

Abstract
BACKGROUND AND PURPOSE
Inhibition of the cGMP-specific phosphodiesterase 5 (PDE5) exerts profound beneficial effects on failing hearts. However, the mechanisms underlying the therapeutic effects of PDE5 inhibition on heart failure are unclear. The purpose of this study was to investigate whether PDE5 inhibition decreases endoplasmic reticulum (ER) stress, a key event in heart failure.
EXPERIMENTAL APPROACH
Heart failure was induced by isoprenaline s.c. injection in Sprague–Dawley rats and transverse aortic constriction (TAC) in mice. PDE5 was inhibited with sildenafil. Heart function was detected by invasive pressure–volume analysis and echocardiography. ER stress markers were analysed by Western blotting. Apoptosis was measured by flow cytometric analysis.
KEY RESULTS
PDE5 inhibition markedly attenuated isoprenaline-induced and TAC-induced cardiac hypertrophy and dysfunction, and reduced ER stress and apoptosis. Further, PDE5 inhibition with sildenafil largely prevented ER stress and reduced apoptosis in isoprenaline- or thapsigargin-treated cardiomyocytes. PKG inhibition markedly prevented the protective effects of sildenafil in vivo and in vitro. To further understand the mechanism of the effect of PDE5 inhibition on ER stress, we demonstrated that PDE5 inhibitor increased sarco-(endo)-plasmic reticulum Ca2+-ATPase activity via phosphorylation of phospholamban at Ser16. This may contribute to the attenuation of ER stress induced by PDE5 inhibition.
CONCLUSION AND IMPLICATIONS
These results suggest that PDE5 inhibition can attenuate ER stress and improve cardiac function in vivo and in vitro. Suppression of ER stress by inhibiting PDE5 may contribute to the therapeutic effects on heart failure.
Keywords: PDE5, ER stress, heart failure, PKG
Introduction
Heart failure remains one of the most important causes of morbidity and mortality in the world and continues to increase in prevalence (Mudd and Kass, 2008). Organelle-mediated stress, particularly endoplasmic reticulum (ER) stress, has emerged as a major pathophysiological process underlying heart failure (Okada et al., 2004; Ni et al., 2011; George et al., 2011a). The ER is a central cellular organelle, with many functions including calcium storage, protein folding, protein maturation and lipid synthesis (Ni et al., 2011). Disturbances in any of these functions, such as dys-regulation of intracellular calcium homeostasis or excessive accumulation of unfolded proteins, can lead to ‘ER stress’ (Kaufman, 2002; Mattson and Chan, 2003). In response to ER stress, ER chaperones such as glucose-regulated protein 78 kDa (GRP78), glucose-regulated protein 94 kDa (GRP94) and calreticulin are up-regulated as a means of restoring normal cell functions (Schroder and Kaufman, 2005). However, when ER stress is excessive and/or prolonged, apoptotic processes are initiated by transcriptional induction of C/EBP homologous protein (CHOP) and/or caspase-12-dependent pathways (Oyadomari et al., 2002; Okada et al., 2004; Szegezdi et al., 2006a). ER stress is involved in many cardiovascular disorders, including atherosclerosis, myocardial ischaemia, hypertension and dilated cardiomyopathy (DCM), which ultimately contribute to heart failure (Schroder and Kaufman, 2005; Myoishi et al., 2007; Ni et al., 2011).
Phosphodiesterase type 5 (PDE5) selectively hydrolyses cGMP. PDE5 is found in human coronary vessels and pulmonary vasculature, while the expression of PDE5 is low in heart under normal physiological conditions (Cheitlin et al., 1999). The importance of PDE5 in the myocardium may have been underestimated to date. Several recent studies have demonstrated that PDE5 expression was up-regulated in myocardium from patients with heart failure (Nagendran et al., 2007; Pokreisz et al., 2009; Lu et al., 2010), and inhibition of PDE5 improved ischaemic cardiomyopathy, doxorubicin-induced cardiomyopathy and pressure overload-induced hypertrophy (Takimoto et al., 2005; Perez et al., 2007; Koka and Kukreja, 2010). PDE5 blockade significantly attenuated cardiomyocyte apoptosis after ischemia/reperfusion injury and improved Ca2+ regulation in transverse aortic constriction (TAC)-induced heart failure in vivo and in vitro (Das et al., 2008; Nagayama et al., 2009). However, the role of PDE5 and the mechanisms by which PDE5 is up-regulated in the pathophysiological process of heart failure remain unknown.
Dys-regulation of intracellular Ca2+ cycling is crucial to the pathogenesis of heart failure, and this likely induces ER stress. Previous studies have shown that the PDE5 inhibitor, sildenafil, could improve Ca2+ cycling in TAC hearts. We thus hypothesized that PDE5 inhibition may protect cardiomyocytes by relieving ER stress. In this study, we examined this hypothesis in cultured cells, in animal models, and in tissues from patients with heart failure.
Methods
Human heart samples
Human heart samples were collected at Tongji Hospital (Wuhan, China). The study was approved by the Ethics Review Board of Tongji Hospital and Tongji Medical College. The study conformed to the principles in the Declaration of Helsinki and the subjects recruited in the study signed written informed consents, or in case of incapacity, were signed by immediate family members. Tissue samples were frozen in liquid nitrogen and then stored at −80°C until use.
Animal models
All animal care and experimental protocols conformed to the ‘Guide for the Care and Use of Laboratory Animals' published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996). The study was approved by the Institutional Animal Research Committee of Tongji Medical College. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 130 animals were used in the experiments described here.
Male Sprague–Dawley (SD) rats (180–200 g) and male C57Bl/6 mice (22–25g) were obtained from the Experimental Animal Center of Tongji Medical College (Wuhan, China). Animals were housed in temperature-controlled cages with a 12 h light–dark cycle and given free access to water and normal chow.
SD rats were randomly divided into seven groups: (i) vehicle control group (saline); (ii) DT3 infusion group (DT3: 500 μg·kg−1·day−1); (iii) isoprenaline injection group; (iv) isoprenaline injection and treatment with sildenafil group; (v) Iso injection and treatment with sildenafil and DT3 (100 μg·kg−1·day−1) group; (vi) isoprenaline injection and treatment with sildenafil and DT3 (500 μg·kg−1·day−1) group; and (vii) isoprenaline injection and treatment with DT3 (500 μg·kg−1·day−1) group. Heart failure was induced by Iso s.c. injection (5 mg·kg−1·day−1 for 2 weeks) (Ni et al., 2011). Treatment with sildenafil at a dose of 50 mg·kg−1·day−1 for 3 weeks was started when rats were exposed to isoprenaline. DT3 was delivered to rats by implanting a micro-osmotic pump (Alzet, Durect, Cupertino, CA, USA). Micro-osmotic pumps were implanted as described previously (Nienaber et al., 2003). DT3 (100or 500 μg·kg−1·day−1) or vehicle (saline) was infused s.c. for 21 days.
In mice, pressure overload was induced by TAC (Takimoto et al., 2005), with sham animals undergoing the same operation without aortic constriction. Mice were randomly divided into three groups: sham, TAC and TAC+sildenafil (100 mg·kg−1·day−1) groups.
