

Abstract
BACKGROUND AND PURPOSE
High-mobility group box 1 (HMGB1), a nuclear protein, is actively or passively released during inflammation. Recombinant human soluble thrombomodulin (rhsTM), a medicine for treatment of disseminated intravascular coagulation (DIC), sequesters HMGB1 and promotes its degradation. Given evidence for involvement of HMGB1 in pain signalling, we determined if peripheral HMGB1 causes hyperalgesia, and then asked if rhsTM modulates the HMGB1-dependent hyperalgesia.
EXPERIMENTAL APPROACH
Mechanical nociceptive threshold and swelling in rat hindpaw were determined by the paw pressure test and by measuring paw thickness, respectively, and HMGB1 levels in rat hindpaw plantar tissue, dorsal root ganglion (DRG) and serum were determined by Western blotting or elisa.
KEY RESULTS
Intraplantar (i.pl.) administration of HMGB1 rapidly evoked paw swelling and gradually caused hyperalgesia in rats. Systemic administration of rhsTM abolished HMGB1-induced hyperalgesia, and partially blocked paw swelling. LPS, administered i.pl., rapidly produced mild paw swelling, and gradually caused hyperalgesia. The anti-HMGB1 neutralizing antibody abolished LPS-induced hyperalgesia, but partially inhibited paw swelling. rhsTM at a high dose, 10 mg kg−1, prevented both hyperalgesia and paw swelling caused by LPS. In contrast, rhsTM at low doses, 0.001–1 mg kg−1, abolished the LPS-induced hyperalgesia, but not paw swelling. HMGB1 levels greatly decreased in the hindpaw, but not DRG. Serum HMGB1 tended to increase after i.pl. LPS in rats pretreated with vehicle, but not rhsTM.
CONCLUSION AND IMPLICATIONS
These data suggest that peripheral HMGB1 causes hyperalgesia, and that rhsTM abolishes HMGB1-dependent hyperalgesia, providing novel evidence for therapeutic usefulness of rhsTM as an analgesic.
Keywords: high-mobility group box 1 (HMGB1), hyperalgesia, edema, pain, thrombomodulin, lipopolysaccharide
Introduction
High-mobility group box 1 (HMGB1) protein, a nuclear architectural chromatin-binding protein, is expressed by almost all mammalian cells, and released actively and/or passively from necrotic or damaged cells and activated macrophages during inflammation, playing pathological roles as one of damage-associated molecular patterns (DAMPs; Scaffidi et al., 2002; Harris and Raucci, 2006; Klune et al., 2008; Sims et al., 2010). HMGB1, once released into the extracellular space, is capable of binding to multiple cell surface receptors such as receptors for advanced glycation end products (RAGE) and toll-like receptor-4 (TLR4), facilitating inflammation and tissue damage (Wang et al., 1999; Schmidt et al., 2001; Scaffidi et al., 2002; Fiuza et al., 2003; Sunden-Cullberg et al., 2006). Accumulating evidence suggests the role of HMGB1 in pain processing. HMGB1 in the spinal cord and/or dorsal root ganglion (DRG) is up-regulated in surgical neuropathic pain models induced by L5 spinal nerve ligation (Shibasaki et al., 2010) or by chronic constriction injury of the sciatic nerve (Kuang et al., 2012) and in a diabetic neuropathic pain model (Ren et al., 2012). Intrathecal administration of the neutralizing antibody against HMGB1 prevents or reverses the neuropathic pain/allodynia (Shibasaki et al., 2010; Ren et al., 2012) and bone cancer pain (Tong et al., 2010). Acute direct application of HMGB1 causes hyperexcitability of dissociated nociceptive DRG neurons isolated from rats (Feldman et al., 2012) and hypersensitivity of sciatic nerves to mechanical stimuli in rats (Chacur et al., 2001).
Interestingly, thrombomodulin (TM), an endothelial anticoagulant cofactor (Esmon, 2005), binds to HMGB1 and promotes its degradation by thrombin, leading to suppression of inflammation (Abeyama et al., 2005; Ito et al., 2008; Ito and Maruyama, 2011). TM is composed of five domains: the N-terminal lectin-like domain [TM domain (TM-D) 1], EGF-like domain (TM-D2), O-glycosylation-rich domain (TM-D3), C-terminal trans-membrane domain (TM-D4) and cytoplasmic domain (TM-D5). TM-D2 plays a critical role in the thrombin-catalysed activation of protein C to activated protein C (APC), while TM-D1 binds to HMGB1 (Abeyama et al., 2005). Recombinant human soluble TM (rhsTM) that lacks TM-D4 and TM-D5 is approved for clinical treatment of disseminated intravascular coagulation (DIC) in Japan (Ito and Maruyama, 2011). Although rhsTM appears to exhibit anti-inflammatory activity in a manner dependent on TM-D1 and TM-D2 in humans, rhsTM is incapable of causing TM-D2-dependent production of protein C in mammals including rodents other than primates (Mohri et al., 1997). Nonetheless, rhsTM exhibits anti-inflammatory activity through TM–D1-dependent sequestration of HMGB1 (Abeyama et al., 2005) and promotion of its degradation in rodents (Ito et al., 2008; Ito and Maruyama, 2011). In this context, we hypothesize that rhsTM might modulate peripheral HMGB1-dependent facilitation of pain signals in rodents.
