

Abstract
BACKGROUND AND PURPOSE
The conversion of plasminogen into plasmin by interstitial urokinase plasminogen activator (uPA) is potentially important in asthma pathophysiology. In this study, the effect of uPA-mediated plasminogen activation on airway smooth muscle (ASM) cell proliferation was investigated.
EXPERIMENTAL APPROACH
Human ASM cells were incubated with plasminogen (0.5–50 μg·mL−1) or plasmin (0.5–50 mU·mL−1) in the presence of pharmacological inhibitors, including UK122, an inhibitor of uPA. Proliferation was assessed by increases in cell number or MTT reduction after 48 h incubation with plasmin(ogen), and by earlier increases in [3H]-thymidine incorporation and cyclin D1 expression.
KEY RESULTS
Plasminogen (5 μg·mL−1)-stimulated increases in cell proliferation were attenuated by UK122 (10 μM) or by transfection with uPA gene-specific siRNA. Exogenous plasmin (5 mU·mL−1) also stimulated increases in cell proliferation. Inhibition of plasmin-stimulated ERK1/2 or PI3K/Akt signalling attenuated plasmin-stimulated increases in ASM proliferation. Furthermore, pharmacological inhibition of cell signalling mediated by the EGF receptor, a receptor trans-activated by plasmin, also reduced plasmin(ogen)-stimulated cell proliferation. Knock down of annexin A2, which has dual roles in both plasminogen activation and plasmin-signal transduction, also attenuated ASM cell proliferation following incubation with either plasminogen or plasmin.
CONCLUSIONS AND IMPLICATIONS
Plasminogen stimulates ASM cell proliferation in a manner mediated by uPA and involving multiple signalling pathways downstream of plasmin. Targeting mediators of plasminogen-evoked ASM responses, such as uPA or annexin A2, may be useful in the treatment of asthma.
Keywords: airway wall remodelling, annexin A2 hetero-tetramer, asthma, α2-antiplasmin, EGF, plasmin
Introduction
Chronic inflammation and hyper-reactivity of the airways are characteristic of the full spectrum of asthma severities. In severe, treatment-refractory asthma, an array of structural changes in the tissue of the airway wall amplifies airway hyper-reactivity (James and Wenzel, 2007). Injury and dysregulated repair processes linked to chronic airway inflammation are likely to contribute to this airway wall remodelling (AWR) (Holgate et al., 2006). Airway smooth muscle (ASM) hyperplasia/hypertrophy and increased deposition of extracellular matrix (ECM) (i.e. collagens I and III and fibronectin) are important elements of AWR (Amin et al., 2000). ASM cells also have an important immunological role in airway patho(physiology), being potent producers of pro-inflammatory mediators (Koziol-White and Panettieri, 2011). An effective therapy that targets ASM cells to reduce AWR is expected to reduce airway reactivity and symptoms in asthma (Camoretti-Mercado, 2009).
During asthma exacerbations, the leakiness of the vascular endothelium enables extravasation of serum proteins, including plasminogen, into airway wall tissue (Khor et al., 2009). Plasminogen is cleaved into plasmin (‘activation’) by one of two main plasminogen activators: urokinase- (uPA) or tissue-type (tPA). The roles and compartmentalization of these activators are different as are the functions of the plasmin they form (Kwaan and McMahon, 2009). Primarily, tPA-generated plasmin cleaves fibrin, which accumulates in the airway lumen in asthma (Wagers et al., 2004). Plasminogen activation associated with airspace fibrin would be beneficial, as fibrin inactivates surfactant (Jarjour and Enhorning, 1999; Wagers et al., 2004). However, uPA-generated plasmin in the interstitium of the inflamed airway wall may contribute to airway dysfunction due to its inflammatory (Zhang et al., 2007), remodelling (Schuliga et al., 2011) and angiogenic actions (Madureira et al., 2011). Sputum levels of uPA (Kowal et al., 2008) and plasma/airway levels of its receptor, uPAR (Chu et al., 2006; Barton et al., 2009), are elevated in asthma. There are multiple single nucleotide polymorphisms found within the promoters of the uPA (Begin et al., 2007) and uPAR (Barton et al., 2009) genes that have been linked to asthma. Furthermore, in a murine model of allergic inflammation, plasminogen gene deletion attenuates airway inflammation (Swaisgood et al., 2007), indicating that plasmin has a net inflammatory role in the airways, notwithstanding the functional benefits of airspace fibrinolysis.
Our laboratory recently showed that human ASM cells in culture convert plasminogen into plasmin in a manner mediated by uPA (Schuliga et al., 2011). It is likely that the plasmin formed has biological activity on the ASM cells. The direct effects of plasmin on other cell types include increases in cytokine production and cell proliferation (Laumonnier et al., 2006; Zhang et al., 2007). Plasmin-induced cell activation occurs via induction of multiple pathways, including the MAPK (ERK1/2 and p38), PI3K/Akt and/or JAK/STAT3 signal transduction pathways. The mechanisms by which plasmin elicits these responses may include: transactivation of heparin bound EGF (Roztocil et al., 2005); activation of the latent TGF-β (Coutts et al., 2001), and the protease-activated receptor-1 (PAR1; for receptor nomenclature see Alexander et al., 2011) (Pendurthi et al., 2002); and the cleavage of extracellular annexin A2 in the annexin A2/S100A10 hetero-tetramer (AIIt) (Laumonnier et al., 2006). The AIIt complex also has a role in plasminogen activation, accelerating the conversion of plasminogen into plasmin (Kassam et al., 1998). Furthermore, soluble extracellular AIIt stimulates macrophage cytokine production by binding the toll-like receptor 4 (TLR4) (Swisher et al., 2010).
In this study, we show that uPA-mediated plasminogen conversion to plasmin stimulates the proliferation of ASM cells, providing further evidence implicating plasminogen in tissue remodelling in airway pathophysiology.
Methods
Cell culture
Human ASM cultures were established using bronchi obtained from macroscopically normal resections of lung transplant patients (Alfred Hospital, Melbourne, Australia) as described previously (Tomlinson et al., 1994). Tissue specimens were obtained with approval from the University of Melbourne's Human Research Ethics Committee (HREC980168X). Briefly, smooth muscle bundles of bronchi were dissected from the surrounding connective tissue. The smooth muscle strips were then immersed in medium and partially digested in the presence of collagenase (3 mg·mL−1, 30 min). The strips were then chopped using a scalpel (∼2 mm3), and digested in medium containing elastase (0.5 mg·mL−1, 2 h), then collagenase (1 mg·mL−1, 12 h). The resulting cell suspension was pelleted and washed twice with PBS before being resuspended in DMEM supplemented with 10% v v−1 heat-inactivated fetal calf serum (FCS), L-glutamine (2 mM), sodium pyruvate (1 mM), non-essential amino acids (1% v v−1, Sigma, St Louis, MO, USA), penicillin-G (100 U·mL−1), streptomycin (100 μg·mL−1) and amphotericin B (2 μg·mL−1). The cells were then seeded into a 25 cm2 tissue culture flask and grown until a confluent monolayer was formed. Under these conditions, the cultures show uniform staining for smooth muscle specific α-actin, a mesenchymal cell marker (Schuliga et al., 2013), but not CD31, an endothelial marker.
For each experiment, separate primary cultures established from a minimum of four different donors (n = 4) were used. The cells were used between the fourth and 10th passage. Cultures were tested for contamination by mycoplasma, and only mycoplasma-free cultures were used. Cells were seeded onto 6, 24 or 96 well plates (2.5 × 104 cells cm-2) in DMEM containing supplements (L-glutamine, sodium pyruvate, non-essential amino acids) and heat-inactivated FCS (5% v v−1) and incubated at 37°C in air containing 5% CO2. Twenty-four hours after seeding, the medium was removed and the cells were then incubated in serum free-DMEM containing BSA (0.25% w v−1) and supplements (L-glutamine, sodium pyruvate and non-essential amino acids) for a further 24 h before the addition of human plasminogen (0.5–50 μg·mL−1, Roche, Indianapolis, IN, USA), plasmin (0.5– mU·mL−1, Roche) or bovine annexin A2 hetero-tetramer (200 ng·mL−1, Biodesign, Saco, ME, USA). In selected experiments, aprotinin (10 KIU·mL−1, Sigma), α2-antiplasmin (0.5 ug·mL−1, Calbiochem, La Jolla, CA, USA) or neutralizing annexin A2 (H-50) or TLR4 (HTA-125) antibodies (2 μg·mL−1, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) were also added. In other experiments, pharmacological inhibitors were added to cell culture medium at a final concentration of 10 μM, 30 min before the addition of plasmin(ogen). The final concentration of DMSO, the diluent for these inhibitors, was 0.1 % v v−1, and all cells were exposed to the same concentration of DMSO. The inhibitors used were: LY294002 for PI3K/Akt; PD98059 for ERK1/2; SB431542 for ALK-5, a TGF-β1 receptor kinase; and UK122 (Calbiochem) for uPA. The EGF receptor (EGFR) kinase inhibitor, AG1478, and the MMP inhibitor, ilomastat, were used at 0.5 and 2.5 μM respectively.