Haemodynamics and echocardiography
Rats were anaesthetized using 1.5% isoflurane and echocardiography was performed using Vevo 2100 Imaging System (Visual Sonics Inc., Toronto, Canada) as previously described (Foster et al., 2009). Echocardiographic parameters, including left ventricular (LV) ejection fraction and fractional shortening of left ventricular diameter, were obtained.
Rats were anaesthetized as described above, and a microtip pressure volume catheter (SPR-838; Millar Instruments, Houston, TX, USA) was inserted through the right carotid artery into the aorta and then into the LV. After stabilization, the readout signals were stored and displayed on a personal computer using an ARIA pressure–volume conductance system (Millar Instruments) coupled to a Powerlab/4SP analogue-to-digital converter (AD Instruments, Mountain View, CA, USA). All pressure–volume loop data were analysed using the PVAN software from Millar Instruments.
Phospholamban (PLB) mutation plasmid construction
pCMV6-XL5-PLB plasmid was purchased from OriGene. The PLB-mutated plasmid (Ser16 → Ala) was constructed using the Fast Mutagenesis System kit (TransGen Biotech, Beijing, China) according to the manufacturer's protocol.
Cell culture, transfection and treatments
H9c2(2-1) cells, which are derived from embryonic BD1X rat heart tissue, were obtained from American Type Culture Collection (CRL-1446). Cells were cultured in DMEM supplemented with 10% FBS and penicillin-streptomycin (100 IU·mL−1). All cell cultures were maintained at 37°C in a humidified incubator containing 95% air and 5% CO2 atmosphere. H9c2(2-1) were transfected with siRNA negative control (siR-con), or siRNA targeting SERCA2 (RiboBio, Guangzhou, China), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's protocol. H9c2(2-1) cells were serum-starved overnight, pre-incubated with 10 μM sildenafil or/and 10 μM KT5823 for 1 h, then incubated with 20 μM isoprenaline and collected 12 h after treatment.
Western blotting
Western blotting was performed as described previously (Wang et al., 2003). Briefly, frozen animal heart tissues and cells were homogenized in ice-cold lysis buffer (500 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP40, 0.02% sodium azide, 0.1% SDS, 100 μg·mL−1 PMSF, 1 μg·mL−1 aprotinin and 0.5% sodium deoxycholate) and then centrifuged at 12 000 g at 4°C for 20 min. Lysates (25 μg protein per lane) were resolved by SDS-PAGE (10%), transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% non-fat dry milk and then incubated with primary antibodies overnight at 4°C, followed by peroxidase-conjugated secondary antibody for 2–3 h. Proteins were visualized by enhanced chemiluminescence (Pierce, Rockford, IL, USA).
Flow cytometry analysis
Cells were plated in 6-well plates and treated with isoprenaline with or without sildenafil. At indicated time points, cells were trypsinized, washed in PBS, and incubated with FITC-conjugated Annexin V and propidium iodide (KeyGen Biotech, Nanjing, China), according to the manufacturer's protocol. Cells were then analysed by flow cytometry.
Immunohistochemistry
Heart samples were dissected, fixed in 4% paraformaldehyde for 16 h, then embedded in paraffin and sectioned into 4 μm slices. Sections were deparaffinized, rehydrated and incubated in 3% hydrogen peroxide for 10 min, then PBS containing 10% goat serum for 30 min, followed by a primary antibody (overnight incubation at 4°C). After rinsing with wash buffer, sections were incubated with a secondary biotinylated antibody for 30 min at room temperature. Subsequently, the slides were incubated with DAB chromogen for 5 min at room temperature and sections were counterstained with haematoxylin and coverslipped.
Activity of sarco-(endo)-plasmic reticulum Ca2+-ATPase (SERCA)
To determine the activity of SERCA, the ER of cardiomyocytes was extracted as previously described (Ikeda et al., 2001), and SERCA activity was measured using a Ca2+-ATPase assay kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions. SERCA activity was normalized to protein concentration.
Statistical analysis
Values were expressed as mean ± SEM. Differences between data groups were compared by Student's t-test for analysis of unpaired data or one-way anova. Values of P < 0.05 were considered significant.
Materials
DMEM and FBS were obtained from Gibco BRL (Life Technologies, Inc., Grand Island, NY, USA). KT5823 and sildenafil were supplied by Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Antibodies against PDE5, spliced form of X-box-binding protein-1 (XBP1s), phospho-protein kinase-like ER kinase (P-PERK), the Lys-Asp-Glu-Leu (KDEL) receptor, GRP78, atrial natriuretic peptide (ANP), SERCA2, phospholamban (PLB), calcineurin and CHOP were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-PLB (Ser16) antibody was purchased from Abcam (Cambridge, MA, USA). HRP-conjugated secondary antibodies were from Pierce Biotechnology (Thermo Fisher Scientific, Rockford, IL, USA). pCMV6-XL5-PLB plasmid was purchased from OriGene (Rockville, MD, USA). All other chemicals and reagents were purchased from Sigma-Aldrich Company, unless otherwise specified.
Results
Induction of ER stress is correlated with increased PDE5 expression in failing human hearts
ER stress activation was detected in heart samples from four heart-transplant patients with DCM and six patients with heart failure undergoing mitral valve replacement, as well as four normal hearts of traffic accident victims. The clinical characteristics of patients are listed in Tables 1 and 2. Several molecular markers of ER stress (Szegezdi et al., 2006b; Zhang and Kaufman, 2008), including GRP78, P-PERK, XBP1s and CHOP, and a marker of heart failure, ANP, were significantly elevated in samples of heart failure (Figure 1A–D). Interestingly, we also found that the expression of PDE5 was significantly higher in failing hearts than in normal hearts (Figure 1A–D) and the increase of myocardial PDE5 was significantly correlated with the increase of ER stress markers (Figure 1E). The results suggested there might be a relevant correlation between PDE5 and ER stress in heart failure.
Table 1.
Clinical characteristics of patients with dilated cardiomyopathy
| Patient | Age | Gender | Diagnosis | NYHA | LVEF (%) | LVEDD (mm) |
|---|---|---|---|---|---|---|
| 1 | 39 | Female | DCM | 3 | 21 | 60 |
| 2 | 30 | Female | DCM | 3 | 31 | 62 |
| 3 | 29 | Male | DCM | 3 | 12 | 65 |
| 4 | 36 | Male | DCM | 4 | 18 | 85 |
DCM, dilated cardiomyopathy; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association function class.
Table 2.
Clinical characteristics of patients with mitral valve replacement
| Patient | Age | Gender | Diagnosis | Diabetes | Hypertension | AF | NYHA | LVEF (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 58 | Female | MS | N | N | N | 3 | 48 |
| 2 | 55 | Female | MS | Y | N | Y | 3 | 55 |
| 3 | 27 | Female | MI | N | N | N | 3 | 66 |
| 4 | 49 | Male | MS, MI | N | N | Y | 4 | 35 |
| 5 | 48 | Female | MS | N | N | N | 4 | 50 |
| 6 | 39 | Male | MI | N | N | N | 3 | 48 |
AF, atrial fibrillation; LVEF, left ventricular ejection fraction; MI, mitral insufficiency; MS, mitral stenosis; NYHA, New York Heart Association function class.