In the present study, we thus tested if peripheral local injection of HMGB1 causes hyperalgesia in rats, and evaluated the effect of systemic administration of rhsTM on the peripheral HMGB1-dependent hyperalgesia. Here we provide evidence for the pronociceptive roles of HMGB1 in the peripheral tissue and novel therapeutic usefulness of rhsTM in relieving various types of clinical pain.
Methods
Animals
Male Wistar rats (7–10 weeks old) were purchased from Kiwa Laboratory Animals Co., Ltd. (Wakayama, Japan). All animals were used with approval by Kinki University School of Pharmacy's Committee for the Care and Use of Laboratory Animals, and all procedures were in accordance with the Guiding Principles approved by The Japanese Pharmacological Society and with Use of Laboratory Animals published by the US National Institutes of Health.
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Drugs and administration schedules
LPS (Escherichia coli Serotype O26:B6) was purchased from Sigma-Aldrich Chemical (St Louis, MO, USA). HMGB1 from bovine thymus, chicken anti-HMGB1 polyclonal antibody (neutralizing antibody) and chicken IgY were obtained from SHINO-TEST Corporation (Kanagawa, Japan). Recombinant human soluble TM (rhsTM; TM alfa) was provided by Asahi Kasei Pharma (Tokyo, Japan). LPS and HMGB1 were dissolved in saline, and rhsTM was in 0.002% Tween 80-containing saline. The neutralizing antibody and chicken IgY were dissolved in 0.2 M PBS. Rats received intraplantar (i.pl.) administration of HMGB1 at 2, 10 or 20 μg per paw or LPS at 1 μg per paw in a volume of 0.1 mL. The HMGB1-neutralizing antibody or chicken IgY at 50 or 100 μg per paw was coadministered i.pl. with LPS at 1 μg per paw in a volume of 0.1 mL. rhsTM in a dose range of 0.001–10 mg kg−1 was administered i.p. to rats 30 min before i.pl. administration of HMGB1 or LPS, considering the previous reports (Abeyama et al., 2005; Shi et al., 2008).
Measurement of mechanical and thermal nociception and of paw thickness
Mechanical nociception was evaluated by the paw pressure test, using an analgesia meter (MK-300, Muromachi Kikai Co., Tokyo, Japan); pressure was applied to the hindpaw of rats at a linearly increasing rate of 30 g s−1. The weight required to elicit nociceptive responses was determined as the nociceptive threshold, and a cut-off value of 500 g was used to avoid damage to the paw (Kawabata et al., 2007). In some experiments, the paw withdrawal latency to thermal stimuli was measured using a thermal analgesia meter (MK-330, Muromachi Kikai), as reported previously (Kawabata et al., 2001). The intensity of the thermal stimulus was adjusted to obtain baseline latencies of approximately 10 s (cut-off: 20 s). Rats were used for experiments after training sessions for approximately 2 weeks. The baseline mechanical threshold, paw withdrawal latency to thermal stimuli and paw thickness were determined before and after drug administration, and are represented as the percentage of the baseline values (% baseline) and/or as the area under the curve for the time course of the nociceptive threshold. The thickness of the hindpaw at the centre was measured, as an indicator of oedema, using a tissue caliper with 0.05 mm accuracy.
Determination of HMGB1 protein levels in the serum, hindpaw plantar tissue and DRG from rats
Under anaesthesia with i.p. administration of urethane at 1.5 g kg−1, the blood was collected from the abdominal aorta in the rats 1 h or 5 h after i.pl. LPS, and the ipsilateral DRG at L4–L6 levels and hindpaw plantar tissues (up to a level of the heel bone and metatarsal bones) were excised from the rats. Serum HMGB1 levels were determined using an elisa kit for HMGB1 (SHINO-TEST Corporation, Kanagawa, Japan). Tissue HMGB1 levels were assessed by Western blotting. Briefly, each sample was homogenized and sonicated in a RIPA containing PBS, 1% Igepal Ca-630 (Sigma-Aldrich Chemical), 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mg mL−1 phenylmethylsulfonyl fluoride, 0.15 U mL−1 aprotinin and 1 mM sodium orthovanadate. After centrifugation, to the supernatant was added the same volume of 2× electrophoresis sample buffer containing 19% glycerol, 5.7% SDS, 240 mM Tris-HCl (pH 6.7). After addition of 2-mercaptoethanol and bromophenol blue, proteins in the samples were denatured at 95–100°C for 5 min and separated by electrophoresis on 12.5% SDS-polyacrylamide gels (Wako Pure Chem., Osaka, Japan) and transferred onto PVDF membrane (Immobilon-P, Millipore Corporation, Billerica, MA, USA). The membrane was blocked with a blocking solution containing 5% skim milk, 137 mM NaCl, 0.1% Tween 20 and 20 mM Tris-HCl (pH 7.6), and then incubated with the affinity-purified anti-HMGB1 rabbit polyclonal antibody (1:1000 dilution; SHINO-TEST Corporation) or the anti-GAPDH rabbit polyclonal antibody (1:3000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunopositive bands for HMGB1 and GAPDH around 29 and 37 kDa, respectively, were visualized by the enhanced chemiluminescence detection reagent (Nacalai Tesque, Inc., Kyoto Japan).