Preparation and transfer of conditioned medium
In selected experiments, the medium of ASM cells was replaced with cell conditioned medium (CM) of the same culture. Both the ‘donor’ and ‘naïve’ cells of the same culture were maintained in serum-free DMEM for 24 h in equivalent size tissue culture plates before the CM was transferred. In some CM transfer experiments, the levels of mRNA for either uPA or annexin A2 in the donor cells were knocked down by transfection with siRNA. For a subset of experiments, the CM from the donor cells was incubated with plasminogen or plasmin in the absence of cells under normal culturing conditions for 6 h before being transferred to the naïve cells. After the transfer of CM, the naïve cells were then maintained for 48 h before cell enumeration.
Cell enumeration
After 48 h of incubation with plasminogen or plasmin, attached cells were dissociated and harvested by incubation with trypsin (0.125 w·v−1)/EDTA (0.02% w·v−1) in PBS. For a selected experiment, detached cells in the culture medium were pelleted by centrifugation. Cells were resuspended in an appropriate volume of 0.25% v·v−1 BSA in PBS containing trypan blue (0.2% w·v−1) and viable cells counted (in duplicate) with the aid of a haemocytometre.
DNA synthesis – [3H]-thymidine incorporation
ASM cells were incubated with plasminogen or plasmin for 24 h in the presence of [3H]-thymidine (1 μCi·mL−1). Harvesting procedures followed the method described by Dicker and Rozengurt (1980). Radioactivity was measured by liquid scintillation counting.
MTT assay
The tetrazolium-based colorimetric MTT assay measures the reduction of yellow 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide. Cells grown in 96 well plates and incubated with plasminogen for 48 h were then incubated for an additional 1 h with MTT (0.25 mg·mL−1). Culture medium was then removed, and the resultant formazan crystals retained within the attached cells were dissolved in DMSO (100 μL) and the absorbance at 450 nm measured.
uPA, EGF receptor and annexin A2 mRNA knockdown
Cells were seeded in 24 well plates (50 000 cells per well) in antibiotic-free serum containing DMEM and transfected 20 h later with either 30 nM uPA, tPA, annexin A2 or negative control Stealth™ siRNA duplex oligonucleotides (Invitrogen, Carlsbad, CA, USA) or EGF receptor) siRNA using RNAiMax Lipofectamine (Invitrogen). The uPA duplex sequences used in this study were 5-GCC CUC CUC UCC UCC AGA AGA AUU A-3' (sequence A) and 5'-CAU CCU ACA CAA GGA CUA CAG CGC U-3' (sequence B). The annexin A2 duplex sequence used was 5'-GCG ACU ACC AGA AAG CGC UGC UGU A-3'. The tPA duplex sequence used was 5'-CCA UGG AAA CCA UGA UGU UUA CAU U-3'. EGFR siRNA duplex used in this study was 5'-CUC CAG AGG AUG UUC AAU ATT-3' and purchased from Genepharma (Shanghai, China). Six hours after the addition of lipofectamine-siRNA complex, cells were then incubated in serum-free DMEM for a further 24 h prior to the addition of plasminogen or plasmin.
Western blot analysis
Lysates of cells grown in 6 well plates were prepared, subjected to electrophoresis and electroblotted as described previously (Schuliga et al., 2010). In the case of uPA immunodetection, culture supernatants instead of cell lysates were resolved by electrophoresis. Following electroblotting, membranes were stained with Ponceau Red to verify uniform protein transfer, and then blocked with 5% skim milk in TBS-T (10 mM Tris; 75 mM NaCl; 0.1% Tween-20; pH 7.4) for 1 h. Membranes were incubated overnight at 4°C with either: anti-phospho-p44/42 MAPK antibody (rabbit polyclonal IgG, 1:1000, Cell Signaling Technology, Danvers, MA, USA); anti-phospho-Akt (rabbit polyclonal IgG, 1:1000, Cell Signaling Technology); anti-annexin A2 (rabbit polyclonal IgG, 1:250, Santa Cruz Biotechnology Inc.) or anti-uPA (rabbit polyclonal IgG, 1:500, Abcam, Cambridge, UK) diluted in 3% BSA in TBS-T. Blots were washed three times with TBS-T prior to incubation with secondary antibody, goat anti-mouse (Chemicon, Melbourne, Victoria, Australia) or anti-rabbit (Cell Signaling Technology) IgG-horse radish peroxidase conjugate, diluted 1:5000 in 5% skim milk/TBS-T) for 1 h at room temperature. After three washes with TBS-T, antigen was detected by enhanced chemiluminescence (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) using a Kodak IS4000 imaging system (Perkin Elmer, Boston, MA, USA). Membranes were then stripped by incubation with 30 mL of 0.1 M glycine solution (pH 2.9) for 1 h at room temperature, blocked and incubated with either primary: anti-p42/44 MAPK (goat polyclonal IgG, 1:1000, Santa Cruz Biotechnology Inc.); anti-Akt (rabbit polyclonal IgG, 1:1000, Cell Signaling Technology) or anti-β-actin (mouse monoclonal antibody, 1:5000, Abcam). Subsequent washes, secondary antibody incubation and chemiluminescence were as described above.
Plasmin assay
Plasmin-like activity was measured as described previously (Schuliga et al., 2011) using a fluorogenic substrate, d-val-leu-lys 7-amido-4- methylcoumarin, which emits an increased level of fluorescence upon cleavage by plasmin; fluorescence excitation 355 nm, emission 460 nm.
RNA extraction and real-time PCR
RNA was purified from cells maintained in either 24 or 96 well culture plates using Trizol (Invitrogen), according to the manufacturer's instructions. Reverse transcription of total RNA and the subsequent real-time PCR using an ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA) with the relevant forward and reverse primers were conducted as previously described (Schuliga et al., 2009). The following primers were used: Human CCND1, 5'-ACT ACC GCC TCA CAC GCT TC-3' (sense) and 5'-CAG TCT GGG TCA CAC TTG ATC AC-3' (antisense); human annexin A2, 5'-ACC TGG TTC AGT GCA TTC AGA A-3' (sense) and 5'-ACA GCC GAT CAG CAA AAT ACA G-3' (antisense); human EGFR, 5'-TTT GCT GAT TCA GGC TTG G-3' (sense) and 5'-AGA AAA CTG ACC ATG TTG CTT G-3' (antisense); and human 18S ribosomal RNA (18S rRNA) 5'-CGC CGC TAG AGG TGA AAT TC-3' (sense) and 5'-TTG GCA AAT GCT TTC GCT C-3' (antisense). The threshold cycle (CT) value determined for each gene of each sample was normalized against that obtained for 18S rRNA, which was included as internal control. For each sample, the level of mRNA for a particular gene is proportional to 2−(ΔCT), where ΔCT is equal to the CT value of the target gene minus the CT value of 18S rRNA.
Measurement of heparin-bound (HB)-EGF levels
Levels of HB-EGF in cell-CM obtained from human ASM cells were measured by specific sandwich ELISA using commercial ELISA kits (R&D, Minneapolis, MN, USA).
Caspase-3 assay
Attached and non-attached cells from 24 well plates were combined and stored at −70°C as cell pellets. Lysates were prepared by adding 125 μL HEPES (25 mM, pH 7.4) buffer containing triton X-100 (1% v v−1), glycerol (10% v v−1), DTT (5 mM) and PMSF (1 mM) to the cells, which were then subjected to two freeze-thaw cycles. Lysates were incubated with the specific caspase-3 substrate, Ac-DEVD-AMC (50 μM), at 37°C for 1 h. Levels of cleaved caspase-3 substrate were monitored at excitation 355 nm and emission 460 nm using a fluorescence plate reader. Caspase-3 activity was expressed as fluorescence units μg−1 of protein h−1. The protein concentrations were determined using the Bio-Rad protein assay kit (Hercules, CA, USA).
Statistical analysis
Data are presented as the mean ± SEM for n individual experiments, each experiment being conducted using cells from a separate donor. All data were statistically analysed using Graphpad Prism 5.0 (Graphpad, San Diego, CA, USA). In most cases, ANOVA with repeated measures was used to analyse the data, and treatment groups were compared with Bonferroni's post hoc tests. A value of P < 0.05 was considered to be statistically significant.
Results
Plasminogen and plasmin stimulate ASM cell proliferation
Incubation with plasminogen (1.5–15 μg·mL−1) for 48 h increased the number of attached ASM cells (P < 0.05, n = 7), corresponding to an increase in mitochondrial MTT reduction (P < 0.05, n = 6–10) (Figure 1A). Furthermore, incubation with 1.5–15 μg·mL−1 plasminogen increased 3H-thymidine incorporation (P < 0.05, n = 6, data not shown). Co-incubation with α2-antiplasmin (0.5 μg·mL−1), an inhibitor of plasmin, attenuated plasminogen (5 μg·mL−1)-stimulated increases in cell number (P < 0.05, n = 8) (Figure 1B). Addition of exogenous plasmin, at concentrations of 1.5–15 mU·mL−1, increased the number of attached cells after 48 h incubation (P < 0.05, n = 6) (Figure 1C). MTT reduction was greater than control at 5 mU·mL−1 plasmin, but less than control at 50 mU·mL−1 (P < 0.05, n = 10) (Figure 1C). It should be noted that at the highest concentration of plasmin (50 mU·mL−1) examined, the number of detached cells detected in the culture supernatant also had increased (P < 0.01, n = 5) (Table 1). Furthermore, plasmin (15 and 50 mU·mL−1) stimulated an increase in the activity of caspase 3, a pro-apoptotic enzyme (P < 0.05, n = 5) (Table 1). Plasmin-stimulated increases in cell number were inhibited by co-incubation with the serine protease inhibitor, aprotinin (10 KIU·mL−1) (P < 0.05, n = 8) (Figure 1D). The levels of the cell cycle protein, cyclin D1, were higher in ASM cells incubated with plasmin (5 mU·mL−1) (Figure 2A, B) for 24 h (P < 0.05, n = 4), as were the levels of cyclin D1 mRNA (Figure 2C) (P < 0.05, n = 5). Plasminogen and bFGF, a mitogen for ASM, also stimulated increases in cyclin D1 mRNA, whereas TGF-β, another ASM cell mitogen, had no effect (Figure 2C).
