC). Left ventricular (LV) end-diastolic dimension (LVDd) (A), left ventricular posterior wall ( LVPW) thickness (B) and percent fractional shortening (%FS) (C) evaluated by echocardiography before and after TAC or sham operation. Values are means ± SEM. *P < 0.01 versus before each operation. (D) Heart weight-to-body weight (HW/BW) ratios 4 weeks after the operation. Values are means ± SEM. *P < 0.01 versus sham-operated mice in each genotype. WT-Sham, wild-type mice subjected to sham operation (n = 37); KO-Sham, AT1aR-KO mice subjected to sham operation (n = 38); WT-TAC, wild-type mice subjected to TAC (n = 45); KO-TAC, AT1aR-KO mice subjected to TAC (n = 35).
Figure 2.
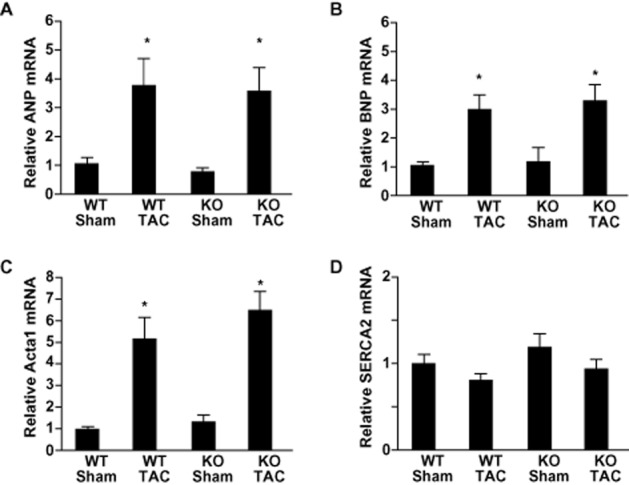
Effects of chronic pressure overload on left ventricular expression of hypertrophic marker genes 4 weeks after transverse aortic constriction (TAC) or sham operation. Relative expression levels of ANP (A), BNP (B), Acta1 (C) and SERCA2 (D) mRNA were normalized to the corresponding GAPDH mRNA levels. The mean relative level of each mRNA in sham-operated wild-type (WT) mice was assigned a value of 1.0. Values are means ± SEM (n = 8 each). *P < 0.01 versus sham-operated mice of each genotype.
Figure 3.
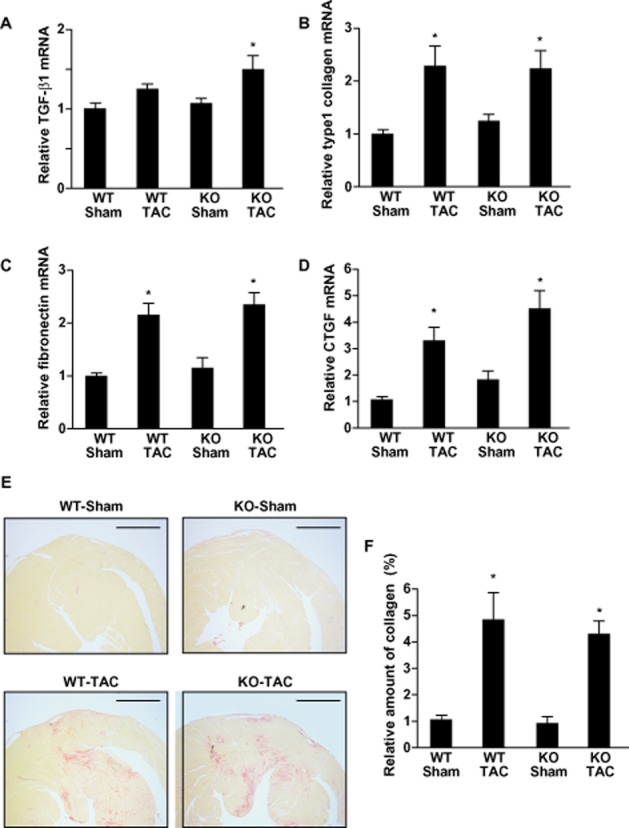
Effects of chronic pressure overload on cardiac fibrosis in wild-type (WT) and angiotensin II type 1a receptor knockout (AT1aR-KO) mice. (A–D) Left ventricular expression of fibrotic marker genes 4 weeks after transverse aortic constriction (TAC) or sham operation. Relative expression levels of TGF-β1 (A), type 1 collagen (B), fibronectin (C) and CTGF (D) mRNA were normalized to corresponding GAPDH mRNA levels. The mean relative level of each mRNA in sham-operated WT mice was assigned a value of 1.0. Values are means ± SEM (n = 8 each). *P < 0.01 versus sham-operated mice of each genotype. (E) Sirius red-stained sections of left ventricle from the both AT1aR-KO and WT mice processed 4 weeks after TAC or sham operation. Scale bars are 1 mm. (F) The relative fibrotic areas in left ventricles from AT1aR-KO and WT mice 4 weeks after TAC or sham operation. The mean value of the relative fibrotic area in left ventricles from sham-operated WT mice was assigned a value of 1.0. Values are means ± SEM (n = 8 each). *P < 0.01 versus sham-operated mice of each genotype.