Determination of mRNA levels of HMGB1, RAGE and TLR4 in the hindpaw plantar tissue and DRG from rats
The excised tissues were stabilized in an RNAlater reagent (Ambion, Austin, TX, USA). Total RNA was extracted from the homogenate of the hindpaw and DRG in the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). After the total RNA was reverse-transcribed, real-time PCR was performed using Light Cycler 480 (Roche Applied Sci., Basel, Switzerland). The volume of each reaction solution was 20 μL, containing 10 μL of 2 × Power SYBR Green PCR Master Mix (Applied Biosystems Japan Ltd., Tokyo, Japan), 100 ng cDNA and 0.2 μM each of forward and reverse primers. The cycling conditions of PCR were: preincubation at 50°C for 2 min followed by 95°C for 10 min, then 45 cycles of 95°C for15 s and 60°C for 60 s. The PCR primers employed for real-time PCR were: 5′-GTAATTTTCCGCGCTTTTGT-3′ (forward) and 5′-TCATCCAGGACTCATGTTCAGT-3′ (reverse) for rat HMGB1; 5′-ACTACCGAGTCCGAGTCTACC-3′ (forward) and 5′-GTAGCTCCCCTCAGACACACA-3′ (reverse) for rat RAGE; 5′-CGGAAAGTTATTGTGGTGGTG-3′ (forward) and 5′-GGACAATGAAGATGATGCCAG-3′ (reverse) for rat TLR4; 5′-GATGGTGAAGGTCGGTGTGAAC-3′ (forward) and 5′-TGACTGTGCCGTTGAACTTGC-3′ (reverse) for rat GAPDH. The product size was 114, 116, 121, and 176 bp for HMGB1, RAGE, TLR4, and GAPDH, respectively. It is to be noted that TLR4 is defined in accordance with the nomenclature of the Guide to Receptors and Channels (Alexander et al., 2011).
Morphological observation of the rat hindpaw tissue
In some experiments, the hindpaw plantar tissue was fixed, embedded in paraffin, sectioned and stained with haematoxylin and eosin for morphological observation.
Statistics
Data are represented as means ± SEM. Statistical analyses were performed by Student's t-test for two-group data and by Tukey's test for multiple comparisons. Differences among experimental groups were considered significant when P < 0.05.
Results
Hyperalgesia induced by i.pl. administration of HMGB1 and its prevention by systemic administration of rhsTM in rats
HMGB1, administered i.pl. at 10 and 20 μg per paw, but not 2 μg per paw, into the left hindpaw, gradually decreased the mechanical nociceptive threshold (Figure 1A). The paw thickness, an indicator of oedema, significantly increased and reached a plateau 1 h after i.pl. HMGB1 in the same dose range (Figure 1B). The paw withdrawal latency in response to thermal stimuli was shortened 5 h after i.pl. HMGB1 at 10–20 μg per paw (Figure 1C). Histological signs of very mild inflammation characterized by infiltration of a few polynuclear leukocytes, but not typical oedema, were observed in the hindpaw 5 h after i.pl. administration of HMGB1 at 20 μg per paw (Figure 1D). rhsTM, preadministered i.p. at 0.1 mg kg−1, completely prevented the development of the HMGB1-induced mechanical hyperalgesia (Figure 1E). On the other hand, rhsTM at relatively higher doses, 0.1–1 mg kg−1, reduced the HMGB1-induced paw swelling (Figure 1F). rhsTM at 10 mg kg−1 altered neither mechanical nociceptive threshold nor paw thickness in naïve rats (n = 4–5, P > 0.05; not shown).
Figure 1.
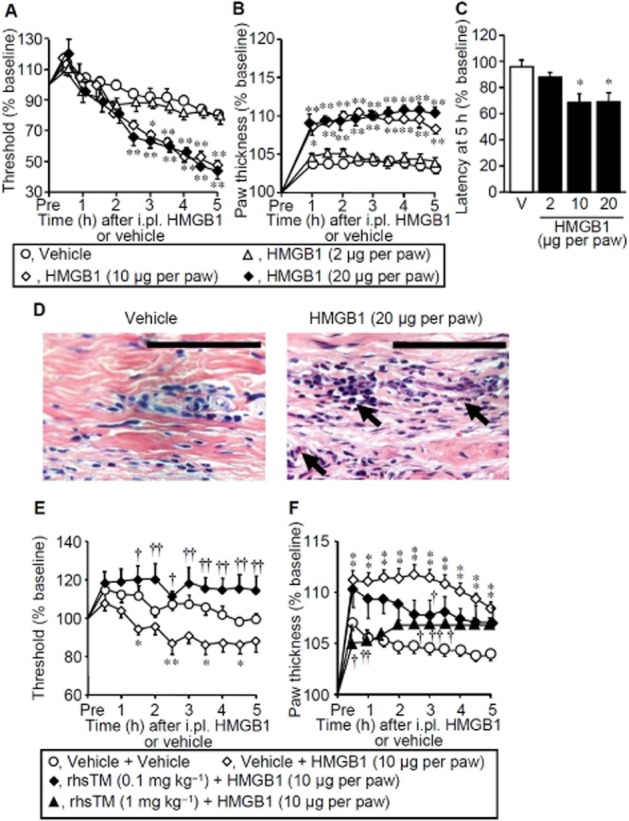
Hyperalgesia induced by exogenously applied HMGB1 and its inhibition by systemic administration of rhsTM in rats. (A–C) Time-courses of mechanical nociceptive threshold (A) and paw thickness (B) following i.pl. administration of HMGB1 at 2, 10 or 20 μg per paw or vehicle, and the paw withdrawal latency in response to thermal stimuli 5 h after i.pl. HMGB1 in rats (C). Data show the mean with SEM for four to five rats. *P < 0.05, **P < 0.01 versus vehicle. (D) Histopathological appearance of the hindpaw tissue 5 h after i.pl. administration of vehicle (left) or HMGB1 at 20 μg per paw (right) in rats. The tissue was stained with haematoxylin-eosin. Arrows show slight infiltration of polynuclear leukocytes. Bars indicate 100 μm. (E,F) Mechanical nociceptive threshold (E) and paw thickness (F) after i.pl. administration of HMGB1 in rats treated with rhsTM. rhsTM at 0.1 or 1 mg kg−1 or vehicle was administered i.p. 30 min before i.pl. administration of HMGB1 at 10 μg per paw or vehicle. Data show the mean with SEM for 5–11 rats. *P < 0.05, **P < 0.01 versus vehicle + vehicle; †P < 0.05, ††P < 0.01 versus vehicle + HMGB1.
