

Abstract
BACKGROUND AND PURPOSE
Two distinct α1-adrenoceptor phenotypes (α1A and α1L) have recently been demonstrated to originate from a single α1A-adrenoceptor gene. Here, we examined the agonist profiles of recombinant α1A and α1L phenotypes and of lower urinary tract (LUT) α1-adrenoceptors.
EXPERIMENTAL APPROACH
A series of drugs (A61603, Ro 115–1240, NS-49, MK017 and ESR1150) originally developed for stress urinary incontinence (SUI) therapy were used to stimulate recombinant α1A- and α1L-adrenoceptor phenotypes, and their potencies and intrinsic activity estimated from Ca2+ responses. Agonist-induced contractions were also examined in LUT tissues of rats and humans and in human mesenteric artery and rat tail artery.
KEY RESULTS
All the drugs were potent agonists of the α1A-adrenoceptor compared with the α1L-adrenoceptor phenotype. Among them, Ro 115–1240 was shown to be an α1A-specific partial agonist that produced partial contractions through α1A-adrenoceptors in rat prostate and tail artery, but not in the other LUT tissues and human mesenteric artery. In contrast, P-come 102 showed full agonist activity at α1A- and α1L-adrenoceptors, but was less selective than noradrenaline for α1A-adrenoceptors. Like noradrenaline, P-come 102 was highly potent at inducing contractions in all of the LUT tissues tested. However, the potency and intrinsic activity of P-come 102 were significantly lower than those of noradrenaline in human mesenteric artery.
CONCLUSIONS AND IMPLICATIONS
The α1A- and α1L-adrenoceptor phenotypes and LUT α1-adrenoceptors were demonstrated to have distinct agonist profiles. As adrenergic contractions in LUT are predominantly mediated through α1L-adrenoceptors, the development of α1L-selective agonists may provide clinically useful drugs for SUI therapy.
Keywords: α1A-adrenoceptor, α1L-adrenoceptor, agonist profile, lower urinary tract
Introduction
Three distinct genotypes (α1A, α1B and α1D; receptor nomenclature follows that of Alexander et al., 2011) of α1-adrenoceptors have been identified, and the pharmacological profiles of these recombinant receptors have been verified to be essentially the same as those of the native receptors (Lomasney et al., 1991; Hieble et al., 1995; Graham et al., 1996; Michelotti et al., 2000; Alexander et al., 2011). Prazosin, a prototypical, specific α1-adrenoceptor antagonist, shows high affinity for α1A, α1B, and α1D-adrenoceptor subtypes (Lomasney et al., 1991; Muramatsu et al., 1995; Ford et al., 1996). With regard to the α1A-adrenoceptor, A61603 and NS-49 were developed as selective agonists (Knepper et al., 1995; Obika et al., 1995), whereas silodosin, 5-methylurapidil, RS-17053 and SNAP5089 have been characterized as selective antagonists (Ford et al., 1996; Murata et al., 1999; Morishima et al., 2007). Of the three classic α1-adrenoceptor subtypes, the α1D-adrenoceptor shows the highest affinity for catecholamines and is selectively antagonized by low concentrations of BMY7378 (Lomasney et al., 1991; Perez et al., 1991; Hieble et al., 1995). In contrast, agonists/antagonists selective for the α1B-adrenoceptor have not yet been identified.
In addition to these classic α1-adrenoceptors, a wide variation in antagonist affinity for prazosin has been demonstrated in native α1-adrenoceptors from different tissues, and the presence of another subtype (designated as α1L-adrenoceptor) has been proposed (Drew, 1985; Flavahan and Vanhoutte, 1986; Muramatsu et al., 1990; Testa et al., 1993; Ford et al., 1996; Argyle and McGrath, 2000; Su et al., 2008). The pharmacological profile of α1L-adrenoceptors is unique and does not match those of classic α1-adrenoceptor subtypes: it has a low affinity for prazosin, 5-methylurapidil, RS-17053 and BMY7378, but high affinity for silodosin and tamsulosin (Muramatsu et al., 1995; Ford et al., 1996; Testa et al., 1997; Murata et al., 1999; 2000). Recently, the α1L-adrenoceptor was demonstrated to be another phenotype originating from the α1A-adrenoceptor gene, in addition to the classic α1A-adrenoceptor subtypes (Gray et al., 2008; Muramatsu et al., 2008). In the lower urinary tract (LUT: prostate, urinary bladder neck and urethra) of mammals including humans, of the three α1-adrenoceptor genes α1A-adrenoceptor mRNA is predominantly expressed (Price et al., 1993; Faure et al., 1994; Nishimune et al., 2012), and it has been proposed that adrenergic contractions are induced by activation of either the α1A- or α1L-adrenoceptor phenotype. Although the identity of these functional phenotypes is still contentious (Marshall et al., 1995; Ford et al., 1996; Testa et al., 1997; Van der Graaf et al., 1997; Michel and Vrydag, 2006; Muramatsu et al., 2008; Nishimune et al., 2012). For some time, α1-adrenoceptor antagonists have been acknowledged to reduce resistance to urinary flow (Andersson, 1993; Ruffolo and Hieble, 1999), and tamsulosin, silodosin, and alfuzosin are currently used to treat urinary problems in male patients with benign prostatic hyperplasia (Andersson, 2002; Michel and Vrydag, 2006). In contrast, α1-adrenoceptor agonists produce contractions in the urethra and bladder neck, resulting in an increase in urinary resistance. Therefore, α1-adrenoceptor agonists may be useful clinically to treat women with stress urinary incontinence (SUI; Taki et al., 1999; Andersson and Wein, 2004). As shown in Figure 1, many α1-adrenoceptor agonists, such as A61603 (Knepper et al., 1995), Ro 115–1240 (Blue et al., 2004; Musselman et al., 2004), NS-49 (Obika et al., 1995), MK017 (Nishimune et al., 2010) and ESR1150 (Matsumaru et al., 2005), have been tested for this purpose, but to date no successful clinical application has been achieved. This may be due to a moderate/poor efficacy of these agonists in the LUT and problems associated with their toxic side effects, which includes their propensity to increase BP. Therefore, we decided to investigate the pharmacology of agonists at α1A- and α1L-adrenoceptor phenotypes and at LUT α1-adrenoceptors.