Figure 1.
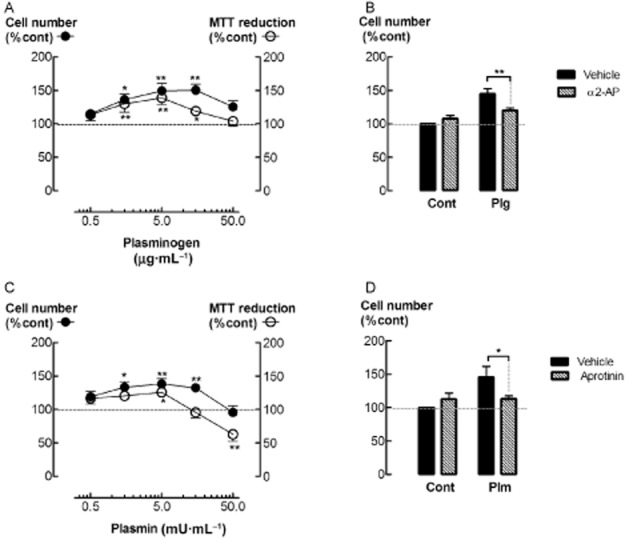
Plasminogen and plasmin increase ASM cell number. (A) Cell number and MTT reduction (%control) in cultures of ASM incubated with plasminogen for 48 h. (B) Cell number (%control) following incubation with plasminogen (Plg, 5 μg·mL−1) and/or α2-antiplasmin (α2-AP, 0.5 μg·mL−1). (C) Cell number and MTT reduction (%control) following incubation with plasmin for 48 h. (D) Cell number (%control) following incubation with plasmin (Plm, 5 mU·mL−1) and/or aprotinin (Apr, 10 KIU·mL−1). *P < 0.05, ** < 0.01, versus control (n = 6–10).
Table 1.
Plasmin increases cell detachment and caspase-3 activity
| Total cell number | Attached cell number | Non-attached cell number | Caspase-3 activity (total cells) | |
|---|---|---|---|---|
| Control | 100 | 97 ± 1 | 3 ± 1 | 100 |
| Plm, 15 mU·mL−1 | 134 ± 3** | 126 ± 3** | 6 ± 1 | 187 ± 20** |
| Plm, 50 mU·mL−1 | 97 ± 6 | 86 ± 6 | 14 ± 2** | 291 ± 86* |
The number of attached and non-attached cells (%control, total cells) in cultures of ASM cells following incubation with plasmin (Plm) for 48 h. Levels of caspase-3 activity (%control, total cells) were also measured using a fluorogenic assay. *P < 0.05, **P < 0.01 versus control (n = 5). For all treatments, the percentage of attached cells that were non-viable was negligible. However between 32 and 58% of the detached cells were non-viable.
Figure 2.
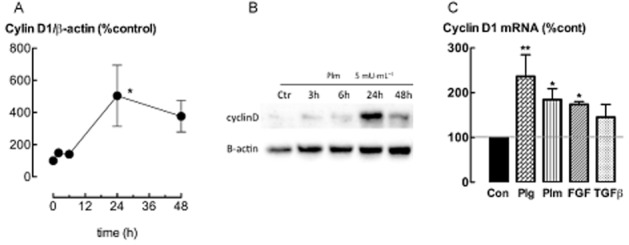
Plasmin increases cyclin D1 expression. (A, B) Levels of cyclin D1 protein (%control) in ASM cells incubated with plasmin (5 mU·mL−1) for 3–48 h. (C) Levels of cyclin D1 mRNA (%control) following incubation with plasminogen (Plg, 5 μg mL−1), plasmin (Plm, 5 mU·mL−1), bFGF (300 pM) or TGF-β (100 pM) for 24 h. *P < 0.05, ** < 0.01 versus control (n = 4–5).
uPA-mediated plasminogen activation regulates ASM cell proliferation
Urokinase (uPA) binds and cleaves plasminogen to form plasmin. The role of uPA in mediating the effects of plasminogen on ASM cell proliferation was investigated by knocking down the levels of uPA mRNA by transfection with gene-silencing RNA (siRNA). uPA siRNA sequence A was more effective than sequence B in reducing the conversion of plasminogen into plasmin (P < 0.05, n = 5) (Figure 3A), and was used in the following experiments. The tPA siRNA had no effect on plasmin formation. Much lower levels of uPA were detected in the culture medium of cells transfected with uPA siRNA as compared to cells transfected with control siRNA (5 μg·mL−1) for 24 h (P < 0.05, n = 5) (Figure 3B). Interestingly, in the presence of plasminogen, the majority of uPA was converted into the low molecular weight form. There was a decrease in cell number and MTT reduction in cultures transfected with uPA siRNA, as compared to control siRNA, following incubation with plasminogen for 48 h (Figure 3C, D) (P < 0.05, n = 6–7). Furthermore, the selective uPA inhibitor, UK122 (10 μM) (Zhu et al., 2007), attenuated increases in plasmin activity, cell number and MTT reduction following incubation with plasminogen (5 μg·mL−1) (P < 0.05, n = 5–8) (Figure 4A–C). Neither uPA knock down nor UK122 attenuated plasmin- or EGF-stimulated increases in ASM cell number (Figures 3 & 4).
Figure 3.
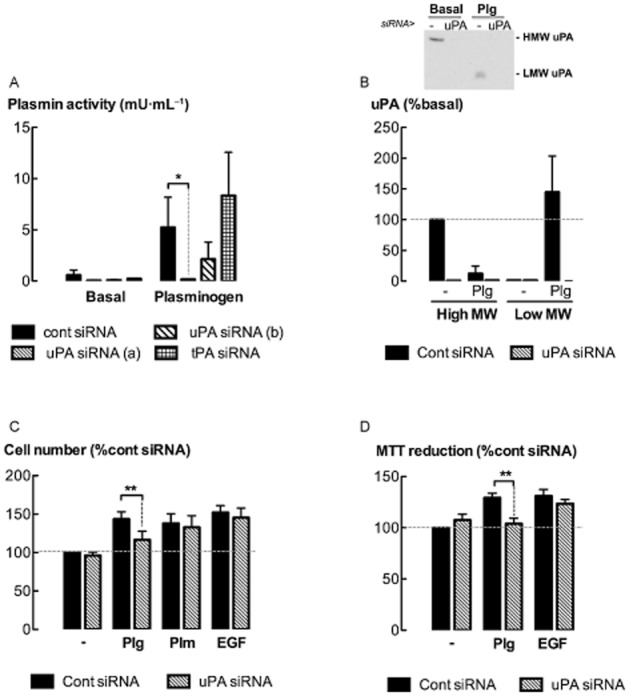
uPA knock down attenuates plasminogen-mediated increases in cell number. (A) Plasmin activity in the medium of ASM cells transfected with uPA (sequences a and b), tPA or cont siRNA, following incubation with plasminogen (5 μg·mL−1) for 8 h. (B) Levels of uPA [high MW (HMW) and low MW (LMW) forms] detected by Western blotting in the supernatants of uPA siRNA transfected cells (sequence a) following incubation with plasminogen for 8 h. Insert shows representative Western blot. (C, D) Cell number and MTT reduction following incubation with plasminogen (Plg, 5 μg·mL−1), plasmin (Plm, 5 mU·mL−1) (cell number only) or EGF (300 pM) for 48 h. *P < 0.05, **P < 0.01 (n = 4–8).
Figure 4.
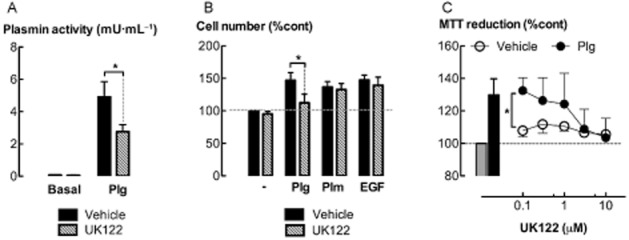
uPA inhibition attenuates plasminogen-mediated increases in cell number. (A) Plasmin activity in culture supernatants following incubation with plasminogen (5 μg·mL−1) and/or UK122 (10 μM) for 24 h. (B) The effect of UK122 on cell number following incubation with plasminogen (Plg, 5 μg·mL−1), plasmin (Plm, 5 mU·mL−1) or EGF (300 pM) for 48 h. (C) The effect of UK122 (0.1–10 μM) on MTT reduction following incubation with plasminogen (5 μg·mL−1) for 48 h. *P < 0.05 (n = 5–8).
Soluble extracellular uPA activates plasminogen
To examine whether soluble extracellular uPA mediates the proliferative effects of plasminogen, CM was incubated with plasminogen for 6 h in the absence of cells. The levels of plasmin formed were less in the CM of uPA siRNA transfected cells than control siRNA transfected cells (P < 0.05, n = 4) (Table 2). The CM-plasminogen incubates were transferred to ‘naive cells’, which were maintained for a further 48 h before cell enumeration. The number of cells maintained in CM incubated with plasminogen was higher than in the absence of plasminogen (P < 0.05, n = 4) (Table 2). However, cell number was less for the plasminogen incubates of CM from uPA knocked down cells (P < 0.05, n = 4).