Diminished induction of ventricular tachycardia in AT1aR-KO mice
Given that AT1aR-KO mice and their WT littermates showed similar structural remodelling in response to chronic pressure overload, we next compared the electrical properties in the two genotypes. To accomplish this, 4 weeks after the sham operation or TAC, we inserted an octapolar 2.0F catheter into the RV via the right jugular vein and assessed the inducibility of ventricular tachycardia (VT) in the mice (Table 1). Figure 4A shows a representative polymorphic VT induced by programmed ventricular stimulation. There was no induction of VT in sham-operated AT1aR-KO or WT mice. Meanwhile, VT was induced in 11 of 15 (73.3%) WT mice subjected to TAC, but in only 4 of 13 (30.8%) AT1aR-KO mice with TAC. Thus, AT1aR deficiency had a significant (P < 0.05) protective effect against induction of VT in hypertrophied hearts.
Figure 4.
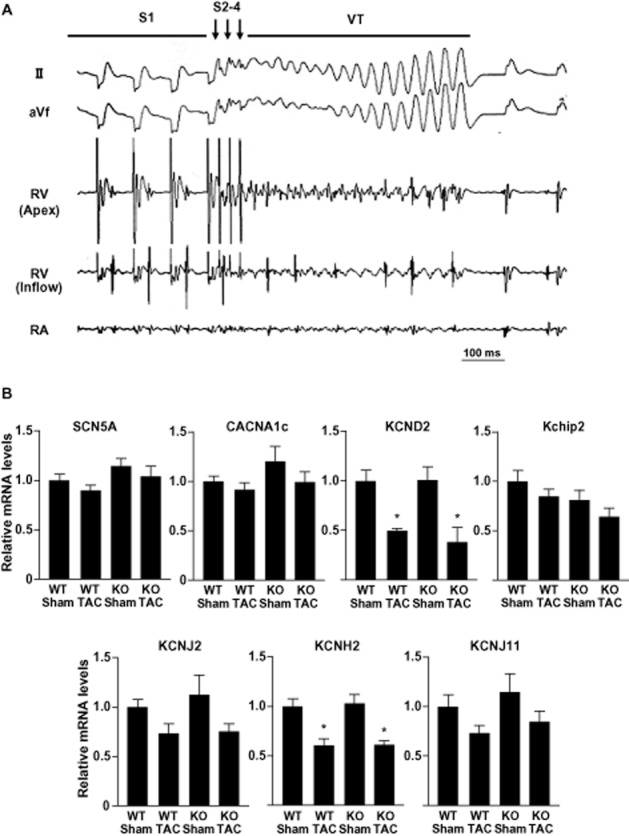
Induction of ventricular arrhythmias in mice with chronic pressure overload. (A) Representative traces showing polymorphic ventricular tachycardia (VT) induced by programmed ventricular stimulation in a mouse subjected to transverse aortic constriction (TAC). RV, right ventricle; RA, right atrium. Ten pacing stimuli (S1; a coupling interval is 80–90 ms) followed by 2–3 extra stimulations (S2-4) were used to induce VT. (B) Effects of chronic pressure overload on left ventricular ion channel gene transcription 4 weeks after TAC or sham operation. Graphs show relative expression levels of SCN5A, CACNA1c, KCND2, Kchip2, KCNJ2, KCNH2 and KCNJ11 mRNA normalized to corresponding GAPDH mRNA levels. The mean relative level of each mRNA in sham-operated WT mice was assigned a value of 1.0. Values are means ± SEM (n = 8 each). *P < 0.01 versus sham-operated mice in each genotype.