The HMGB1-neutralizing antibody inhibits the hyperalgesia induced by i.pl. LPS in rats
To clarify the role of endogenous HMGB1 in the development of inflammatory pain, we used a rat model for inflammatory hyperalgesia caused by LPS, known to trigger release of HMGB1 (Yanai et al., 2012). LPS, administered i.pl. at 1 μg per paw, caused some histological signs of mild inflammation characterized by slight oedema and polynuclear leukocyte infiltration (Figure 2A). The paw thickness, an indicator of oedema, rapidly increased within 2 h after i.pl. LPS (Figure 2C), whereas the mechanical nociceptive threshold gradulally decreased, reaching a plateau around 4–5 h after i.pl. LPS (Figure 2B), in agreement with our previous study (Kawabata et al., 2007). Shortening of the paw withdrawal latency in response to thermal stimuli was also observed 5 h after i.pl. LPS (Figure 2D). The chicken anti-HMGB1 polyclonal antibody, a HMGB1-neutralizing antibody, when co-administered i.pl. at 50 and 100 μg, but not 5 μg per paw with LPS, clearly prevented the development of the LPS-induced mechanical hyperalgesia (Figure 2B). The HMGB1-neutralizing antibody at 50 μg per paw failed to reduce the LPS-induced paw swelling, whereas a larger dose, 100 μg per paw, of the antibody significantly attenuated the paw swelling (Figure 2C). It was also confirmed that the LPS-induced thermal hyperalgesia was blocked by the HMGB1-neutralizing antibody (Figure 2D).
Figure 2.
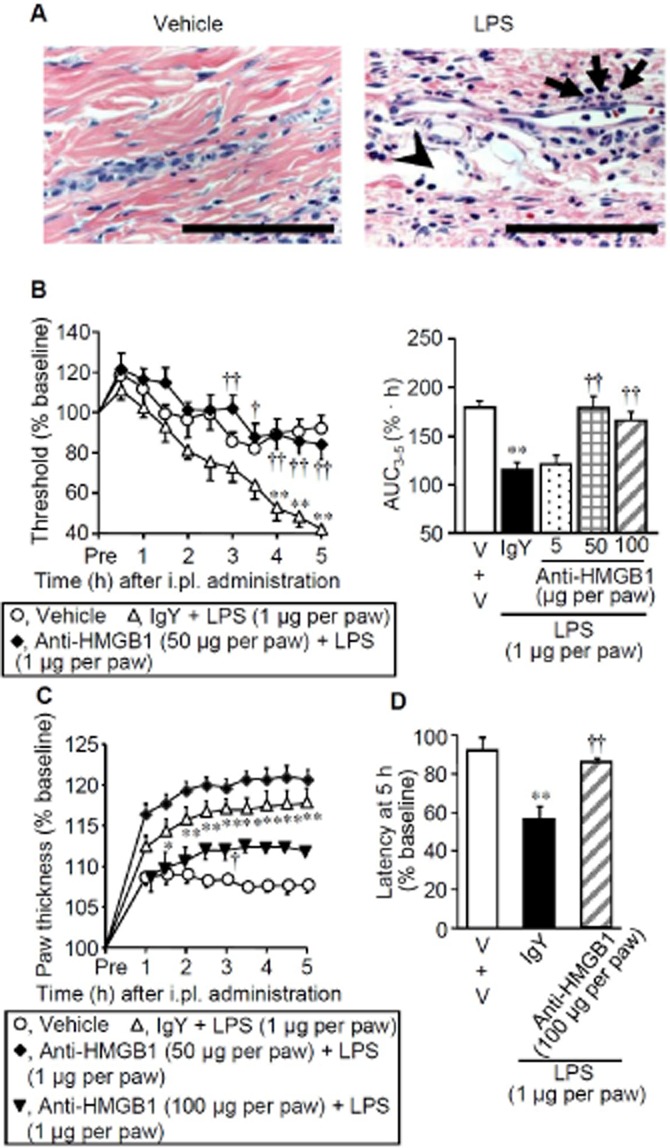
Inflammatory hyperalgesia induced by LPS and its inhibition by the HMGB1-neutralizing antibody in rats. (A) Histopathological appearance of the hindpaw tissue 5 h after i.pl. administration of vehicle (left) or LPS at 1 μg per paw (right) in rats. The tissue was stained with haematoxylin-eosin. Arrows and arrow heads show slight infiltration of polynuclear leukocytes and oedematous lesion, respectively. Bars indicate 100 μm. (B–D) Mechanical nociceptive threshold (B), paw thickness (C) and paw withdrawal latency in response to thermal stimuli (D) after i.pl. combined administration of LPS and IgY or the HMGB1-neutralizing antibody in rats. Chicken anti-HMGB1 polyclonal antibody (neutralizing antibody) at 5, 50 or 100 μg per paw or chicken IgY at 50 μg per paw was co-administered i.pl. with LPS at 1 μg per paw. Area under the curve3–5 was calculated from the time-threshold curve between 3 and 5 h after i.pl. LPS (B, right), and the latency was determined 5 h i.pl. LPS (D). V, vehicle. Data show the mean with SEM for 4–10 rats. *P < 0.05, **P < 0.01 versus Vehicle; †P < 0.05, ††P < 0.01 versus IgY + LPS.