Table 2.
Fluid phase plasminogen activation is uPA-dependent and stimulates an increase in cell number
| Control siRNA | uPA siRNA | |||
|---|---|---|---|---|
| Basal | Plasminogen | Basal | Plasminogen | |
| Plasmin activity (mU·mL−1) | 0.1 ± 0.1 | 7.2 ± 2.9 | 0.1 ± 0.1 | 0.1 ± 0.1** |
| Cell number (%Control) | 100 | 131 ± 5∧ | 109 ± 5 | 113 ± 5* |
In the absence of cells, CM from uPA or Cont siRNA-transfected cells was incubated with plasminogen (5 μg·mL−1) for 6 h (when plasmin activity was assessed). Plasminogen-naïve cells were then maintained in the plasminogen-treated CM for 48 h before cell enumeration. ∧P < 0.05 compared to basal + control siRNA. *P < 0.05, **P < 0.01 compared to plasminogen + control siRNA (n = 4).
Plasmin-mediated increases in cell number are regulated by PI3K/Akt and MAPK signal-transduction
Incubation of ASM cells with exogenous plasmin (5 mU·mL−1) for 30 min increased the phosphorylation of Akt and ERK1/2 (P < 0.05, n = 4) (Figure 5A, B). Inhibitors of Akt and ERK1/2 phosphorylation, LY294002 (10 μM) and PD98059 (10 μM) respectively, attenuated plasmin-stimulated increases in ASM cell number (P < 0.05, n = 5) (Figure 5c). The selectivity and effectiveness of these inhibitors in attenuating plasmin-stimulated phosphorylation of ERK1/2 and Akt was shown by Western blotting (Figure 5A, B).
Figure 5.
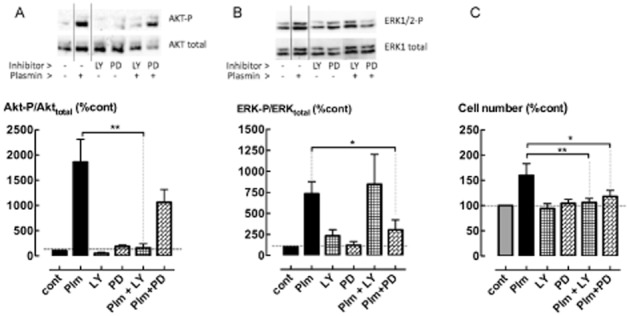
Plasmin-mediated increases in cell number are regulated by the PI3K/Akt and ERK1/2 MAPK signalling pathways. Phosphorylation of (A) Akt- and (B) ERK1/2 following incubation with plasmin (5 mU·mL−1) for 30 min in the presence of LY294002 (LY, 10 μM) or PD98059 (PD, 10 μM). Inserts show representative Western blots. (C) The effect of LY294002 and PD98059 on cell number following incubation with plasmin (5 mU·mL−1) for 48 h. *P < 0.05, **P < 0.01 (n = 4–6).
EGFR mediates the stimulatory action of plasmin
Plasmin biological activity can be mediated by proteolytic-activation of MMPs, which in turn release latent, ECM-bound EGF and TGF-β (Coutts et al., 2001; Roztocil et al., 2005). The MMP- and EGF receptor kinase inhibitors, ilomastat (2.5 μM) and AG1478 (0.5 μM) respectively, inhibited both plasminogen and plasmin-stimulated increases in ASM cell number and MTT reduction (P < 0.05, n = 8) (Figure 6A, B). However, the TGF-β-receptor kinase inhibitor, SB431542 (10 μM), had no effect (P > 0.05). Our laboratory has shown that ilomastat is pharmacologically active under these conditions (Schuliga et al., 2010). Moreover, the attenuation of EGF-stimulated cell proliferation by AG1478 demonstrates the activity of this inhibitor (Figure 6A, B). Inhibition of TGF-β-stimulated increases in ASM cell contractile protein gene expression (Schuliga et al., 2013) demonstrates SB431542 pharmacological activity under the conditions. Knock down of EGF receptor mRNA by siRNA transfection (Figure 6C) also attenuated plasmin-stimulated increases in cell number (P < 0.05, n = 5) (Figure 6D). The level of HB-EGF detected in the culture supernatant of ASM cells increased after incubation with plasmin (1.5–50 mU·mL−1) for 48 h (P < 0.05, n = 5) (Figure 6E).
Figure 6.
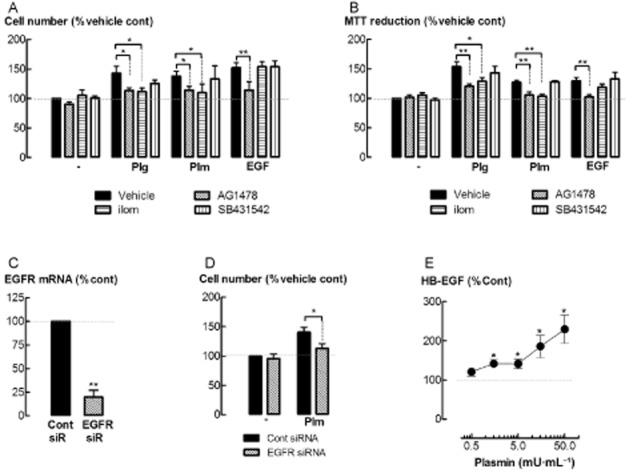
EGF-receptor (EGFR) kinase inhibition and EGF receptor knock down attenuates plasminogen-mediated increases in cell number. (A) Cell number and (B) MTT reduction following incubation with plasminogen (Plg, 5 μg·mL−1), plasmin (Plm, 5 mU·mL−1) or EGF (300 pM) with either ilomastat (ilom, 2.5 μM), AG1478 (0.5 μM) or SB431542 (10 μM) for 48 h. (C) Levels of EGF receptor mRNA in cells transfected with EGF receptor siRNA. (D) Cell number in EGF receptor siRNA transfected cells following incubation with plasmin (5 mU·mL−1) for 48 h. (E) Levels of HB-EGF detected in the culture supernatants of cells incubated with plasmin for 48 h. *P < 0.05, **P < 0.01 (n = 4–8).
Annexin A2 also mediates plasmin-stimulated proliferation
The role of annexin A2 as a mediator of plasminogen biological activity in ASM cells was investigated by annexin A2 knock down. Lower levels of annexin A2 protein in annexin A2 siRNA transfected cells were associated with a reduction in plasmin activity following incubation with plasminogen (5 μg·mL−1) for 24 h (P < 0.05, n = 4–5) (Figure 7A, B). Increases in cell number following incubation with either plasminogen or plasmin were attenuated by transfection with annexin A2 siRNA (P < 0.05, n = 9) (Figure 7C). However, annexin A2 knock down had no effect on thrombin or bFGF-stimulated increases in ASM cell number (data not shown). Similarly, cell number was less in cultures maintained in CM obtained from annexin A2 siRNA transfected cells, as compared to control siRNA transfected cells, following incubation with either plasminogen or plasmin (P < 0.05, n = 5) (Figure 7D).
Figure 7.
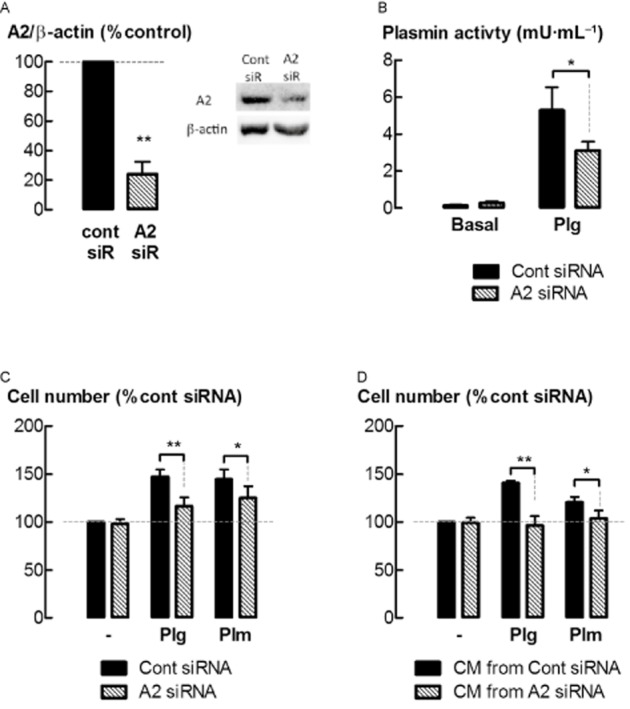
Annexin A2 regulates the conversion of plasminogen into plasmin and subsequent increases in cell number. Levels of annexin A2 (A2) in ASM cells were knocked down by transfection with gene-silencing RNA (siRNA). (A) Levels of A2 protein in siRNA-transfected ASM cells. (B) Plasmin activity in the medium of cells following siRNA transfection and incubation with plasminogen (5 μg·mL−1) for 24 h. (C) Cell number following siRNA transfection and incubation with plasminogen (5 μg·mL−1) or plasmin (5 mU·mL−1) for 48 h. (D) Cell number in cultures replenished with CM from siRNA transfected cells, and incubated with plasminogen (5 μg·mL−1) or plasmin (5 mU·mL−1) for 48 h. *P < 0.05, **P < 0.01 (n = 5–9).