Comparable ion channel gene expression in AT1aR-KO and WT mice
The ventricular expression of genes encoding ion channels has been shown to be altered during cardiac hypertrophy (Tomaselli and Marban, 1999). To test whether a difference in ion channel gene expression in the ventricular myocardium contributes to the difference in susceptibility to VT seen in AT1aR-KO and WT mice with TAC, we used real-time PCR to compare the transcription levels of several ion channel genes in the two genotypes, including those responsible for the INa (SCN5A), ICa-L (CACNA1C), ITO (KCND2 and Kchip2), IK1 (KCNJ2), IKr (KCNH2) and IK-ATP (KCNJ11). We found no significant difference in the levels of any of the corresponding mRNAs between AT1aR-KO and WT mice subjected to TAC or sham operation (Figure 4B).
Attenuated tyrosine phosphorylation and preserved levels of connexin 43 protein in AT1aR-KO ventricles
Gap junctions are clusters of intercellular channels that mediate electrical communication between adjacent cells. Connexin 43(Cx43) is the predominant ventricular gap junction protein, and under various conditions, it is phosphorylated at multiple serine residues by PKC and MAPK, and at two tyrosine residues by c-src (Lampe and Lau, 2004). Phosphorylation of Cx43 affects its tissue level, intracellular distribution and function (Toyofuku et al., 2001; Warn-Cramer and Lau, 2004). The changes in gap junctions caused by Cx43 phosphorylation likely contribute to an overall reduction in conduction velocity and increased dispersion of action potential duration and refractory properties, which combine to form the substrate for potentially lethal arrhythmias (Gutstein et al., 2001; Danik et al., 2004; Poelzing and Rosenbaum, 2004; van Rijen et al., 2004; King et al., 2013). Indeed, a recent study showed the relationship between reduced conduction velocity and arrhythmogenicity in mouse hearts (King et al., 2013). Mice with cardiac-restricted deletion of Cx43 showed slow conduction velocity in the heart, resulting in sudden arrhythmic death (Gutstein et al., 2001). Furthermore, angiotensin II signalling reportedly activates kinases that modulate Cx43 phosphorylation (Sadoshima and Izumo, 1996; Zou et al., 1998; Lampe and Lau, 2004; Sovari et al., 2011). Particularly, it has been recently reported that angiotensin II induces c-src TK-mediated remodelling of Cx43, which leads to the increase in sudden arrhythmic death (Sovari et al., 2011). We therefore examined the tyrosine phosphorylation status and protein amount of Cx43 in TAC- and sham-operated AT1aR-KO and WT mice. We observed prominent tyrosine phosphorylation of Cx43 and a substantial reduction in tissue Cx43 protein levels in WT mice subjected to TAC (Figure 5A and B). Both the TAC-induced increase in tyrosine phosphorylation of Cx43 (Figure 5A and B) and the reduction in the levels of Cx43 protein were diminished in AT1aR-KO mice (Figure 5A and C). AT1aR-mediated signalling thus appears to play a key role in a pressure overload-induced increase in the tyrosine phosphorylation of Cx43 that leads to diminished tissue levels of Cx43 protein (Toyofuku et al., 2001). It may be that the preservation of Cx43 underlies the reduced arrhythmogenicity of chronic pressure overload in AT1aR-KO mice.
Figure 5.