Antihyperalgesic effect of rhsTM in rats treated with LPS
Systemic (i.p.) preadministration of rhsTM at 10 mg kg−1, a relatively high dose, abolished the LPS-induced hyperalgesia (Figure 3A) and largely prevented the paw swelling in rats (Figure 3B). In contrast, as did the HMGB1-neutralizing antibody (Figure 2B,C), rhsTM at 0.01–1 mg kg−1, lower doses, almost completely inhibited the LPS-induced hyperalgesia, but not paw swelling (Figure 3C,D). Boiled rhsTM at 10 mg kg−1 unaffected the LPS-induced mechanical hyperalgesia (Figure 3E) or paw swelling (Figure 3F).
Figure 3.
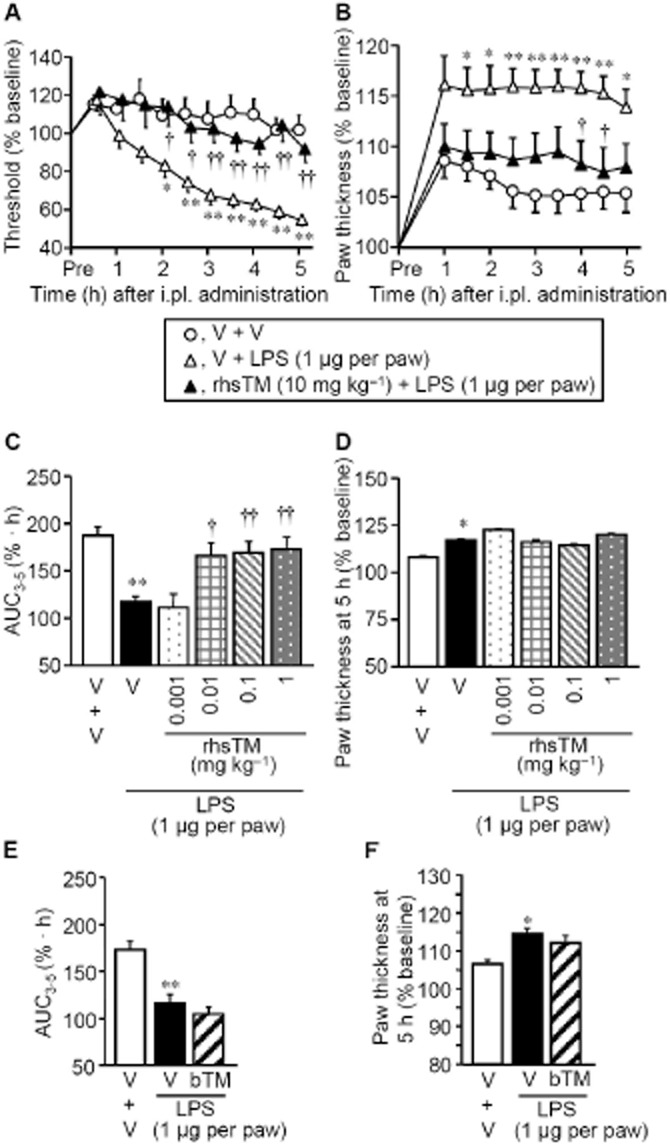
Effect of rhsTM on LPS-induced inflammatory hyperalgesia in rats. (A,B) A high dose of rhsTM abolishes both LPS-induced mechanical hyperalgesia (A) and paw swelling (B). rhsTM at 10 mg kg−1 or vehicle was administered i.p. 30 min before i.pl. administration of LPS at 1 μg per paw or vehicle. (C,D) Low doses of rhsTM prevent the LPS-induced hyperalgesia (C), but not paw swelling (D), in rats. rhsTM at 0.001–1.0 mg kg−1 or vehicle was administered i.p. 30 min before i.pl. administration of LPS at 1 μg per paw or vehicle. (E,F) Lack of effect of boiled rhsTM (bTM) on LPS-induced hyperalgesia (E) or paw swelling (F) in rats. bTM was prepared by boiling rhsTM for 10 min, and administered i.p. at 10 mg kg−1, 30 min before i.pl. LPS. Area under the curve3–5 was calculated from the time-threshold curve between 3 and 5 h after i.pl. LPS (C,E), and paw swelling was evaluated by measuring paw thickness 5 h after i.pl. LPS (D,F). V, vehicle. Data show the mean with SEM for 8–12 (A and B) or 4–9 (C, D, E and F) rats. *P < 0.05, **P < 0.01 versus vehicle + vehicle; †P < 0.05, ††P < 0.01 versus vehicle + LPS.