Figure 1.

Increased PDE5 expression in left ventricular samples from patients with heart failure (HF) is correlated with increased ventricular ER stress markers. Several ER stress markers, including phosphorylated PERK (P-PERK), GRP78, XBP1s and CHOP, were measured. (A, B) ER stress was increased in failing hearts from heart transplant recipients compared with normal hearts. DCM, dilated cardiomyopathy. (C, D) Left ventricular ANP, PDE5, P-PERK, GRP78, XBP1s and CHOP were increased in hearts of patients with heart failure undergoing mitral valve replacement and (E) the increase in ventricular PDE5 was significantly correlated with the increase in ventricular P-PERK, GRP78, XBP1s and CHOP. p1–p6, heart samples of six patients undergoing mitral valve replacement; N1 and N2, normal human hearts. *P < 0.05 versus normal hearts.
PDE5 inhibition attenuated cardiac hypertrophy and heart dysfunction in failing hearts
To investigate the effects of PDE5 inhibition on ER stress in heart failure, we used a heart failure model with continuous infusion of the β-adrenoceptor agonist isoprenaline for 2 weeks. Chronic isoprenaline infusion induced cardiac hypertrophy, determined by cardiomyocyte cross-sectional diameter and the ratio of heart weight to body weight (HW/BW) compared with the saline group, while sildenafil treatment prevented the cardiac hypertrophy induced by isoprenaline (Figure 2A–C). Heart function was assayed by invasive pressure–volume analysis (Table 3) and echocardiography (Figure 2D,E), showing that PDE5 inhibition markedly improved heart function in isoprenaline-induced heart failure.
Figure 2.
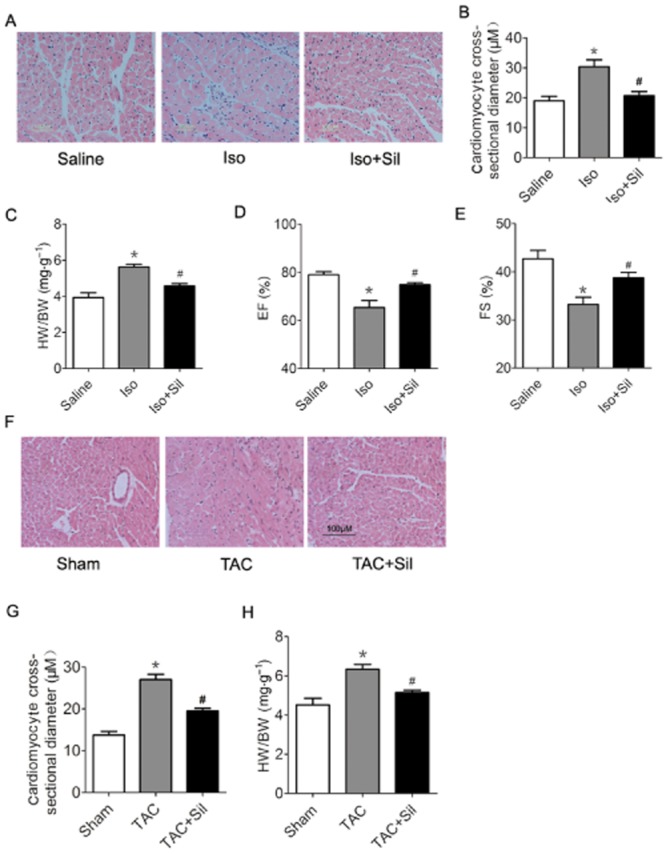
PDE5 inhibition attenuated cardiac hypertrophy and improved function in failing hearts. (A) to (E) show that sildenafil attenuated cardiac hypertrophy and dysfunction in rats induced by isoprenaline (Iso). (A) and (B) HE staining of rat hearts and cardiomyocyte cross-sectional diameter (μm). Scale bar: 100 μm. (C) Ratio of heart weight to body weight. *P < 0.05 versus saline. #P < 0.05 versus Iso. (D) and (E) Echocardiography parameters in isoprenaline rats and isoprenaline rats with sildenafil treatment. EF: ejection fraction, FS: fractional shortening. *P < 0.05 versus saline. #P < 0.05 versus isoprenaline. (F) to (H) show sildenafil attenuated cardiac hypertrophy and dysfunction induced by transverse aortic constriction transverse aortic constriction (TAC) in mice. (F) and (G) HE staining of mice hearts and cardiomyocyte cross-sectional diameter (μm). Scale bar, 100 μm. (H) Heart/body weight ratios of TAC mice. *P < 0.05 versus sham. #P < 0.05 versus TAC.
Table 3.
Haemodynamic parameters in isoprenaline-treated rats (Iso) and isoprenaline-treated rats with the PDE5 inhibitor sildenafil (Iso + Sil)
| Control | Iso | Iso + Sil | |
|---|---|---|---|
| N | 6 | 5 | 6 |
| HR (bpm) | 213 ± 13.6 | 209 ± 17.7 | 203 ± 15.8 |
| PES (mmHg) | 143.6 ± 10.6 | 139.9 ± 50.1 | 147.7 ± 48.9 |
| PED (mmHg) | 7.6 ± 1.7 | 10.9 ± 4.1 | 8.9 ± 3.9 |
| dP/dtmax (mmHg·s−1) | 5291 ± 626 | 4047 ± 991* | 5123 ± 761# |
| dP/dtmin (mmHg·s−1) | –6915 ± 1251 | –4959 ± 1087* | –6411.3 ± 1122 # |
Data are mean ± SD.
P < 0.05 versus control.
P < 0.05 versus Iso.
dP/dtmax, maximal slope of systolic pressure increment; dP/dtmin, maximal slope of diastolic pressure decrement; HR, heart rate; PED, end-diastolic pressure; PES, end-systolic pressure.
To further support this finding, we used another animal model of heart failure, induced by TAC for 3 weeks in mice, concurrently treating them with sildenafil or vehicle. TAC induced significant cardiac hypertrophy, and sildenafil treatment blunted the TAC-induced heart hypertrophy (Figure 2F–H). Cardiac function was also markedly improved as assessed by echocardiography (Table 4). These data suggested that PDE5 inhibition markedly attenuated cardiac hypertrophy and improved heart function in mice with heart failure.
Table 4.
Cardiac functional data of mice under control conditions (sham), after TAC, after TAC plus sildenafil (Sil)
| Sham | TAC | TAC + Sil | |
|---|---|---|---|
| N | 6 | 8 | 8 |
| HR (bpm) | 565 ± 13.6 | 530 ± 17.7 | 553 ± 15.8 |
| LVEF (%) | 89 ± 6.5 | 70 ± 10 | 82 ± 1.8 |
| LVFS (%) | 58 ± 10.8 | 40 ± 6.3* | 48 ± 1.8# |
| LVIDd (mm) | 2.38 ± 0.11 | 3.01 ± 0.33* | 2.48 ± 0.09# |
| LVIDs (mm) | 1.00 ± 0.28 | 1.70 ± 0.45* | 1.28 ± 0.08# |
| LVPWd (mm) | 0.74 ± 0.15 | 1.07 ± 0.18* | 0.85 ± 0.10# |
| LVPWs (mm) | 1.33 ± 0.06 | 1.42 ± 0.09* | 1.43 ± 0.07 |
| LV Mass (mg) | 61.15 ± 10.74 | 120.09 ± 33.18* | 72.99 ± 14.24# |
P < 0.05 versus sham.