Phosphorylation status and tissue level of Cx43 protein in the left ventricles of angiotensin II type 1a receptor knockout (AT1aR-KO) and wild-type (WT) mice 4 weeks after transverse aortic constriction (TAC) or sham operation. (A) Protein obtained from left ventricles was immunoprecipitated (IP) with anti-Cx43 antibody. The panels show the immunocomplexes separated by electrophoresis and blotted with the indicated antibodies (Blot). Crude lysates were analysed by Western blotting to control for variation in protein expression (Input). (B) The ratios of phospho-Cx43 to total Cx43 in immunoprecipitation assays evaluated by quantitative densitometry. The mean value of the phospho-Cx43/total Cx43 ratios in sham-operated WT mice was assigned a value of 1.0. Values are means ± SEM (n = 3 each). *P < 0.05. (C) The results of quantitative densitometric analysis of left ventricular Cx43. The mean relative levels (corrected by β-actin level) of total Cx43 in sham-operated WT mice was assigned a value of 1.0. Values are means ± SEM (n = 3 each). *P < 0.05.
Acute blockade of AT1R signalling decreased induced ventricular tachyarrhythmias in mice with chronic pressure overload
Finally, we tested whether acute pharmacological blockade of AT1R signalling would decrease tachyarrhythmias induced in mice with chronic pressure overload. We compared the effects of vehicle to those of 0.3 mg·kg−1 EXP-3174, an active metabolite of the AT1R blocker losartan, which reduced systolic BP as much as 10.2 mmHg in WT mice (Figure 6A). Intravenous administration of EXP-3174 or vehicle to mice after 4 weeks of TAC did not significantly affect heart rates 60 min after administration under anaesthesia (Figure 6B). In addition, cardiac hypertrophy evaluated based on heart weight-to-body weight ratios was similar in the two groups (Figure 6C and Table 2). Meanwhile, the induction rate of VT in the mice acutely administered EXP-3174 was significantly lower than in the mice administered vehicle (Figure 6D and Table 2). The CX43 protein level was decreased in mice subjected to TAC with vehicle treatment (Figure 6E, left lane), compared with sham-operated mice with vehicle treatment (Figure 6, right lane). Acute EXP-3174 treatment restored the decreased CX43 protein level in mice subjected to TAC (Figure 6E, middle lane). The relative amount of Tyr265–phosphorylated Cx43 to total Cx43 was tended to be increased in mice subjected to TAC with vehicle treatment (Figure 6E, left lane) compared with that in sham-operated mice (Figure 6E, right lane) and that in mice subjected to TAC with EXP-3174 treatment (Figure 6E, middle lane). The result is consistent with the results obtained in the experiments using AT1aR-KO mice and suggests that acute EXP-3174 treatment affected tyrosine phosphorylation of Cx43 and then restored Cx43 protein levels, thereby reducing the increased arrhythmogenicity in mice subjected to TAC. These results suggest that acute inhibition of AT1R-mediated signalling can provide a significant protective effect against induction of VT in hypertrophied hearts, and further supports our hypothesis that AT1R-mediated signalling directly modulates arrhythmogenicity in hypertrophied ventricles.
Figure 6.

Effects of acute pharmacological blockade of AT1R signalling on the inducibility of ventricular tachycardia (VT) in mice with chronic pressure overload. (A) Systolic BP before (Pre) or 60 min after injection of control wild-type (WT) mice with vehicle or 0.3 mg·kg−1 of EXP-1374 (n = 6 each). *P < 0.05. (B) Heart rates recorded after 4 weeks of transverse aortic constriction (TAC) in mice treated with vehicle (n = 11) or EXP-3174 (n = 12) under anaesthesia. (C) Heart weight-to-body weight (HW/BW) ratios after 4 weeks of TAC in mice treated with vehicle (n = 11) or EXP-3174 (n = 12) treatment. (D) Representative traces recorded during programmed ventricular stimulation in a mouse subjected to 4 weeks of TAC and then acutely injected with vehicle (left panel) or EXP-3174 (right panel). RV, right ventricle; VT, ventricular tachyarrhythmias. Ten pacing stimuli (S1; a coupling interval is 80 ms) followed by 2 extra stimulations (S2, S3) were used to induce VT. (E) Representative Western blots showing Tyr265-phosphorylated Cx43, total Cx43 and GAPDH protein in a mouse subjected to 4 weeks of TAC or sham-operation and then acutely injected with vehicle (Sham-vehicle and TAC-vehicle) or EXP-3174 (TAC-EXP). Protein obtained from left ventricles was separated by electrophoresis and blotted with the indicated antibodies (Blot). Two different experiments gave essentially identical results.