Soluble extracellular annexin A2 mediates the proliferative effects of plasmin
To further evaluate the role of soluble extracellular annexin A2 in plasmin-stimulated proliferation, CM in the absence of cells was incubated with plasmin and/or anti-annexin A2 IgG for 6 h. Anti-annexin A2 IgG attenuated increases in cell number for cultures that were maintained in plasmin treated CM for 48 h (P < 0.05, n = 5) (Table 3). To neutralize the direct effects of plasmin, α2-anti-plasmin (α2-AP, 1 μg·mL−1) was added to the CM 30 min before being transferred to naïve cells. Despite the presence of α2-AP, the number of cells after 48 h was still greater for CM treated with plasmin than CM alone (P < 0.05, n = 4) (Table 3).
Table 3.
Anti-annexin A2 IgG attenuates plasmin-stimulated increases in cell number
| Control IgG | Annexin A2 IgG | |||
|---|---|---|---|---|
| Basal | Plasmin | Basal | Plasmin | |
| Vehicle | 100 | 140 ± 9 | 90 ± 12 | 111 ± 0.4** |
| α2-AP | 104 ± 6 | 127 ± 7∧∧ | 88 ± 13 | 93 ± 13* |
CM of untreated cells was incubated with plasmin (5 mU·mL) for 6 h in the presence of either anti-annexin A2 or control IgG (2 μg·mL−1). The medium of naïve cells was then replenished by the plasmin-treated CM and the cells were maintained for a further 48 h before cell enumeration. *P < 0.05, **P < 0.01 compared to plasmin + control IgG; ∧∧P < 0.01 compared to basal + control IgG (n = 5).
Soluble extracellular annexin A2 tetramer stimulates proliferation
Soluble extracellular annexin A2, in the form of the annexin A2 tetramer (AIIt), binds the TLR4 to mediate biological activity (Swisher et al., 2010). The addition of exogenous AIIt stimulated an increase in cell number after 48 h of incubation (P < 0.05, n = 5) (Figure 8A). AIIt-stimulated increases in cell number were attenuated by co-incubation with anti-TLR4 monoclonal IgG antibody (1 μg·mL−1) (P < 0.05, n = 4) (Figure 8B). LPS at 10 ng·mL−1, a TLR4 agonist, did not have a significant effect on cell number.
Figure 8.
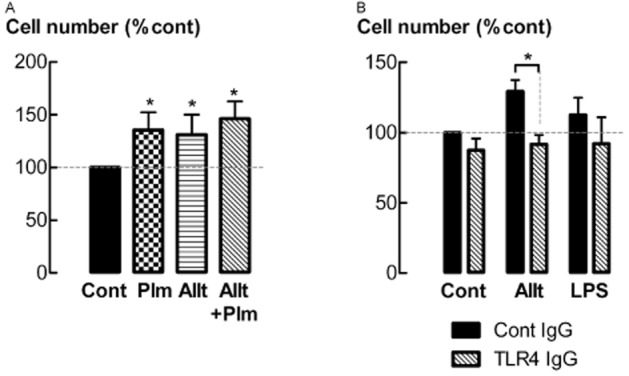
The annexin A2 hetero-tetramer (AIIt) stimulates increases in cell number via binding to TLR4. (A) Cell number following incubation with AIIt (200 ng·mL−1) and/or plasmin (Plm, 5 mU·mL−1) for 48 h. (B) Cell number following incubation with AIIt (200 ng·mL−1) or LPS (10 ng·mL−1), with or without anti-TLR4 IgG (1 μg·mL−1) for 48 h. *P < 0.05 (n = 3–5).
Discussion
ASM hyperplasia is an important contributor to AWR in severe asthma (Hassan et al., 2010). ASM cells convert plasminogen into plasmin (Schuliga et al., 2011), a protease with mitogenic activity (Nicholl et al., 2005). In this study, plasminogen was shown to regulate ASM function by stimulating a larger increase in cell proliferation, in a manner requiring plasmin formation. uPA regulates the mitogenic actions of plasminogen by cleaving it into plasmin. The resulting plasmin in turn activates signal transduction pathways involved in cell proliferation, including the ERK1/2 and PI3K/Akt pathways. MMPs, EGFR and annexin A2 facilitate plasminogen-stimulated ASM proliferation (Figure 9). Our observations suggest that plasminogen may be an important regulator of ASM function, complementing existing evidence for the potential relevance of plasminogen in airway (patho)physiology.
Figure 9.
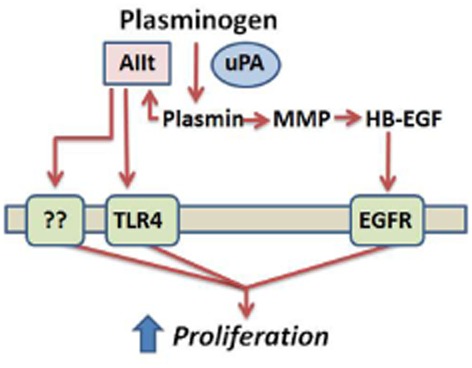
Urokinase and annexin A2 regulate plasminogen-stimulated ASM cell proliferation. The proposed effects of soluble, extracellular uPA and annexin A2 (as a component of AIIt) on plasminogen-stimulated cell proliferation occurs in the fluid phase and involves plasmin-activated signalling. The latter potentially involves MMP and annexin A2 activation in the fluid phase and subsequent receptor (i.e. EGF receptor and TLR4) activation.
As a consequence of vascular leak, plasminogen extravasates into the airway wall interstitium during injury and inflammation. Levels of plasminogen in the airways increase during asthma exacerbation (Brims et al., 2010) and are likely to be (patho)physiologically relevant. In this study, concentrations of plasminogen, well below those present in plasma (>100 μg·mL−1) (Cederholm-Williams, 1981), stimulated the proliferation of ASM cells in vitro. Proliferation was measured by cell enumeration and further evidenced by increases in: 3H-thymidine incorporation; MTT reduction; and the levels of cyclin D1, a cell cycle protein. While the MTT assay is not a definitive cell proliferation assay, as mitochondrial activity per unit cell can be influenced by many factors, including cell viability, this assay was used routinely throughout the study to complement cell enumeration data. The observation that plasminogen stimulates ASM cell proliferation extends our previous finding that ASM cells activate plasminogen via the activity of uPA (Schuliga et al., 2011). There have been few investigations that have examined the potential role of interstitial plasmin formation in the context of airway disease and injury. However, in vascular disease and injury, it is considered that extravascular plasminogen activation contributes to tissue remodelling in the vascular wall (Fay et al., 2007). Plasminogen is thought to have an important role in the proliferation and migration of vascular smooth muscle cells in neointima formation following vascular injury (Levin and Loskutoff, 1979).
In this study, the proliferative response of ASM cells was mediated by plasmin, as α2-AP attenuated the mitogenic actions of plasminogen. Furthermore, exogenous plasmin also stimulated cell proliferation. The effects of exogenous plasmin occurred at low mU·mL−1 activity range (1.5–15 mU·mL−1), comparable to the range of levels of plasmin activity detected in cultures of ASM cells incubated with 5–15 μg·mL−1 plasminogen for 24 h (Schuliga et al., 2011). The mitogenic effect of plasmin is likely to require enzymatically active plasmin, as the serine protease inhibitor, aprotinin, inhibited this response. Interestingly, at the highest concentration of exogenous plasmin examined (50 mU·mL−1), there was a decrease in MTT metabolism below basal, possibly due to decreased cell viability and/or cell loss. The latter is supported by our observations that plasmin, at 50 mU·mL−1 increased the number of detached cells in culture, the majority of which (>50%) were non-viable. This increase in cell detachment corresponded with an increase in cellular activity of caspase 3, a pro-apoptotic enzyme. It has been reported that plasminogen stimulates fibroblast apoptosis in vitro by proteolytic degradation of secreted fibronectin, which in turn leads to cell detachment and anoikis (Horowitz et al., 2008). The potential effect of plasmin on cell detachment and/or viability may explain why both exogenous plasminogen and plasmin exhibited bell-shaped concentration-response curves in regard to cell number. It seems that plasminogen at lower concentrations (1.5–15 μg·mL−1) is mitogenic, whereas at higher concentrations, plasminogen is pro-apoptotic.