P < 0.05 versus TAC.
HR, heart rate; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVIDd and LVIDs, left ventricular internal dimension at diastole and systole, respectively; LVPWd and LVPWs, posterior wall thickness at diastole and systole, respectively; LV Mass, left ventricular mass.
PDE5 inhibition attenuated ER stress and ER stress-triggered apoptosis in failing hearts
To determine whether inhibition of PDE5 attenuates ER stress, we examined the expression of several markers of ER stress in failing hearts. As shown in Figure 3A and B, ER stress was induced in the failing hearts, as assessed by increased expression of P-PERK, GRP78 and XBP1s. Treatment with the PDE5 inhibitor sildenafil markedly suppressed ER stress in the hearts. Immunohistochemical analysis for the ER stress marker KDEL further confirmed the above results (Figure 3C). Severe or prolonged ER stress triggers apoptosis. We examined a characteristic marker of ER stress-induced apoptosis, CHOP, and found that the expression iof CHOP increased dramatically after isoprenaline or TAC, and that sildenafil treatment abolished expression of CHOP (Figure 3D,E). TUNEL staining showed that the sildenafil treatment significantly attenuated the number of apoptotic cells in isoprenaline-induced heart failure (Figure 3F). Taken together, our results suggested that PDE5 inhibition suppressed ER stress and cardiomyocyte apotosis in this heart failure model.
Figure 3.

PDE5 inhibition suppressed ER stress and ER stress-mediated apoptosis in hypertrophic and failing hearts. (A) PDE5 inhibition by sildenafil (Sil) significantly attenuated the isoprenaline (Iso)-induced increase in ANP, PDE5, P-PERK, GRP78 and XBP1s in rats. Proteins were normalized to GAPDH. *P < 0.05 versus saline. #P < 0.05 versus isoprenaline. (B) Sildenafil markedly attenuated the TAC-induced ER stress in mice. *P < 0.05 versus sham. #P < 0.05 versus TAC. (C) Immunohistochemical analyses of KDEL in rat hearts. Scale bar: 100 μm. (D) CHOP was increased in isoprenaline rats, and sildenafil treatment reduced expression of CHOP. CHOP was normalized to GAPDH. *P < 0.05 versus saline. #P < 0.05 versus Iso. (E) Sildenafil treatment reduced the TAC-induced increase of CHOP. *P < 0.05 versus sham. #P < 0.05 versus TAC. (F) Representative images of TUNEL staining showing cardiomyocyte apoptosis and quantitative analysis of TUNEL-positive myocardial cells in rats. Scale bar: 50 μm. Data are expressed as mean ± SEM (n = 5). *P < 0.05 versus saline. #P < 0.05 versus isoprenaline.
PDE5 inhibition suppressed ER stress and ER stress-mediated apoptosis in cardiomyocytes
To further examine the action of PDE5 inhibition on ER stress, we pretreated H9c2(2-1) cells with isoprenaline to increase expression of GRP78, P-PERK, ATF4 and CHOP (Supplementary Information Figure S1). Pretreatment with sildenafil markedly decreased the expression of ER stress markers and reduced expression of the ER stress-mediated pro-apoptotic protein CHOP, induced by isoprenaline (Figure 4A,B). To assess if PDE5 inhibition attenuated ER stress generally, we pretreated H9c2(2–1) cells with sildenafil before exposing them to thapsigargin (TG), an agent commonly used to induce ER stress (Urano et al., 2000; Hiroi et al., 2005; Ni et al., 2011). Sildenafil significantly decreased the overexpression of TG-induced ER stress markers including GRP78, XBP1s and CHOP (Figure 4C,D). Consistent with these results, sildenafil pretreatment significantly reduced cell apoptosis induced by isoprenaline and TG as determined by Annexin V/PI staining with flow cytometric analysis (Figure 4E,F). These results indicated that PDE5 inhibition attenuated ER stress and subsequent apoptosis in cardiomyocytes.
Figure 4.
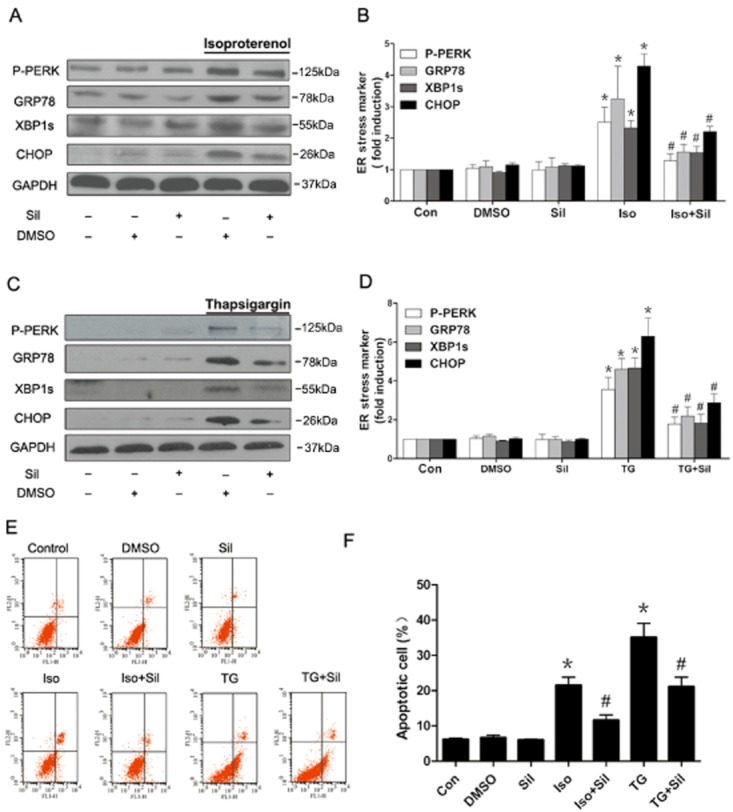
PDE5 inhibition alleviated ER stress and ER stress-mediated apoptosis induced in cardiomyocytes. (A, B) Sildenafil (Sil) alleviated isoprenaline (Iso)-induced ER stress in H9c2(2–1) cells. Cells were pretreated with sildenafil for 1 h, and then exposed to isoprenaline (20 μM) for 12 h (n = 3). (C, D) Sildenafil alleviated TG-induced ER stress in cardiomyocytes. H9c2(2–1) cells were pretreated with sildenafil for 1 h, and then exposed to TG (5 μM) for 24 h (n = 3). (E, F) Flow cytometric analysis of H9c2(2–1) cells incubated with isoprenaline (20 μM) or TG (5 μM), and treated with sildenafil (10 μM) for 24 h using Annexin V-FITC and propidium iodide staining. Results shown are mean ± SEM, (n = 3). *P < 0.05 versus control. #P < 0.05 versus isoprenaline or TG.