Discussion and conclusions
The evidence suggests that angiotensin II signalling contributes to adverse electrical remodelling in patients with heart failure. It has been shown that angiotensin II signalling plays an important role in the structural remodelling of the heart (e.g. cardiomyocyte hypertrophy and cardiac fibrosis), which can contribute to the increase in arrhythmogenicity, during the development and progression of cardiomyopathy. However, it remained unclear whether this signalling makes a direct contribution to the altered electrical properties that increase cardiac arrhythmogenicity independently of structural remodelling. In the present study, we compared the susceptibility to arrhythmia of WT and AT1aR-deficient mice following induction of cardiac hypertrophy through chronic pressure overload. We found that AT1aR-KO mice with TAC developed cardiac hypertrophy in exactly the same manner as WT mice with TAC, as was seen previously in a study of AT1aR-KO mice subjected to abdominal aortic banding (Harada et al., 1998b). Nevertheless, induction of VT by programmed stimulation was significantly diminished in AT1aR-KO mice, as compared to WT mice. The overall ventricular levels of several ion channel genes, including those responsible for the INa, ICa-L, ITO, IK1, IKr and IK-ATP, were comparable in the two genotypes, although the protein levels of these ion channels and their spatial distribution throughout the ventricles were not assessed in detail. We found that there was significantly less tyrosine phosphorylation of Cx43 in AT1aR-KO than WT mice subjected to TAC, and that levels of Cx43 protein were better preserved in AT1aR-KO mice, which would be expected to ameliorate the functional deterioration of junctional conductance caused by loss of Cx43. Collectively, these results demonstrate that AT1aR-mediated signalling makes a direct contribution to the increase in arrhythmogenicity in hypertrophied hearts independently of structural remodelling, in addition to its potential effects on the increased susceptibility to arrhythmias by promoting structural remodelling.
Harada et al. (1998a) previously showed that the incidence of arrhythmias is lower in AT1aR-KO than WT mice following ischaemia-reperfusion. Although the mechanism involved in reperfusion-induced arrhythmias, which occur in an acute setting, may differ from the one underlying the arrhythmogenicity induced by chronic pressure overload, there is the possibility that a common molecular mechanism underlies the anti-arrhythmogenic effects of AT1aR inhibition under both acute and chronic pathological conditions. In this regard, the observed reduction in tyrosine phosphorylation of ventricular Cx43 in AT1aR-KO mice is intriguing. That acute blockade of AT1R signalling by EXP-3174, an active metabolite of AT1R blocker losartan, significantly reduced the induction rate of VT in mice with chronic pressure overload supports this notion. Lynch et al. (1999) also reported the acute anti-arrhythmic effect of EXP-3174 in canine model of acute myocardial ischaemia. In this report, however, the authors could not preclude the possible existence of AT1R-independent mechanisms underlying the anti-arrhythmic effect of EXP-3174 (Lynch et al., 1999). Further studies are necessary to assess the possible contribution of AT1R-independent mechanism to the anti-arrhythmic effect of EXP-3174 observed in this study. In addition, EXP-3174 reduced systolic BP as much as 10.2 mmHg in WT mice (Figure 6A). There is another possibility that acute reduction of systemic BP may influence the effect of EXP-3174 on mice subjected to TAC.