Protein levels of HMGB1 in the hindpaw plantar tissue, DRG and serum of the rats after i.pl. LPS
HMGB1 protein levels in the ipsilateral (left) hindpaw tissue significantly decreased 5 h, but not 1 h, after i.pl. administration of LPS in the left hindpaw (Figure 4A,B). In DRG, HMGB1 levels did not change by LPS treatment (Figure 4C). Preadministration of rhsTM did not prevent the LPS-induced decrease in the hindpaw HMGB1 levels (Figure 4B). Serum HMGB1 levels tended to increase 5 h after i.pl. administration of LPS in the rats treated with vehicle, but not rhsTM at 10 mg kg−1 (Figure 4E), while no change in serum HMGB1 levels was detected 1 h after i.pl. LPS (Figure 4D). rhsTM at 10 mg kg−1 itself did not affect HMGB1 levels in the hindpaw (Figure 4F), but significantly decreased serum HMGB1 levels (Figure 4G).
Figure 4.

HMGB1 protein levels in the hindpaw, DRG and serum after i.pl. administration of LPS in rats. rhsTM at 10 mg kg−1 or vehicle was administered i.p. 30 min before i.pl. administration of LPS at 1 μg per paw. (A–C) Expression of HMGB1 protein in the hindpaw plantar tissue 1 h (A) and 5 h (B) after i.pl. LPS, and in DRG at L4-L6 levels (C) 5 h after i.pl. LPS. The rats pretreated with rhsTM or not received i.pl. administration of LPS the left hindpaw, and HMGB1 levels in the left (L) and/or right (R) hindpaws (A,B) and in the left DRG (C) were assessed by Western blotting. (D,E) Serum HMGB1 levels 1 h (D) and 5 h (E) after i.pl. LPS in rats, as assessed by ELISA. (F,G) HMGB1 levels in the hindpaw (F) and serum (G) 5 h after i.p. administration of rhsTM at 10 mg kg−1 or vehicle in naïve rats. Photographs show typical examples of Western blotting for HMGB1 and GAPDH. Levels of each protein are quantified by densitometry in Western blotting. Data show the mean with SEM for 4 (A, B, C, D, F, G) or 5–8 (E) rats. *P < 0.05 versus vehicle + vehicle, or vehicle.
mRNA levels for HMGB1, RAGE and TLR4 in the hindpaw plantar tissue and DRG after i.pl. LPS in the rats
There was no significant change in mRNA levels for HMGB1, RAGE and TLR4 in the hindpaw or DRG 5 h after i.pl. administration of LPS in rats (Figure 5).
Figure 5.
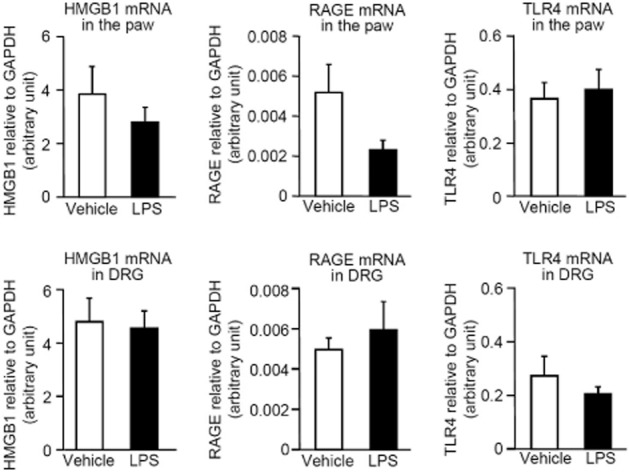
Levels of mRNA for HMGB1, RAGE and TLR4 in the hindpaw and DRG after i.pl. administration of LPS in the rats. The ipsilateral hindpaw and DRG at L4–L6 levels were collected 5 h after i.pl. administration of LPS at 1 μg per paw in rats, and mRNAs were reverse-transcribed and quantified by the real-time PCR method. Data show the mean with SEM for four rats.
Discussion and conclusions
Our findings that a single i.pl. administration of HMGB1 caused mechanical and thermal hyperalgesia and that the HMGB1-neutralizing antibody abolished the LPS-induced hyperalgesia, suggest that peripheral HMGB1 plays a major role in nociceptor sensitization during inflammation. Further, our study demonstrates that rhsTM, known to sequester HMGB1 and promote its degradation, actually blocks the hyperalgesia caused by HMGB1 or LPS, implying novel therapeutic usefulness of rhsTM as an analgesic, in addition to a medicine for treatment of DIC.