In the extravascular compartment, uPA-generated plasmin is associated with cell-mediated proteolysis and cell activation in inflammation and cancer (Cook et al., 2010; Henneke et al., 2010). There is also growing recognition that urokinase has a role in airway inflammation in conditions such as asthma (Brooks et al., 2006; Chu et al., 2006; Begin et al., 2007; Kowal et al., 2008; Barton et al., 2009; Schuliga et al., 2011). In this study, knock down or inhibition of uPA in cultures of ASM cells attenuated increases in both plasmin formation and cell proliferation following incubation with plasminogen. However, neither uPA knock down nor inhibition influenced proliferation in response to exogenous plasmin. Therefore, it seems that uPA-mediated plasmin formation is required for the mitogenic actions of plasminogen on ASM cells in vitro, supporting the notion that interstitial uPA may contribute to AWR in asthma. uPA also has roles in ASM cell migration (Carlin et al., 2005) and airway inflammatory cell infiltration (Brooks et al., 2006). The observation that inhaled aerosolized uPA attenuates AWR in allergen-challenged mice (Kuramoto et al., 2009) has perhaps diminished the perceived importance of uPA in asthma. However, this effect of supraphysiological concentrations of uPA is more likely to be a consequence of beneficial airspace fibrinolysis, than a result of any effect of uPA in the interstitium. Fibrinolysis in the airway lumen is a physiological role of tPA, which unlike uPA, shows fibrin-enhanced proteolytic activity (Wagers et al., 2004). Studies involving uPA gene deletion or uPA inhibition in animal models of allergic airway inflammation are required to provide greater insight into the role of uPA in asthma pathophysiology. Interestingly, uPA gene deletion in mice reduces endotoxin-induced inflammation (Abraham et al., 2003) and hypoxia-induced pulmonary hypertension and vascular remodelling (Levi et al., 2001).
uPA is a secreted, soluble extracellular protein that can also associate with the cell surface via interaction with its membrane-tethered receptor, uPAR (Stewart et al., 2012). Hence, uPA-generated plasmin can be formed by both fluid-phase and cell surface localized systems. However, we have shown previously that plasminogen activation by ASM cells in vitro occurs primarily in the culture supernatant (fluid-phase), and not at the cell surface (Schuliga et al., 2011). Furthermore, it has been reported for human carcinoma cell lines in vitro, that the majority of uPA-generated plasmin formation occurs in the fluid-phase compartment (Deryugina and Quigley, 2012). In the current study, experiments using ASM cell-CM show that plasminogen activation in the fluid phase is mediated by soluble, extracellular uPA. The conversion of plasminogen into plasmin was lower in CM obtained from uPA siRNA-transfected cells as compared to control siRNA-transfected cells. When naïve cells were incubated with CM from uPA knock-down cells, plasminogen-stimulated increases in cell number were attenuated. This observation suggests that extracellular uPA-generated plasmin stimulates ASM cell proliferation.
As previously described, uPA-mediated activation of plasminogen by ASM cells in vitro is an extracellular phenomenon. However, the extracellular compartment of the cell culture system used in this study is not truly representative of that in vivo. In ASM bundles, cells are tightly packed together, where the volume of interstitial fluid in relation to the cell surface area is much lower than the equivalent ratio for the cell culture model. Within the ASM bundle and its immediate surroundings, uPA-generated plasmin would occur in vicinity close to the cell surface. Consequently, cell surface plasminogen and uPARs may have a more prominent role, localizing the plasmin formed at the cell membrane or in close proximity. There are a number of membrane-associated proteins with C-terminal lysines that bind to the kringle domain of plasminogen (i.e. α-enolase, S100A10 and the plasminogen receptor, Plg-Rkt). The actions of surface-generated plasmin and fluid phase-generated plasmin would differ because the range and effective concentration of substrates for plasmin would be different in these distinct compartments. When plasmin formation is localized at the cell surface, membrane receptor substrates of plasmin, such as PAR-1 (Pendurthi et al., 2002), are likely to assume much greater significance for the signalling effects of plasmin. In our study, exogenous plasmin was added to ASM cell cultures to investigate the consequences of uPA-mediated plasmin formation. However, for the aforementioned reasons, the array of effects of exogenous plasmin in the fluid phase as compared to that generated at the cell surface would not be expected to be the same, even though overlap is likely.
Plasmin is a potent activator of both inflammatory and mesenchymal cells. ERK1/2 and PI3K/Akt pathways are reported to be rapidly activated by plasmin in macrophages (Laumonnier et al., 2006; Zhang et al., 2007) and vascular smooth muscle cells (Roztocil et al., 2005). In ASM cells, the activation of these pathways by incubation with plasmin for 30 min was shown by phosphorylation of intermediates. Furthermore, the experiments using pharmacological inhibitors of these pathways (PD98059 for ERK1/2 and LY294002 for PI3K/Akt) revealed their roles in regulating plasmin-stimulated ASM cell proliferation. As in vascular smooth muscle, these pathways regulate plasmin-stimulated ASM proliferation (Nicholl et al., 2005; Roztocil et al., 2005).
Plasminogen-mediated cellular activity can occur by multiple mechanisms. Plasmin catalyzes the proteolytic activation of MMPs, which release the otherwise latent forms of EGF and TGF-β (Coutts et al., 2001; Roztocil et al., 2005). Plasmin is involved in the activation of a number of MMPs including MMP-1, MMP-2, MMP-3, MMP-9, MMP-13 and MMP14 (Deryugina and Quigley, 2012). The activation of MMP-1, an efficient interstitial collagenase, is a well-known example of the importance of plasmin as an MMP-activating enzyme. Our laboratory has shown that MMP-1, MMP-2 and MMP-14 are expressed by human ASM cells in culture (Schuliga et al., 2010). Plasminogen activation by ASM cells is associated with MMP activation and increases in collageneolytic activity (Schuliga et al., 2011). ASM cell proliferation has been reported to be stimulated by both EGF (Gosens et al., 2006) and TGF-β (Chen and Khalil, 2006). In this study, the inhibitor of the EGF receptor kinase, AG1478, and the MMP inhibitor ilomastat, attenuated plasmin(ogen)-stimulated ASM cell proliferation, as did EGF receptor knock down. Furthermore, incubation with plasmin stimulated an increase in the levels of HB-EGF detected in the culture supernatant. HB-EGF is an important ligand of the EGF receptor, and is released from heparin by the proteolytic actions of MMPs. HB-EGF is expressed in the airway epithelium and ASM in situ (Hirota et al., 2012). Our observations suggest that EGF liberation via MMP activation, but not mobilization of TGF-β, contributes to the proliferative effects of plasmin(ogen) on ASM cells. A lack of evidence for a role of TGF-β in plasmin-mediated ASM proliferation may be because the mitogenic actions of TGF-β occur indirectly via bFGF production (Bosse et al., 2006). The effects of TGF-β-induced bFGF autocrine signalling in response to plasmin would take longer than the direct effects of EGF, and therefore may not be as readily evident following a 48 h incubation with plasmin(ogen).
This study provides evidence that the annexin A2 hetero-tetramer (AIIt) also mediates the effects of plasminogen on ASM cell proliferation. AIIt is a protein complex comprised of both annexin A2 and S100A10 (p11), which can found as a soluble form in the extracellular compartment or in association with the extracellular side of the cell membrane. AIIt binds plasminogen and plasminogen activators to accelerate the conversion of plasminogen into plasmin (Kassam et al., 1998). In this study, annexin A2 knock down attenuated plasminogen activation by ASM and plasminogen-stimulated increases in cell proliferation. Furthermore, knock-down of annexin A2 attenuated increases in cell number following incubation with exogenous plasmin. Annexin A2 knock down had no effect on the proliferative response of other mitogens examined, suggesting that the effects of annexin A2 are selective for plasmin-stimulated proliferation. AIIt is also a receptor for plasmin, mediating proinflammatory responses in human macrophages (Laumonnier et al., 2006). However, the mechanism by which AIIt acts as a signal transducer of plasmin remains unclear. There is evidence that plasmin binds to and cleaves the N-terminal end of annexin A2 in the process of plasmin-evoked signalling (Laumonnier et al., 2006). Our data support a signal transducer role for soluble extracellular annexin A2 in ASM cell proliferation. When plasmin(ogen) naïve cells were replenished with ASM cell-CM from annexin A2 knock-down cells, plasmin-stimulated proliferation was attenuated. Responses to plasmin were similarly reduced in cells replenished in CM that had been pre-incubated with an anti-annexin A2 IgG that binds the bioactive, N-terminus of annexin A2. These observations suggest that soluble, extracellular annexin A2 has a role in plasmin-activated signalling, possibly being cleaved at the N-terminus by plasmin in the process (Figure 9). This notion is supported by the observation that plasmin neutralization using α2-AP did not inhibit the mitogenic effect of plasmin when incubated with CM for 6 h.
Soluble AIIt stimulates cytokine release from macrophages in a TLR4-dependent manner (Swisher et al., 2010). In the current study, TLR4 was also shown to have a role in mediating the effects of AIIt on ASM proliferation. However, further investigation is required to evaluate the mechanism by which AIIt action involves TLR4, and the involvement of plasmin in this process. Interestingly, Katherine Hajjar and colleagues have reported that plasmin induces the dissociation of annexin A2 from AIIt in cultures of endothelial cells, allowing annexin A2 to activate TLR4 (He et al., 2011). LPS, an activator of TLR4, has been shown to stimulate ASM proliferation (Pera et al., 2010). However LPS, at the concentration used in this study (10 ng·mL−1) had no significant effect on cell proliferation, unlike AIIt (200 ng·mL−1). Suggestions that annexin A2 is a key mediator of asthma pathophysiology are supported by evidence implicating annexin A2 involvement in disease (Hedhli et al., 2012). Similar to uPA, the level of annexin A2 is elevated in many cancers (Inokuchi et al., 2009) and is considered to have important roles in tumour progression and tissue invasion (Sharma et al., 2006). Exploring the role of a plasminogen/annexin A2 axis in AWR may identify new strategies to treat asthma.