Phosphorylation of Cx43 affects the function of gap junctions (Warn-Cramer and Lau, 2004) largely by facilitating the degradation of Cx43, which leads to functional deterioration of the junction (Saffitz et al., 1999; Toyofuku et al., 2001; Warn-Cramer and Lau, 2004). Such gap junctional dysfunction is thought to be involved in the increased arrhythmogenicity seen in models of chronic cardiac hypertrophy and heart failure (Danik et al., 2004; Poelzing and Rosenbaum, 2004; van Rijen et al., 2004). Altered Cx43 phosphorylation is also reportedly involved in the increased arrhythmogenicity seen after ischaemia-reperfusion, although the precise molecular mechanisms linking Cx43 phosphorylation and arrhythmogenicity appear to differ in acute and chronic disease models. Nonetheless, Cx43 may be the common target of AT1aR-mediated signalling leading to increased arrhythmogenicity in both acute and chronic pathological conditions. Consistent with that idea, cardiac overexpression of ACE in mice was shown to reduce Cx43 expression via c-src TK-mediated pathways and increase susceptibility to cardiac arrhythmias and sudden death in the absence of structural remodelling (Xiao et al., 2004; Kasi et al., 2007; Sovari et al., 2011). In addition, transgenic rats overexpressing human renin and angiotensinogen also died from lethal arrhythmias, and Cx43 disorganization was detected in the ventricles of those rats (Fischer et al., 2007). All of these findings implicate angiotensin II signalling in post-translational modification of Cx43 that increases the arrhythmogenicity of diseased hearts.
Acknowledgments
We thank Ms Y. Kubo for her excellent secretarial works. We also thank Ms A. Fujishima and Ms A. Abe for their excellent technical assistance. EXP-3174 was kindly provided from Merck & Co., Inc.
This research was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; a grant from the Japanese Ministry of Health (to K. K., H. K. and K. N.), Labor and Welfare (to N. K.); a Japan Heart Foundation/Pfizer Pharmaceuticals Inc. Grant on Cardiovascular Disease Research; a Japan Heart Foundation/Novartis Grant for Research Award on Molecular and Cellular Cardiology; the grant for scientific research from MSD K.K. and grants from the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Uehara Memorial Foundation, the Astellas Foundation for Research on Metabolic Disorders, the Mitsubishi Foundation, the Suzuken Memorial Foundation, the Kanae Foundation for the Promotion of Medical Science, the Takeda Medical Research Foundation, the Takeda Science Foundation, the Hoh-ansha Foundation and the SENSHIN Medical Research Foundation (to K. K.).
Glossary
- WT
wild-type
- AT1aR
angiotensin II type 1a receptor
- AT1aR-KO
angiotensin II type 1a receptor knockout
- TAC
transverse aortic constriction
- LV
left ventricular
- LVDd
left ventricular end-diastolic dimension
- LVPW
left ventricular posterior wall
- %FS
percent fractional shortening
- RV
right ventricle
- Cx43
connexin43
- AT2R
angiotensin type 2 receptor
- VT
ventricular tachycardia
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Adachi Y, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, et al. Angiotensin II type 2 receptor deficiency exacerbates heart failure and reduces survival after acute myocardial infarction in mice. Circulation. 2003;107:2406–2408. doi: 10.1161/01.CIR.0000072763.98069.B4. [DOI] [PubMed] [Google Scholar]
- Chang RS, Lotti VJ, Chen TB, O'Malley SS, Bendesky RJ, Kling PJ, et al. In vitro pharmacology of an angiotensin AT1 receptor antagonist with balanced affinity for AT2 receptors. Eur J Pharmacol. 1995;294:429–437. doi: 10.1016/0014-2999(95)00563-3. [DOI] [PubMed] [Google Scholar]
- Cleland JG, Erhardt L, Murray G, Hall AS, Ball SG. Effect of ramipril on morbidity and mode of death among survivors of acute myocardial infarction with clinical evidence of heart failure. A report from the AIRE study investigators. Eur Heart J. 1997;18:41–51. [PubMed] [Google Scholar]
- Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–2356. doi: 10.1001/jama.292.19.2350. [DOI] [PubMed] [Google Scholar]