HMGB1, a nuclear, non-histone DNA-binding protein, is expressed in extensive cells including neurons, and released actively or passively during inflammation (Kim et al., 2008; Klune et al., 2008). There is evidence for involvement of exogenous and endogenous HMGB1 in pain processing (Chacur et al., 2001; Tong et al., 2010; Feldman et al., 2012). In particular, HMGB1 present in the DRG and spinal cord appears to participate in the pathogenesis of neuropathic pain (Shibasaki et al., 2010; Kuang et al., 2012; Ren et al., 2012). Our present study provides novel evidence for involvement of exogenous and endogenous HMGB1 in the development of peripheral hyperalgesia. rhsTM sequesters HMGB1 (Abeyama et al., 2005) and thereafter accelerates proteolytic degradation of HMGB1 by thrombin that takes 1–2 h in vitro (Ito et al., 2008). Therefore, our results that the development of paw swelling preceded the onset of hyperalgesia after i.pl. administration of HMGB1, might interpret why rhsTM exerted relatively minor inhibitory effect on the HMGB1-induced paw swelling, compared with its strong effect on the hyperalgesia (see Figure 1A,B). HMGB1 is considered to greatly participate in the LPS-induced hyperalgesia, but moderately contribute to the paw swelling, considering the extent of the effects of the HMGB1-neutralizing antibody (see Figure 2). This is in agreement with our results that rhsTM at relatively low doses prevented the development of the LPS-induced hyperalgesia, but not paw swelling (see Figure 3C,D). It is noteworthy that slight oedema, in addition to polynuclear leukocyte infiltration, was observed in the hindpaw tissues following i.pl. LPS, but not HMGB1 (see Figures 1D and 2A). The LPS-induced oedema or swelling in the hindpaw would thus appear to involve other pro-inflammatory mediators in addition to endogenous HMGB1. Nonetheless, rhsTM at 10 mg kg−1, a high dose, abolished both LPS-induced hyperalgesia and paw swelling (see Figure 3A,B). This might be explained by the previous evidence that TM also sequesters LPS through the TM-D1 at relatively high concentrations (Shi et al., 2008). The possibility cannot be ruled out that the different timing paradigm for administration of the anti-HMGB1 antibody and rhsTM that were coadministered and preadministered, respectively, might affect the extent of their inhibitory effects on the paw swelling.
Our data show that LPS treatment significantly and clearly decreased the protein levels of HMGB1 in the hindpaw plantar tissue, but not DRG, an effect resistant to rhsTM (see Figure 4). It is now known that pro-inflammatory stimuli including LPS challenge cause active and passive release of HMGB1 from various tissues/cells (Taniguchi et al., 2003; Oyama et al., 2010; Tsoyi et al., 2011; Nadatani et al., 2012; Wu et al., 2012), which might result in the decreased tissue HMGB1 levels, as shown in the present study. Although systemic LPS is known to increase serum HMGB1 levels (Lamkanfi et al., 2010; Yanai et al., 2012), i.pl. administration of LPS only slightly increased serum HMGB1 levels in rats treated with vehicle, but not rhsTM, in the present study. It is likely that a small amount of HMGB1 released persistently from the hindpaw tissue in response to i.pl. administration of LPS might enter the blood stream and quickly disappear because of degradation by endothelial TM-thrombin complexes (Ito et al., 2008). In future studies, it would be necessary to detect extracellular HMGB1 released locally from the cells using a perfusion system.
HMGB1 interacts with multiple cell surface receptors including RAGE and TLR4, a receptor for LPS (Wang et al., 1999; Schmidt et al., 2001; Scaffidi et al., 2002; Fiuza et al., 2003; Sunden-Cullberg et al., 2006). RAGE is also expressed in neurons, modulates neuronal functions, and may be involved in diabetic neuropathy (Brett et al., 1993; Chou et al., 2004; Sugimoto et al., 2008). Our finding that the HMGB1-neutralizing antibody completely blocked the LPS-induced hyperalgesia, but exerted only partial inhibition of paw swelling (see Figure 2B–D), may suggest that endogenous HMGB1 causes hyperalgesia via activation of RAGE or other receptors distinct from TLR4, although neither RAGE nor TLR4 was up-regulated at mRNA levels (see Figure 5). What molecules HMGB1 targets for induction of hyperalgesia are still open to question. In the present study, the anti-HMGB1 neutralizing antibody was injected locally to the plantar region of the hindpaw, so that it would act locally within the paw tissues, considering the effective dose of the antibody that is not enough to be distributed at effective concentrations in the blood. However, the possibility cannot be ruled out that the anti-hyperalgesic effect of systemically administered rhsTM involves its actions on tissues/cells out of the paw tissue, including the brain and spinal cord, although rhsTM is not considered to easily penetrate into the CNS in consideration of its large molecular size.
In conclusion, our data suggest that peripheral HMGB1 plays important roles in the development of inflammatory hyperalgesia and that rhsTM is available as a novel analgesic for treatment of HMGB1-mediated inflammatory pain/hyperalgesia.
Acknowledgments
This work was supported in part by ‘Antiaging Center Project’ for Private Universities from Ministry of Education, Culture, Sports, Science and Technology, 2008–2012.
Glossary
- APC
activated protein C
- DRG
dorsal root ganglion
- HMGB1
high-mobility group box 1
- RAGE
receptors for advanced glycation end products
- rhsTM
recombinant human soluble thrombomodulin
- TLR4
toll-like receptor-4
- TM
thrombomodulin
- TM-D
thrombomodulin domain
Conflict of interest
There are no conflicts of interest.