PDE5 inhibitor suppresses ER stress via the PKG pathway
Next, we evaluated whether PKG mediated some part of the anti-ER stress effect of sildenafil. Results showed that the PKG inhibitors, KT5823 and DT3, blunted the reversal of the isoprenaline-induced ER stress by sildenafil in cardiomyocytes (Figure 5A–D), suggesting that the PKG pathway was involved in the protective effects of sildenafil on ER stress. To confirm this finding, we also carried out PKG inhibition experiments in rats, and the results showed that DT3 (500 μg·kg−1·day−1) markedly inhibited the protective effects of sildenafil in isoprenaline-induced heart failure in rats (Figure 5E–G and Table 5).
Figure 5.
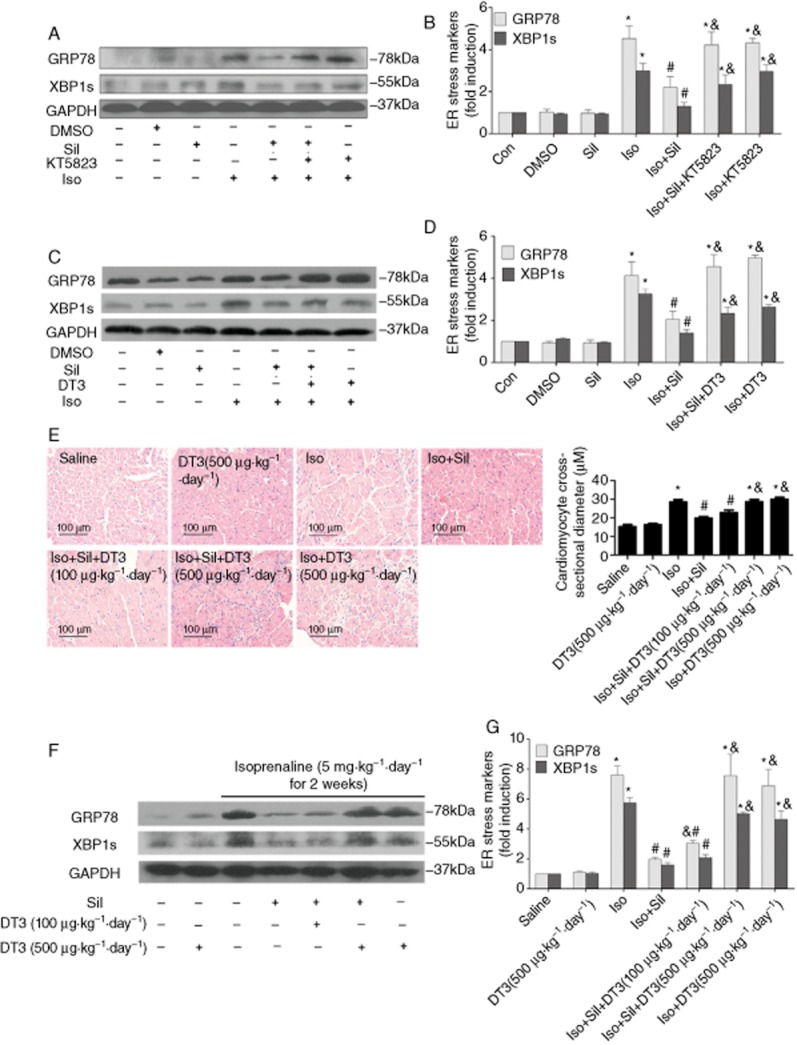
PDE5 inhibitor-mediated suppression of cardiac hypertrophy and ER stress are dependent on PKG pathway. H9c2(2–1) cells were pretreated with sildenafil (Sil; 10 μM) and (A, B) KT5823 (M) or (C, D) DT3 (1 μM) for 1 h, then exposed to isoprenaline (Iso; 20 μM) for 24 h. ER stress marker expression was measured by Western blot (n = 3). (E–G) Sildenafil-attenuated cardiac hypertrophy and ER stress are dependent on PKG pathway in rats. (E) HE staining of rat hearts and cardiomyocyte cross-sectional diameter (μm). Scale bar: 100 μm. (F, G) ER chaperone GRP78 and XBP-1 s were increased in isoprenaline-induced rats. Sildenafil treatment (50 mg·kg−1·day−1 for 3 weeks) reduced the expression of GRP78 and XBP-1 s, and PKG antagonist DT3 (500μg·kg−1·day−1 for 3 weeks) significantly inhibited this effect. *P < 0.05 versus control. #P < 0.05 versus isoprenaline. &P < 0.05 versus isoprenaline + sildenafil. Data are expressed as mean ± SEM, n = 4.
Table 5.
Cardiac functional data of rats under control conditions (saline), after isoprenaline ( Iso) injection, and after isoprenaline injection plus sildenafil (Sil) and/or DT3
| Saline | DT3 (500μM·kg−1·day−1) | Iso | Iso + Sil | Iso + Sil + DT3 (100 μM·kg−1·day−1) | Iso + Sil + DT3 (500 μM·kg−1·day−1) | Iso + DT3 (500 μM·kg−1·day−1) | |
|---|---|---|---|---|---|---|---|
| N | 6 | 8 | 5 | 9 | 6 | 8 | 8 |
| HR (bpm) | 208 ± 10.3 | 211 ± 15.6 | 219 ± 8.9 | 201 ± 13.6 | 215 ± 15.1 | 220 ± 13.2 | 204.56 ± 11.6 |
| LVEF (%) | 79 ± 2.5 | 78 ± 2.8 | 65 ± 5.0* | 74.8 ± 1.4# | 71 ± 6.5 | 64 ± 4.5*& | 65.03 ± 3.52*& |
| LVFS (%) | 43 ± 3.0 | 44 ± 2.9 | 33 ± 3.3* | 39 ± 2.2# | 38 ± 5.8 | 32 ± 4.0*& | 30.14 ± 2.89*& |
| LVPWd (mm) | 2.05 ± 0.1 | 2.02 ± 0.15 | 2.17 ± 0.9 | 1.97 ± 0.09 | 1.94 ± 0.08 | 2.12 ± 0.08 | 2.4 ± 0.55 |
| LVPWs (mm) | 2.23± 0.5 | 2.4 ± 0.1 | 3 ± 0.25 * | 2.56 ± 0.25# | 2.7 ± 0.26 | 3.06 ± 0.15*& | 3.17 ± 0.15*& |
P < 0.05 versus saline.
P < 0.05 versus Iso.
P < 0.05 versus Iso + Sil.
HR, heart rate; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; LVPWd and LVPWs, posterior wall thickness at diastole and systole, respectively.