- Domenighetti AA, Boixel C, Cefai D, Abriel H, Pedrazzini T. Chronic angiotensin II stimulation in the heart produces an acquired long QT syndrome associated with IK1 potassium current downregulation. J Mol Cell Cardiol. 2007;42:63–70. doi: 10.1016/j.yjmcc.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Fischer R, Dechend R, Gapelyuk A, Shagdarsuren E, Gruner K, Gruner A, et al. Angiotensin II-induced sudden arrhythmic death and electrical remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H1242–H1253. doi: 10.1152/ajpheart.01400.2006. [DOI] [PubMed] [Google Scholar]
- Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- Gardin JM, Lauer MS. Left ventricular hypertrophy: the next treatable, silent killer? JAMA. 2004;292:2396–2398. doi: 10.1001/jama.292.19.2396. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Berul CI. Cardiac electrophysiology in genetically engineered mice. J Cardiovasc Electrophysiol. 2000;11:354–368. doi: 10.1111/j.1540-8167.2000.tb01806.x. [DOI] [PubMed] [Google Scholar]
- Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–1459. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- Harada K, Komuro I, Hayashi D, Sugaya T, Murakami K, Yazaki Y. Angiotensin II type 1a receptor is involved in the occurrence of reperfusion arrhythmias. Circulation. 1998a;97:315–317. doi: 10.1161/01.cir.97.4.315. [DOI] [PubMed] [Google Scholar]
- Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation. 1998b;97:1952–1959. doi: 10.1161/01.cir.97.19.1952. [DOI] [PubMed] [Google Scholar]
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, et al. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- Howard PA, Cheng JW, Crouch MA, Colucci VJ, Kalus JS, Spinler SA, et al. Drug therapy recommendations from the 2005 ACC/AHA guidelines for treatment of chronic heart failure. Ann Pharmacother. 2006;40:1607–1617. doi: 10.1345/aph.1H059. [DOI] [PubMed] [Google Scholar]
- Izumi T, Saito Y, Kishimoto I, Harada M, Kuwahara K, Hamanaka I, et al. Blockade of the natriuretic peptide receptor guanylyl cyclase-A inhibits NF-kappaB activation and alleviates myocardial ischemia/reperfusion injury. J Clin Invest. 2001;108:203–213. doi: 10.1172/JCI12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB. Vital epidemiologic clues in heart failure. J Clin Epidemiol. 2000;53:229–235. doi: 10.1016/s0895-4356(99)00135-3. [DOI] [PubMed] [Google Scholar]
- Kasi VS, Xiao HD, Shang LL, Iravanian S, Langberg J, Witham EA, et al. Cardiac-restricted angiotensin-converting enzyme overexpression causes conduction defects and connexin dysregulation. Am J Physiol Heart Circ Physiol. 2007;293:H182–H192. doi: 10.1152/ajpheart.00684.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JH, Zhang Y, Lei M, Grace AA, Huang CL, Fraser JA. Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol (Oxf) 2013;207:308–323. doi: 10.1111/apha.12006. [DOI] [PubMed] [Google Scholar]
- Kober L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, et al. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. N Engl J Med. 1995;333:1670–1676. doi: 10.1056/NEJM199512213332503. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Saito Y, Takano M, Arai Y, Yasuno S, Nakagawa Y, et al. NRSF regulates the fetal cardiac gene program and maintains normal cardiac structure and function. EMBO J. 2003;22:6310–6321. doi: 10.1093/emboj/cdg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer MS, Larson MG, Evans JC, Levy D. Association of left ventricular dilatation and hypertrophy with chronotropic incompetence in the Framingham Heart Study. Am Heart J. 1999;137:903–909. doi: 10.1016/s0002-8703(99)70415-1. [DOI] [PubMed] [Google Scholar]
- Levy D, Anderson KM, Savage DD, Balkus SA, Kannel WB, Castelli WP. Risk of ventricular arrhythmias in left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol. 1987;60:560–565. doi: 10.1016/0002-9149(87)90305-5. [DOI] [PubMed] [Google Scholar]
- Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- Li Y, Kishimoto I, Saito Y, Harada M, Kuwahara K, Izumi T, et al. Guanylyl cyclase-A inhibits angiotensin II type 1A receptor-mediated cardiac remodeling, an endogenous protective mechanism in the heart. Circulation. 2002;106:1722–1728. doi: 10.1161/01.cir.0000029923.57048.61. [DOI] [PubMed] [Google Scholar]
- Lynch JJ, Jr, Stump GL, Wallace AA, Painter CA, Thomas JM, Kusma SE, et al. EXP3174, the AII antagonist human metabolite of losartan, but not losartan nor the angiotensin-converting enzyme inhibitor captopril, prevents the development of lethal ischemic ventricular arrhythmias in a canine model of recent myocardial infarction. J Am Coll Cardiol. 1999;34:876–884. doi: 10.1016/s0735-1097(99)00253-3. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Olson EN. Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J Clin Invest. 2005;115:538–546. doi: 10.1172/JCI24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, Kuwahara K, Harada M, Takahashi N, Yasuno S, Adachi Y, et al. Class II HDACs mediate CaMK-dependent signaling to NRSF in ventricular myocytes. J Mol Cell Cardiol. 2006;41:1010–1022. doi: 10.1016/j.yjmcc.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Okin PM, Devereux RB, Liu JE, Oikarinen L, Jern S, Kjeldsen SE, et al. Regression of electrocardiographic left ventricular hypertrophy predicts regression of echocardiographic left ventricular mass: the LIFE study. J Hum Hypertens. 2004;18:403–409. doi: 10.1038/sj.jhh.1001707. [DOI] [PubMed] [Google Scholar]
- Poelzing S, Rosenbaum DS. Nature, significance, and mechanisms of electrical heterogeneities in ventricle. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:1010–1017. doi: 10.1002/ar.a.20103. [DOI] [PubMed] [Google Scholar]
- van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. The heterotrimeric G q protein-coupled angiotensin II receptor activates p21 ras via the tyrosine kinase-Shc-Grb2-Sos pathway in cardiac myocytes. EMBO J. 1996;15:775–787. [PMC free article] [PubMed] [Google Scholar]
- Saffitz JE, Schuessler RB, Yamada KA. Mechanisms of remodeling of gap junction distributions and the development of anatomic substrates of arrhythmias. Cardiovasc Res. 1999;42:309–317. doi: 10.1016/s0008-6363(99)00023-1. [DOI] [PubMed] [Google Scholar]
- Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–2339. doi: 10.1016/j.jacc.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya T, Nishimatsu S, Tanimoto K, Takimoto E, Yamagishi T, Imamura K, et al. Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem. 1995;270:18719–18722. doi: 10.1074/jbc.270.32.18719. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–341. doi: 10.1067/mhj.2001.113218. [DOI] [PubMed] [Google Scholar]
- Warn-Cramer BJ, Lau AF. Regulation of gap junctions by tyrosine protein kinases. Biochim Biophys Acta. 2004;1662:81–95. doi: 10.1016/j.bbamem.2003.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CM, Moskowitz IP, Arno S, Branco DM, Semsarian C, Bernstein SA, et al. Somatic events modify hypertrophic cardiomyopathy pathology and link hypertrophy to arrhythmia. Proc Natl Acad Sci U S A. 2005;102:18123–18128. doi: 10.1073/pnas.0509145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao HD, Fuchs S, Campbell DJ, Lewis W, Dudley SC, Jr, Kasi VS, et al. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am J Pathol. 2004;165:1019–1032. doi: 10.1016/S0002-9440(10)63363-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannad F, Briancon S, Juilliere Y, Mertes PM, Villemot JP, Alla F, et al. Incidence, clinical and etiologic features, and outcomes of advanced chronic heart failure: the EPICAL Study. Epidemiologie de l'Insuffisance Cardiaque Avancee en Lorraine. J Am Coll Cardiol. 1999;33:734–742. doi: 10.1016/s0735-1097(98)00634-2. [DOI] [PubMed] [Google Scholar]
- Zou Y, Komuro I, Yamazaki T, Kudoh S, Aikawa R, Zhu W, et al. Cell type-specific angiotensin II-evoked signal transduction pathways: critical roles of Gbetagamma subunit, Src family, and Ras in cardiac fibroblasts. Circ Res. 1998;82:337–345. doi: 10.1161/01.res.82.3.337. [DOI] [PubMed] [Google Scholar]