Selective targeting of uPA is a potential therapeutic strategy to treat chronic respiratory diseases. Small molecule inhibitors such as analogues of 4-oxazolidinone (i.e. UK122), p-aminobenzamidine (i.e. CJ-463) and 3-aminophenylalanine (i.e. WX-UK1) exhibit high selectively for uPA and have been validated in cell and animal studies to assess uPA inhibition as a strategy to treat cancer (Zhu et al., 2007; Henneke et al., 2010; Schmitt et al., 2011). In clinical trial programmes, uPA inhibitors have been shown to be well-tolerated and have provided promising results, greatly increasing their translational potential for the treatment of other diseases including asthma. Tumour cell invasion and hepatic fibrinolysis in vivo are inhibited by uPA and uPAR functional blocking antibodies (Botkjaer et al., 2011; Lund et al., 2012), providing evidence for the feasibility of targeting uPA using antibody-based strategies. Furthermore, in a mouse model of acute lung injury, uPA antibodies were shown to reduce inflammation and oedema (Wang et al., 2006). Additionally, the systemic administration of annexin A2 antibody inhibits tumour growth/metastasis in murine cancer models in vivo without detectable toxicity (Sharma et al., 2012; Zheng and Jaffee, 2012).
We have shown that plasminogen stimulates the proliferation of human ASM cells in a manner mediated by plasmin, uPA and annexin A2. As ASM hyperplasia has an important role in AWR, these findings suggest that uPA, and downstream mediators of plasmin (i.e. annexin A2) are potential targets for drug treatment for asthma and other chronic airway diseases.
Acknowledgments
We thank the Departments of Respiratory Medicine, Surgery, and Anatomical Pathology, Alfred Hospital, Australia and Professor Catriona MacClean for assistance in obtaining human biopsies. This work was supported by the NHMRC (Australia) research grant 1022048.
Glossary
- ASM
airway smooth muscle
- AIIt
annexin A2 tetramer
- AWR
airway wall remodelling
- PAR
protease-activated receptor
- TLR4
toll-like receptor 4
- tPA
tissue-type plasminogen activator
- uPA
urokinase plasminogen activator
Conflict of interest
None.
References
- Abraham E, Gyetko MR, Kuhn K, Arcaroli J, Strassheim D, Park JS, et al. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J Immunol. 2003;170:5644–5651. doi: 10.4049/jimmunol.170.11.5644. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin K, Ludviksdottir D, Janson C, Nettelbladt O, Bjornsson E, Roomans GM, et al. Inflammation and structural changes in the airways of patients with atopic and nonatopic asthma. BHR Group. Am J Respir Crit Care Med. 2000;162:2295–2301. doi: 10.1164/ajrccm.162.6.9912001. [DOI] [PubMed] [Google Scholar]
- Barton SJ, Koppelman GH, Vonk JM, Browning CA, Nolte IM, Stewart CE, et al. PLAUR polymorphisms are associated with asthma, PLAUR levels, and lung function decline. J Allergy Clin Immunol. 2009;123:1391–1400. doi: 10.1016/j.jaci.2009.03.014. e1317. [DOI] [PubMed] [Google Scholar]
- Begin P, Tremblay K, Daley D, Lemire M, Claveau S, Salesse C, et al. Association of urokinase-type plasminogen activator with asthma and atopy. Am J Respir Crit Care Med. 2007;175:1109–1116. doi: 10.1164/rccm.200607-1012OC. [DOI] [PubMed] [Google Scholar]
- Bosse Y, Thompson C, Stankova J, Rola-Pleszczynski M. Fibroblast growth factor 2 and transforming growth factor beta1 synergism in human bronchial smooth muscle cell proliferation. Am J Respir Cell Mol Biol. 2006;34:746–753. doi: 10.1165/rcmb.2005-0309OC. [DOI] [PubMed] [Google Scholar]
- Botkjaer KA, Fogh S, Bekes EC, Chen Z, Blouse GE, Jensen JM, et al. Targeting the autolysis loop of urokinase-type plasminogen activator with conformation-specific monoclonal antibodies. Biochem J. 2011;438:39–51. doi: 10.1042/BJ20110129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brims FJ, Chauhan AJ, Higgins B, Shute JK. Up-regulation of the extrinsic coagulation pathway in acute asthma – a case study. J Asthma. 2010;47:695–698. doi: 10.3109/02770901003682802. [DOI] [PubMed] [Google Scholar]
- Brooks AM, Bates ME, Vrtis RF, Jarjour NN, Bertics PJ, Sedgwick JB. Urokinase-type plasminogen activator modulates airway eosinophil adhesion in asthma. Am J Respir Cell Mol Biol. 2006;35:503–511. doi: 10.1165/rcmb.2006-0113OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camoretti-Mercado B. Targeting the airway smooth muscle for asthma treatment. Transl Res. 2009;154:165–174. doi: 10.1016/j.trsl.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin SM, Resink TJ, Tamm M, Roth M. Urokinase signal transduction and its role in cell migration. FASEB J. 2005;19:195–202. doi: 10.1096/fj.04-1644com. [DOI] [PubMed] [Google Scholar]
- Cederholm-Williams SA. Concentration of plasminogen and antiplasmin in plasma and serum. J Clin Pathol. 1981;34:979–981. doi: 10.1136/jcp.34.9.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Khalil N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir Res. 2006;7:2. doi: 10.1186/1465-9921-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu EK, Cheng J, Foley JS, Mecham BH, Owen CA, Haley KJ, et al. Induction of the plasminogen activator system by mechanical stimulation of human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2006;35:628–638. doi: 10.1165/rcmb.2006-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook AD, De Nardo CM, Braine EL, Turner AL, Vlahos R, Way KJ, et al. Urokinase-type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther. 2010;12:R37. doi: 10.1186/ar2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts A, Chen G, Stephens N, Hirst S, Douglas D, Eichholtz T, et al. Release of biologically active TGF-beta from airway smooth muscle cells induces autocrine synthesis of collagen. Am J Physiol Lung Cell Mol Physiol. 2001;280:L999–1008. doi: 10.1152/ajplung.2001.280.5.L999. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Quigley JP. Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol. 2012;2012:564259. doi: 10.1155/2012/564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicker P, Rozengurt E. Phorbol esters and vasopressin stimulate DNA synthesis by a common mechanism. Nature. 1980;287:607–612. doi: 10.1038/287607a0. [DOI] [PubMed] [Google Scholar]
- Fay WP, Garg N, Sunkar M. Vascular functions of the plasminogen activation system. Arterioscler Thromb Vasc Biol. 2007;27:1231–1237. doi: 10.1161/ATVBAHA.107.140046. [DOI] [PubMed] [Google Scholar]
- Gosens R, Grootte Bromhaar MM, Maarsingh H, ten Damme A, Meurs H, Zaagsma J, et al. Bradykinin augments EGF-induced airway smooth muscle proliferation by activation of conventional protein kinase C isoenzymes. Eur J Pharmacol. 2006;535:253–262. doi: 10.1016/j.ejphar.2006.01.065. [DOI] [PubMed] [Google Scholar]
- Hassan M, Jo T, Risse PA, Tolloczko B, Lemiere C, Olivenstein R, et al. Airway smooth muscle remodeling is a dynamic process in severe long-standing asthma. J Allergy Clin Immunol. 2010;125:1037–1045. doi: 10.1016/j.jaci.2010.02.031. e1033. [DOI] [PubMed] [Google Scholar]
- He KL, Sui G, Xiong H, Broekman MJ, Huang B, Marcus AJ, et al. Feedback regulation of endothelial cell surface plasmin generation by PKC-dependent phosphorylation of annexin A2. J Biol Chem. 2011;286:15428–15439. doi: 10.1074/jbc.M110.185058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedhli N, Falcone DJ, Huang B, Cesarman-Maus G, Kraemer R, Zhai H, et al. The annexin A2/S100A10 system in health and disease: emerging paradigms. J Biomed Biotechnol. 2012;2012:406273. doi: 10.1155/2012/406273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneke I, Greschus S, Savai R, Korfei M, Markart P, Mahavadi P, et al. Inhibition of urokinase activity reduces primary tumor growth and metastasis formation in a murine lung carcinoma model. Am J Respir Crit Care Med. 2010;181:611–619. doi: 10.1164/rccm.200903-0342OC. [DOI] [PubMed] [Google Scholar]
- Hirota N, Risse PA, Novali M, McGovern T, Al-Alwan L, McCuaig S, et al. Histamine may induce airway remodeling through release of epidermal growth factor receptor ligands from bronchial epithelial cells. FASEB J. 2012;26:1704–1716. doi: 10.1096/fj.11-197061. [DOI] [PubMed] [Google Scholar]
- Holgate ST, Holloway J, Wilson S, Howarth PH, Haitchi HM, Babu S, et al. Understanding the pathophysiology of severe asthma to generate new therapeutic opportunities. J Allergy Clin Immunol. 2006;117:496–506. doi: 10.1016/j.jaci.2006.01.039. quiz 507. [DOI] [PubMed] [Google Scholar]