PDE5 inhibition restores sarcoplasmic reticulum (SR) calcium handing proteins and phosphorylation in isoprenaline- and TAC-induced heart failure
SR handling proteins closely correlate with calcium homeostasis and ER stress. We further examined whether SR handling proteins were also modified by therapy in the failing heart. The results showed that SERCA2 and PLB expression decreased in the failing heart, and sildenafil treatment restored SERCA2 expression and the P-PLB-Ser16 / PLB ratio (a measure of SERCA activity), compared with the corresponding values in isoprenaline- or TAC-treated animals (Figure 6).
Figure 6.
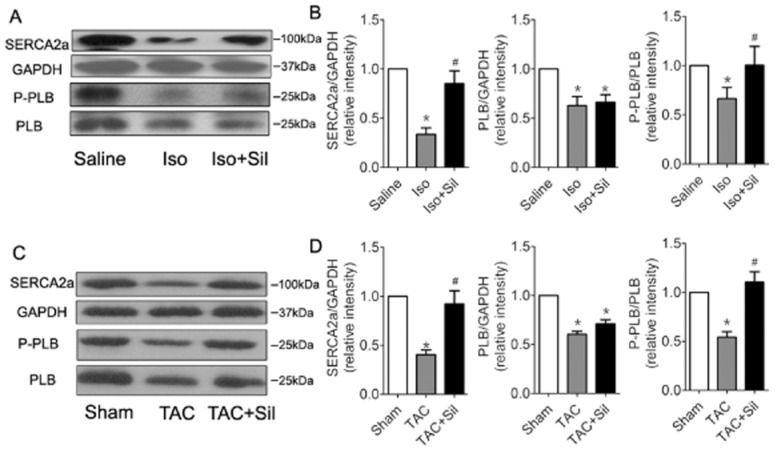
Sildenafil (Sil) treatment in failing hearts restores SR calcium-handling proteins and phosphorylation. Western blots of SERCA2, total PLB (PLB) and phosphorylated PLB (P-PLB) expression in isoprenaline( Iso)-induced rats (A, B) and in TAC mice (C, D) *P < 0.05 versus saline or sham. #P < 0.05 versus Iso or TAC. Results shown are mean ± SEM; n = 3.
Phospholamban phosphorylation at Ser16 plays a crucial role in sildenafil-mediated inhibition of ER stress
Results showed that sildenafil increased the P-PLB-Ser16 / PLB ratio and SERCA activity in cardiomyocytes (Figure 7 and Supporting Information Figure S7A) and that this effect of PDE5 inhibition on P-PLB and SERCA activity was dependent on the PKG pathway (Figure 7 and Supporting Information Figure S7A). To further explore the role of P-PLB-Ser16 in the anti-ER stress effect of PDE5 inhibition, we constructed a wild-type PLB (WT PLB) plasmid and a mutated PLB plasmid (Ser16 → Ala). As Figure 7 indicated, sildenafil markedly prevented the ER stress induced by the WT PLB plasmid but could not suppress ER stress induced by the mutated PLB. This demonstrated that the anti-ER stress effect mediated via PDE5 inhibition was dependent on sildenafil-induced PLB phosphorylation at Ser16.
Figure 7.
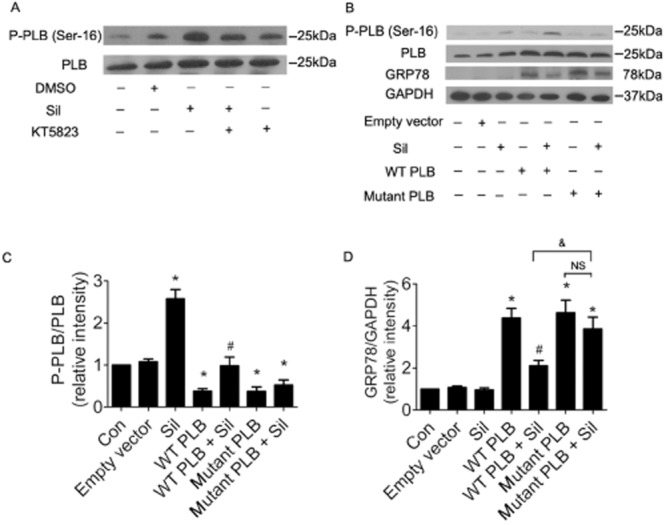
The anti-ER stress effect of PDE5 inhibition depends on sildenafil-induced phospholamban phosphorylation at Ser16. (A) H9c2(2–1) cells were treated with sildenafil (Sil; 10 μM) and PKG inhibitor, KT5823 (2 μM), for 24 h. P-PLB-Ser16 and total PLB expression was determined by Western blot (n = 3). (B) H9c2(2–1) cells were transfected with empty vector, WT PLB or PLB mutation and were treated with sildenafil (10 μM). P-PLB-Ser16, PLB, GRP78 and GAPDH were determined by Western-blot (n = 3). (C) P-PLB-Ser16 / PLB ratio. *P < 0.05 versus control. #P < 0.05 versus WT PLB. (C) GRP78/GAPDH. *P < 0.05 versus control. #P < 0.05 versus WT PLB. Data are expressed as mean ± SEM.
Discussion
To our knowledge, this represents the first demonstration that chronic PDE5 inhibition attenuated ER stress in cardiac hypertrophy and heart failure. In this study, we first showed that induction of ER stress in human heart failure was significantly correlated with increased myocardial PDE5 expression. We subsequently found that chronic PDE5 inhibition reduced ER stress and corresponding apoptosis in isoprenaline- and TAC-induced heart failure, and the studies in cultured cardiomyocytes showed similar results. Moreover, we found that PDE5 inhibition restored SERCA2a expression and the ratio of P-PLB/PLB in failing hearts. Finally, we demonstrated that the anti-ER stress effect of PDE5 inhibition with sildenafil may partly depend on elevated SERCA activity in cardiomyocytes by up-regulating PLB phosphorylation. Together, our study demonstrated that PDE5 inhibition in heart failure might contribute to attenuation of ER stress through increased SERCA activity and restoration of intracellular Ca2+ balance.
Several forms of myocardial injury, including ischaemia, abnormal Ca2+ regulation, nutrient depletion, oxidative stress and accumulation of unfolded proteins, can impair cardiomyocyte and heart function to produce heart failure, and those stressors also induce ER stress (Minamino and Kitakaze, 2010). ER stress is involved in heart failure (Okada et al., 2004; Ni et al., 2011; George et al., 2011b) and cardiomyocytes isolated from explanted human hearts with heart failure, as well as from hearts of animals with experimentally induced heart failure, exhibit diminished systolic shortening, altered short-frequency response and decreased force of contraction (Davies et al., 1995; Maltsev et al., 1998; George et al., 2011b). This indicates an abnormal Ca2+ regulation in those failing cardiomyocytes. Many previous studies have shown that abnormal expression of calcium-handling proteins, including reduced SERCA2a, increased NCX1 and RyR2, may contribute to impaired Ca2+ cycling in failing hearts (George et al., 2011b). The ER is a central cellular organelle, and as a major Ca2+ storage site, disturbances in Ca2+ homeostasis can result in ER dysfunction, ER stress and ultimately lead to apoptosis associated with ER stress. Therefore, abnormalities in intracellular Ca2+ cycling in cardiomyocytes may contribute to ER stress induction in failing hearts.