References
- Abeyama K, Stern DM, Ito Y, Kawahara K, Yoshimoto Y, Tanaka M, et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115:1267–1274. doi: 10.1172/JCI22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, et al. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–244. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- Chou DK, Zhang J, Smith FI, McCaffery P, Jungalwala FB. Developmental expression of receptor for advanced glycation end products (RAGE), amphoterin and sulfoglucuronyl (HNK-1) carbohydrate in mouse cerebellum and their role in neurite outgrowth and cell migration. J Neurochem. 2004;90:1389–1401. doi: 10.1111/j.1471-4159.2004.02609.x. [DOI] [PubMed] [Google Scholar]
- Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- Feldman P, Due MR, Ripsch MS, Khanna R, White FA. The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J Neuroinflammation. 2012;9:180. doi: 10.1186/1742-2094-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–778. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Maruyama I. Thrombomodulin: protectorate God of the vasculature in thrombosis and inflammation. J Thromb Haemost. 2011;9(Suppl 1):168–173. doi: 10.1111/j.1538-7836.2011.04319.x. [DOI] [PubMed] [Google Scholar]
- Ito T, Kawahara K, Okamoto K, Yamada S, Yasuda M, Imaizumi H, et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler Thromb Vasc Biol. 2008;28:1825–1830. doi: 10.1161/ATVBAHA.107.150631. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Kawao N, Kuroda R, Tanaka A, Itoh H, Nishikawa H. Peripheral PAR-2 triggers thermal hyperalgesia and nociceptive responses in rats. Neuroreport. 2001;12:715–719. doi: 10.1097/00001756-200103260-00020. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, et al. Hydrogen sulfide as a novel nociceptive messenger. Pain. 2007;132:74–81. doi: 10.1016/j.pain.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86:1125–1131. doi: 10.1002/jnr.21555. [DOI] [PubMed] [Google Scholar]
- Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: endogenous danger signaling. Mol Med. 2008;14:476–484. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang X, Huang Y, Gu HF, Zu XY, Zou WY, Song ZB, et al. Effects of intrathecal epigallocatechin gallate, an inhibitor of Toll-like receptor 4, on chronic neuropathic pain in rats. Eur J Pharmacol. 2012;676:51–56. doi: 10.1016/j.ejphar.2011.11.037. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri M, Gonda Y, Oka M, Aoki Y, Gomi K, Kiyota T, et al. The antithrombotic effects of recombinant human soluble thrombomodulin (rhsTM) on tissue factor-induced disseminated intravascular coagulation in crab-eating monkeys (Macaca fascicularis. Blood Coagul Fibrinolysis. 1997;8:274–283. doi: 10.1097/00001721-199707000-00003. [DOI] [PubMed] [Google Scholar]
- Nadatani Y, Watanabe T, Tanigawa T, Machida H, Okazaki H, Yamagami H, et al. High mobility group box 1 promotes small intestinal damage induced by nonsteroidal anti-inflammatory drugs through Toll-like receptor 4. Am J Pathol. 2012;181:98–110. doi: 10.1016/j.ajpath.2012.03.039. [DOI] [PubMed] [Google Scholar]
- Oyama Y, Hashiguchi T, Taniguchi N, Tancharoen S, Uchimura T, Biswas KK, et al. High-mobility group box-1 protein promotes granulomatous nephritis in adenine-induced nephropathy. Lab Invest. 2010;90:853–866. doi: 10.1038/labinvest.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren PC, Zhang Y, Zhang XD, An LJ, Lv HG, He J, et al. High-mobility group box 1 contributes to mechanical allodynia and spinal astrocytic activation in a mouse model of type 2 diabetes. Brain Res Bull. 2012;88:332–337. doi: 10.1016/j.brainresbull.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Shi GY, Hsiao SM, Kao YC, Kuo KL, Ma CY, et al. Lectin-like domain of thrombomodulin binds to its specific ligand Lewis Y antigen and neutralizes lipopolysaccharide-induced inflammatory response. Blood. 2008;112:3661–3670. doi: 10.1182/blood-2008-03-142760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki M, Sasaki M, Miura M, Mizukoshi K, Ueno H, Hashimoto S, et al. Induction of high mobility group box-1 in dorsal root ganglion contributes to pain hypersensitivity after peripheral nerve injury. Pain. 2010;149:514–521. doi: 10.1016/j.pain.2010.03.023. [DOI] [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Yasujima M, Yagihashi S. Role of advanced glycation end products in diabetic neuropathy. Curr Pharm Des. 2008;14:953–961. doi: 10.2174/138161208784139774. [DOI] [PubMed] [Google Scholar]
- Sunden-Cullberg J, Norrby-Teglund A, Treutiger CJ. The role of high mobility group box-1 protein in severe sepsis. Curr Opin Infect Dis. 2006;19:231–236. doi: 10.1097/01.qco.0000224816.96986.67. [DOI] [PubMed] [Google Scholar]
- Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–981. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- Tong W, Wang W, Huang J, Ren N, Wu SX, Li YQ. Spinal high-mobility group box 1 contributes to mechanical allodynia in a rat model of bone cancer pain. Biochem Biophys Res Commun. 2010;395:572–576. doi: 10.1016/j.bbrc.2010.04.086. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Jang HJ, Nizamutdinova IT, Kim YM, Lee YS, Kim HJ, et al. Metformin inhibits HMGB1 release in LPS-treated RAW 264.7 cells and increases survival rate of endotoxaemic mice. Br J Pharmacol. 2011;162:1498–1508. doi: 10.1111/j.1476-5381.2010.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Wu CX, Sun H, Liu Q, Guo H, Gong JP. LPS induces HMGB1 relocation and release by activating the NF-kappaB-CBP signal transduction pathway in the murine macrophage-like cell line RAW264.7. J Surg Res. 2012;175:88–100. doi: 10.1016/j.jss.2011.02.026. [DOI] [PubMed] [Google Scholar]
- Yanai H, Ban T, Taniguchi T. High-mobility group box family of proteins: ligand and sensor for innate immunity. Trends Immunol. 2012;33:633–640. doi: 10.1016/j.it.2012.10.005. [DOI] [PubMed] [Google Scholar]