- Horowitz JC, Rogers DS, Simon RH, Sisson TH, Thannickal VJ. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am J Respir Cell Mol Biol. 2008;38:78–87. doi: 10.1165/rcmb.2007-0174OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuchi J, Narula N, Yee DS, Skarecky DW, Lau A, Ornstein DK, et al. Annexin A2 positively contributes to the malignant phenotype and secretion of IL-6 in DU145 prostate cancer cells. Int J Cancer. 2009;124:68–74. doi: 10.1002/ijc.23928. [DOI] [PubMed] [Google Scholar]
- James AL, Wenzel S. Clinical relevance of airway remodelling in airway diseases. Eur Respir J. 2007;30:134–155. doi: 10.1183/09031936.00146905. [DOI] [PubMed] [Google Scholar]
- Jarjour NN, Enhorning G. Antigen-induced airway inflammation in atopic subjects generates dysfunction of pulmonary surfactant. Am J Respir Crit Care Med. 1999;160:336–341. doi: 10.1164/ajrccm.160.1.9806155. [DOI] [PubMed] [Google Scholar]
- Kassam G, Choi KS, Ghuman J, Kang HM, Fitzpatrick SL, Zackson T, et al. The role of annexin II tetramer in the activation of plasminogen. J Biol Chem. 1998;273:4790–4799. doi: 10.1074/jbc.273.8.4790. [DOI] [PubMed] [Google Scholar]
- Khor YH, Teoh AK, Lam SM, Mo DC, Weston S, Reid DW, et al. Increased vascular permeability precedes cellular inflammation as asthma control deteriorates. Clin Exp Allergy. 2009;39:1659–1667. doi: 10.1111/j.1365-2222.2009.03349.x. [DOI] [PubMed] [Google Scholar]
- Kowal K, Zukowski S, Moniuszko M, Bodzenta-Lukaszyk A. Plasminogen activator inhibitor-1 (PAI-1) and urokinase plasminogen activator (uPA) in sputum of allergic asthma patients. Folia Histochem Cytobiol. 2008;46:193–198. doi: 10.2478/v10042-008-0029-0. [DOI] [PubMed] [Google Scholar]
- Koziol-White CJ, Panettieri RA., Jr Airway smooth muscle and immunomodulation in acute exacerbations of airway disease. Immunol Rev. 2011;242:178–185. doi: 10.1111/j.1600-065X.2011.01022.x. [DOI] [PubMed] [Google Scholar]
- Kuramoto E, Nishiuma T, Kobayashi K, Yamamoto M, Kono Y, Funada Y, et al. Inhalation of urokinase-type plasminogen activator reduces airway remodeling in a murine asthma model. Am J Physiol Lung Cell Mol Physiol. 2009;296:L337–L346. doi: 10.1152/ajplung.90434.2008. [DOI] [PubMed] [Google Scholar]
- Kwaan HC, McMahon B. The role of plasminogen-plasmin system in cancer. Cancer Treat Res. 2009;148:43–66. doi: 10.1007/978-0-387-79962-9_4. [DOI] [PubMed] [Google Scholar]
- Laumonnier Y, Syrovets T, Burysek L, Simmet T. Identification of the annexin A2 heterotetramer as a receptor for the plasmin-induced signaling in human peripheral monocytes. Blood. 2006;107:3342–3349. doi: 10.1182/blood-2005-07-2840. [DOI] [PubMed] [Google Scholar]
- Levi M, Moons L, Bouche A, Shapiro SD, Collen D, Carmeliet P. Deficiency of urokinase-type plasminogen activator-mediated plasmin generation impairs vascular remodeling during hypoxia-induced pulmonary hypertension in mice. Circulation. 2001;103:2014–2020. doi: 10.1161/01.cir.103.15.2014. [DOI] [PubMed] [Google Scholar]
- Levin EG, Loskutoff DJ. Comparative studies of the fibrinolytic activity of cultured vascular cells. Thromb Res. 1979;15:869–878. doi: 10.1016/0049-3848(79)90195-6. [DOI] [PubMed] [Google Scholar]
- Lund IK, Rasch MG, Ingvarsen S, Pass J, Madsen DH, Engelholm LH, et al. Inhibitory monoclonal antibodies against mouse proteases raised in gene-deficient mice block proteolytic functions in vivo. Front Pharmacol. 2012;3:122. doi: 10.3389/fphar.2012.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madureira PA, Surette AP, Phipps KD, Taboski MA, Miller VA, Waisman DM. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood. 2011;118:4789–4797. doi: 10.1182/blood-2011-06-334672. [DOI] [PubMed] [Google Scholar]
- Nicholl SM, Roztocil E, Galaria, Davies MG. Plasmin induces smooth muscle cell proliferation. J Surg Res. 2005;127:39–45. doi: 10.1016/j.jss.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Pendurthi UR, Ngyuen M, Andrade-Gordon P, Petersen LC, Rao LV. Plasmin induces Cyr61 gene expression in fibroblasts via protease-activated receptor-1 and p44/42 mitogen-activated protein kinase-dependent signaling pathway. Arterioscler Thromb Vasc Biol. 2002;22:1421–1426. doi: 10.1161/01.atv.0000030200.59331.3f. [DOI] [PubMed] [Google Scholar]
- Pera T, Gosens R, Lesterhuis AH, Sami R, van der Toorn M, Zaagsma J, et al. Cigarette smoke and lipopolysaccharide induce a proliferative airway smooth muscle phenotype. Respir Res. 2010;11:48. doi: 10.1186/1465-9921-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roztocil E, Nicholl SM, Galaria, Davies MG. Plasmin-induced smooth muscle cell proliferation requires epidermal growth factor activation through an extracellular pathway. Surgery. 2005;138:180–186. doi: 10.1016/j.surg.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Schmitt M, Harbeck N, Brunner N, Janicke F, Meisner C, Muhlenweg B, et al. Cancer therapy trials employing level-of-evidence-1 disease forecast cancer biomarkers uPA and its inhibitor PAI-1. Expert Rev Mol Diagn. 2011;11:617–634. doi: 10.1586/erm.11.47. [DOI] [PubMed] [Google Scholar]
- Schuliga M, Ong SC, Soon L, Zal F, Harris T, Stewart AG. Airway smooth muscle remodels pericellular collagen fibrils: implications for proliferation. Am J Physiol Lung Cell Mol Physiol. 2010;298:L584–L592. doi: 10.1152/ajplung.00312.2009. [DOI] [PubMed] [Google Scholar]
- Schuliga M, Harris T, Stewart AG. Plasminogen activation by airway smooth muscle is regulated by type I collagen. Am J Respir Cell Mol Biol. 2011;44:831–839. doi: 10.1165/rcmb.2009-0469OC. [DOI] [PubMed] [Google Scholar]
- Schuliga M, Javeed A, Harris T, Xia Y, Qin C, Wang Z, et al. Transforming growth factor-beta-induced differentiation of airway smooth muscle cells is inhibited by fibroblast growth factor-2. Am J Respir Cell Mol Biol. 2013;48:346–353. doi: 10.1165/rcmb.2012-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuliga MJ, See I, Ong SC, Soon L, Camoretti-Mercado B, Harris T, et al. Fibrillar collagen clamps lung mesenchymal cells in a nonproliferative and noncontractile phenotype. Am J Respir Cell Mol Biol. 2009;41:731–741. doi: 10.1165/rcmb.2008-0361OC. [DOI] [PubMed] [Google Scholar]
- Sharma M, Blackman MR, Sharma MC. Antibody-directed neutralization of annexin II (ANX II) inhibits neoangiogenesis and human breast tumor growth in a xenograft model. Exp Mol Pathol. 2012;92:175–184. doi: 10.1016/j.yexmp.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Sharma MR, Rothman V, Tuszynski GP, Sharma MC. Antibody-directed targeting of angiostatin's receptor annexin II inhibits Lewis Lung Carcinoma tumor growth via blocking of plasminogen activation: possible biochemical mechanism of angiostatin's action. Exp Mol Pathol. 2006;81:136–145. doi: 10.1016/j.yexmp.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Stewart CE, Nijmeh HS, Brightling CE, Sayers I. uPAR regulates bronchial epithelial repair in vitro and is elevated in asthmatic epithelium. Thorax. 2012;67:477–487. doi: 10.1136/thoraxjnl-2011-200508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaisgood CM, Aronica MA, Swaidani S, Plow EF. Plasminogen is an important regulator in the pathogenesis of a murine model of asthma. Am J Respir Crit Care Med. 2007;176:333–342. doi: 10.1164/rccm.200609-1345OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher JF, Burton N, Bacot SM, Vogel SN, Feldman GM. Annexin A2 tetramer activates human and murine macrophages through TLR4. Blood. 2010;115:549–558. doi: 10.1182/blood-2009-06-226944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson PR, Wilson JW, Stewart AG. Inhibition by salbutamol of the proliferation of human airway smooth muscle cells grown in culture. Br J Pharmacol. 1994;111:641–647. doi: 10.1111/j.1476-5381.1994.tb14784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J Appl Physiol. 2004;96:2019–2027. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Bdeir K, Yarovoi SV, Cines DB, Fang W, Abraham E. Involvement of the urokinase kringle domain in lipopolysaccharide-induced acute lung injury. J Immunol. 2006;177:5550–5557. doi: 10.4049/jimmunol.177.8.5550. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhou ZH, Bugge TH, Wahl LM. Urokinase-type plasminogen activator stimulation of monocyte matrix metalloproteinase-1 production is mediated by plasmin-dependent signaling through annexin A2 and inhibited by inactive plasmin. J Immunol. 2007;179:3297–3304. doi: 10.4049/jimmunol.179.5.3297. [DOI] [PubMed] [Google Scholar]
- Zheng L, Jaffee EM. Annexin A2 is a new antigenic target for pancreatic cancer immunotherapy. Oncoimmunology. 2012;1:112–114. doi: 10.4161/onci.1.1.18017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Gokhale VM, Szabo L, Munoz RM, Baek H, Bashyam S, et al. Identification of a novel inhibitor of urokinase-type plasminogen activator. Mol Cancer Ther. 2007;6:1348–1356. doi: 10.1158/1535-7163.MCT-06-0520. [DOI] [PubMed] [Google Scholar]