Previous studies have implicated the induction of ER stress in heart failure, but the origin of this ER stress in failing hearts is still unclear. Our present studies suggest that PDE5 up-regulation may be an important mechanism underlying ER stress induction in cardiac hypertrophy and heart failure. Lu et al., (2010) found PDE5 induction in patients with DCM. Treatment with the PDE5 inhibitor, sildenafil, in TAC-induced heart failure in mice, is known to attenuate myocardial injury and remodelling, and to improve ventricular function (Takimoto et al., 2005; Nagayama et al., 2009). Sildenafil treatment restored expression of SERCA2 and increased PLB phosphorylation (at Ser16) in a TAC-induced model, consistent with improved calcium handling (Nagayama et al., 2009). Thus, PDE5 expression may regulate Ca2+ homeostasis and through this mechanism affect ER stress in the failing heart. In the present study, we detected up-regulation of PDE5 in patients with end-stage heart failure and found unexpectedly that there was a significant correlation between increased PDE5 expression and activation of ER stress in failing hearts. The attenuation of ER stress by PDE5 inhibition was subsequently found in isoprenaline-stimulated cardiomyocytes and animal models of heart failure. These results confirm our hypothesis and indicate that chronic PDE5 inhibition suppressed ER stress in cardiac hypertrophy and heart failure. In order to demonstrate that the PDE5 inhibitors can alleviate directly induced ER stress, we used the ER stress-specific activator TG and found that sildenafil markedly prevented TG-induced ER stress and apoptosis.
Earlier work (Takimoto et al., 2005) has suggested that PDE5 inhibition deactivated many hypertrophy signalling pathways, including the calcineurin/NFAT, PI3K/Akt and ERK1/2 signalling pathways. Our data also showed that sildenafil markedly suppressed calcineurin and Akt pathways in isoprenaline-injected rats (Supporting Information Figure S3). However, PDE5 inhibition does not attenuate hypertrophy induced by overexpression of calcineurin in vitro and AKT in vivo (Takimoto et al., 2005; Hsu et al., 2009), suggesting that other mechanisms are involved in these pathways. What, then, is the source of activation of these pathways? The anti-ER stress effect with PDE5 inhibition may provide an explanation to this question. Recent work by Jiang et al. (2009) has demonstrated that ER stress-induced activation of Akt was mediated by XBP-1. In addition, Hu et al. (2007) obtained similar results indicating that ER stress activated the Akt pathway in a zebrafish embryonic cell line. Therefore, the protection against ER stress obtained with PDE5 inhibition may, at least in part, be the underlying mechanism by which PDE5 inhibition attenuated cardiac hypertrophy.
PDE5 is the predominant enzyme responsible for cGMP hydrolysis, and inhibition of its activity by sildenafil, a potent specific PDE5 inhibitor, ultimately increases intracellular cGMP concentrations and activates cGMP-dependent kinase (PKG). In this study, the effect of PDE5 inhibition on reduced ER stress could be reversed by the PKG inhibitor, KT5823 or DT3, indicating that PDE5 inhibition attenuated ER stress in a PKG-dependent manner.
Based on this overview, we know that disturbances in Ca2+ homeostasis induced ER stress and that PDE5 inhibition or PKG action attenuated ER stress, in failing hearts. However, whether there is a link between Ca2+ homeostasis and the anti-ER stress of PDE5 inhibition has not been reported. In this study, we utilized isoprenaline-stimulated rats and TAC mice to explore this link. Here, we found that isoprenaline or TAC significantly reduced SERCA2a, phospholamban phosphorylation at Ser16 (P-PLB-Ser16) and the ratio P-PLB-Ser16 /PLB in failing hearts. We also clearly illustrated that PDE5 inhibition restored SERCA2 expression and phosphorylation of PLB at Ser16. Therefore, we hypothesized that the alleviation of heart failure-induced ER stress by PDE5 inhibition may partly depend on the restoration of SR calcium handing proteins and phosphorylation and restoration of intracellular Ca2+ balance. To confirm this hypothesis, we used SERCA2a siRNA in cardiomyocytes. The results showed that PDE5 inhibition could attenuate SERCA2a siRNA-induced ER stress, increased P-PLB-Ser16 and partly restored SERCA activity compared with SERCA2a siRNA-transformed cardiomyocytes. This indicated that an increased P-PLB-Ser16 may be a crucial component of the effects of sildenafil. To further confirm the importance of P-PLB-Ser16 on sildenafil-mediated ER stress inhibition, we constructed a WT PLB plasmid and a mutated PLB plasmid (Ser16 → Ala). The mutant PLB could not be phosphorylated at Ser16. As Figure 7 indicated, sildenafil markedly prevented WT PLB plasmid-induced ER stress, but could not suppress the ER stress induced by the mutated PLB. This demonstrated that the anti-ER stress effect of sildenafil depended on phosphorylation of PLB at Ser16, following inhibition of PDE5.
In summary, the current study demonstrated that PDE5 played a crucial role in the induction of ER stress in heart failure, and that chronic PDE5 inhibition markedly relieved ER stress and ER-mediated apoptosis in cardiomyocytes and failing hearts. Thus, alleviation of ER stress may be an important mechanism underlying the therapeutic effect of PDE5 inhibitors in heart failure.
Acknowledgments
We thank Xiaolan Li for her help in analysing the flow cytometry results. This study was supported by 973 program (No. 2012CB518004), NSFC grant (Nos. 31130031 and 30930039).
Glossary
- ANP
atrial natriuretic peptide
- CHOP
C/EBP homologous protein
- DCM
dilated cardiomyopathy
- ER
endoplasmic reticulum
- GRP78
glucose regulated protein 78 kDa
- KDEL
Lys-Asp-Glu-Leu
- PDE5
cGMP-specific phosphodiesterase 5
- PERK
protein kinase-like ER kinase
- PKG
cGMP-dependent kinase
- PLB
phospholamban
- SD
Sprague–Dawley
- SERCA
sarco-(endo)-plasmic reticulum Ca2+-ATPase
- TAC
transverse aortic constriction
- TG
thapsigargin
- XBP1s
spliced form of X-box-binding protein-1
Authors contributions
W. G., Q. D. and D. W. W.: Participated in research design.
W. G.: Conducted experiments.
W. G. and D. W. W.: Performed data analysis.
W. G., C. C., Z. C., K. C. and D. W. W.: Wrote or contributed to the writing of the manuscript.
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12346
Figure S1 Isoprenaline induced ER stress in a dose- and time-dependent manner in cardiomyocytes. ER stress marker expressions after isoprenaline treatment for the indicated time and dose. Data are expressed as mean ± SEM, n = 3, *P < 0.05 versus 0 μM or 0 h.
Figure S2 Isoprenaline injection did not directly cause ER stress. (A) and (B) Western blots of GRP78 expression in isoprenaline-injected rats (n = 4).
Figure S3 The PDE5 blocker sildenafil suppressed increased activation of calcineurin and Akt in rat failing hearts. Results shown are mean ± SEM; n = 3, *P < 0.05 versus control. #P < 0.05 versus isoprenaline (Iso).
Figure S4 PDE5 inhibitor sildenafil did not affect the level of cAMP in isoprenaline- or TAC-induced failing hearts (n = 4). #P < 0.05 versus saline or sham.
Figure S5 Effect of isoprenaline (Iso) on phospholamban phosphorylation. H9c2 cells were treated with isoprenaline (20 μM) in a time-course, and PLB phosphorylation was assayed (n = 4). Data are expressed as mean ± SEM. *P < 0.05 versus 0 h.
Figure S6 Effect of PKG inhibition on ER stress. H9c2 cells were treated with a range of concentrations of KT5823, for 24 h, and GRP78 expression was assayed. Data are expressed as mean ± SEM (n = 3). *P < 0.05 versus 0 μM.
Figure S7 PDE5 inhibition attenuated ER stress by up-regulating the SERCA2 activation-dependent PKG pathway. (A) SERCA activity. H9c2(2–1) cells were incubated with sildenafil (Sil; 10 μM) and KT5823 (2 μM) for 24 h and activities were then measured. Data expressed are mean ± SEM (n = 6). *P < 0.05 versus control. #P < 0.05 versus sildenafil. (B) H9c2(2–1) cells were treated with SERCA2 specific siRNA (100 nM) for 24h. SERCA2 expression was determined by Western blot (n = 3). (C) to (G) H9c2(2–1) cells were treated with SERCA2 specific siRNA (100nM) and sildenafil (10 μM) for 24 h. (C) and (D) ER stress marker expression was measured by Western blot (n = 3). (E) and (F) P-PLB-Ser16 and PLB expression was determined by Western blot (n = 3). (G) SERCA activity. *P < 0.05 versus control. #P < 0.05 versus SERCA2 specific siRNA. Data are expressed as mean ± SEM.
References
- Cheitlin MD, Hutter AM, Jr, Brindis RG, Ganz P, Kaul S, Russell RO, Jr, et al. ACC/AHA expert consensus document. Use of sildenafil (Viagra) in patients with cardiovascular disease. American College of Cardiology/American Heart Association. J Am Coll Cardiol. 1999;33:273–282. doi: 10.1016/s0735-1097(98)00656-1. [DOI] [PubMed] [Google Scholar]
- Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283:29572–29585. doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92:2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- Foster FS, Mehi J, Lukacs M, Hirson D, White C, Chaggares C, et al. A new 15-50 MHz array-based micro-ultrasound scanner for preclinical imaging. Ultrasound Med Biol. 2009;35:1700–1708. doi: 10.1016/j.ultrasmedbio.2009.04.012. [DOI] [PubMed] [Google Scholar]
- George I, Mekahli D, Rayyan M, Levtchenko E, Allegaert K. Postnatal trends in creatinemia and its covariates in extremely low birth weight (ELBW) neonates. Pediatr Nephrol. 2011a;26:1843–1849. doi: 10.1007/s00467-011-1883-0. [DOI] [PubMed] [Google Scholar]
- George I, Sabbah HN, Xu K, Wang N, Wang J. beta-adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovasc Res. 2011b;91:447–455. doi: 10.1093/cvr/cvr106. [DOI] [PubMed] [Google Scholar]
- Hiroi T, Wei H, Hough C, Leeds P, Chuang DM. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: roles of intracellular calcium, GRP78 and Bcl-2. Pharmacogenomics J. 2005;5:102–111. doi: 10.1038/sj.tpj.6500296. [DOI] [PubMed] [Google Scholar]
- Hsu S, Nagayama T, Koitabashi N, Zhang M, Zhou L, Bedja D, et al. Phosphodiesterase 5 inhibition blocks pressure overload-induced cardiac hypertrophy independent of the calcineurin pathway. Cardiovasc Res. 2009;81:301–309. doi: 10.1093/cvr/cvn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Gong HY, Lin GH, Hu SY, Chen MH, Huang SJ, et al. XBP-1, a key regulator of unfolded protein response, activates transcription of IGF1 and Akt phosphorylation in zebrafish embryonic cell line. Biochem Biophys Res Commun. 2007;359:778–783. doi: 10.1016/j.bbrc.2007.05.183. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Gohra H, Hamano K, Zempo N, Ueyama T, Ohkusa T, et al. Effects of cardioplegic arrest and reperfusion on rabbit cardiac ryanodine receptors. Jpn Circ J. 2001;65:330–334. doi: 10.1253/jcj.65.330. [DOI] [PubMed] [Google Scholar]
- Jiang CC, Wroblewski D, Yang F, Hersey P, Zhang XD. Human melanoma cells under endoplasmic reticulum stress are more susceptible to apoptosis induced by the BH3 mimetic obatoclax. Neoplasia. 2009;11:945–955. doi: 10.1593/neo.09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koka S, Kukreja RC. Attenuation of Doxorubicin-induced cardiotoxicity by tadalafil: a long acting phosphodiesterase-5 inhibitor. Mol Cell Pharmacol. 2010;2:173–178. [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Xu X, Hu X, Lee S, Traverse JH, Zhu G, et al. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation. 2010;121:1474–1483. doi: 10.1161/CIRCULATIONAHA.109.906818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Tanimura M, Lesch M, Goldstein S, Undrovinas AI. Relationship between action potential, contraction-relaxation pattern, and intracellular Ca2+ transient in cardiomyocytes of dogs with chronic heart failure. Cell Mol Life Sci. 1998;54:597–605. doi: 10.1007/s000180050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, et al. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Hsu S, Zhang M, Koitabashi N, Bedja D, Gabrielson KL, et al. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J Am Coll Cardiol. 2009;53:207–215. doi: 10.1016/j.jacc.2008.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116:238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- Ni L, Zhou C, Duan Q, Lv J, Fu X, Xia Y, et al. β-AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. Plos ONE. 2011;6:e27294. doi: 10.1371/journal.pone.0027294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, et al. Inhibition of receptor-localized PI3K preserves cardiac beta-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 2003;112:1067–1079. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- Perez NG, Piaggio MR, Ennis IL, Garciarena CD, Morales C, Escudero EM, et al. Phosphodiesterase 5A inhibition induces Na+/H+ exchanger blockade and protection against myocardial infarction. Hypertension. 2007;49:1095–1103. doi: 10.1161/HYPERTENSIONAHA.107.087759. [DOI] [PubMed] [Google Scholar]
- Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C, et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. 2009;119:408–416. doi: 10.1161/CIRCULATIONAHA.108.822072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Duffy A, O'Mahoney ME, Logue SE, Mylotte LA, O'Brien T, et al. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem Biophys Res Commun. 2006a;349:1406–1411. doi: 10.1016/j.bbrc.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006b;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Wang H, Lin L, Jiang J, Wang Y, Lu ZY, Bradbury JA, et al. Up-regulation of endothelial nitric-oxide synthase by endothelium-derived hyperpolarizing factor involves mitogen-activated protein kinase and protein kinase C signaling pathways. J Pharmacol Exp Ther. 2003;307:753–764. doi: 10.1124/jpet.103.052787. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.