Figure 1.
Chemical structures of the representative drugs developed for SUI therapy and tested in the present study. A61603, N-(5-[4,5-dihydro-1H-imidazol-2-y]-2-hydroxy-5,6,7,8-tetrahydronaphtalen-1-yl)methanesulfonamide hydrobromide; Ro 115–1240 (Dabuzalgron, R450), N-[6-chloro-3-[ (4,5-dihydro-1H-imidazol-2yl)methoxy]-2-methylphenyl]methanesulphonamide; NS-49, (R)-(–)-3′-(2-amino-1-hydroxyethyl)-4′-fluoromethanesulphonanilide; ESR1150, 4-bromo-N3-imidazolidin-2-ylidine-2,N1,N1-trimethyl-benzene-1,3-diamine hydrochloride; MK017, 2-[ (5-chloro-3-isopropyl-2-methylphenyl)methyl]-4,5-dihydro-1H-imidazole hydrochloride; P-come 102, 2-[ (6-dimethylamino-3-isopropyl-2-methyphenyl)methyl]-4,5-dihydro-1H-imidazole hydrochloride.
Recently, our group identified a cysteine-rich epidermal growth factor-like domain 1α (CRELD1α) as an α1A-adrenoceptor-interacting protein and found that this protein down-regulates the expression of the α1A-adrenoceptor phenotype (Nishimune et al., 2010). By overexpressing this CRELD1α in α1A-adrenoceptor-transfected cells, we obtained a cell line that predominantly expresses the α1L-adrenoceptor phenotype (US patent no. 8173378). In the present study, we compared the agonist profiles of the recombinant α1A- and α1L-adrenoceptors and then examined the effects of a few representative agonists on LUT α1-adrenoceptors.
Methods
Cell lines expressing α1-adrenoceptors
CHO cells expressing human α1A, α1B and α1D-adrenoceptors (CHO-α1A, CHO-α1B and CHO-α1D cells, respectively), and co-expressing human α1A- and α1L-adrenoceptors (CHO-α1AL cells, US patent no. 8173378) were purchased from Pharmacome LCC (Eiheiji-Matsuoka, Fukui, Japan). The densities (fmol·mg−1 protein) of each α1-adrenoceptor expressed were 3000 in CHO-α1A cells, 550 in CHO-α1B cells, 150 in CHO-α1D cells, and 120 (α1A) and 120 (α1L) in CHO-α1AL−1 cells. These CHO cells were cultured and maintained as reported previously (Nishimune et al., 2010).
Measurement of the intracellular Ca2+ concentration
The intracellular calcium concentration ( [Ca2+]i) was measured as described previously (Nishimune et al., 2010). Briefly, CHO cells were loaded with 5 μM Fura-2 AM (Dojindo, Kumamoto, Japan) together with 0.02% pluronic F-127 and 1.4 mM probenecid for 45 min, washed, and then resuspended in Ca2+ assay buffer with 1.4 mM probenecid and 3% FBS. The measurements were done at 37°C with Fura-2 ratio fluorometry using a CAF-110 fluorescence spectrophotometer (JASCO, Tokyo, Japan). The cells were exposed to single concentrations of the drugs tested, and the change in [Ca2+]i was measured for 5 min. At the end of the measurement, the maximum and minimum fluorescence ratios were determined by subsequent addition of Triton X-100 and EGTA respectively. The [Ca2+]i concentration was calculated using the formula [Ca2+]i (nM) = Kd × [(R − Rmin) / (Rmax − R)] × (Sf2 / Sb2), where Kd (224 nM at 37°C) is the dissociation constant of Fura-2 for Ca2+; R is the ratio of fluorescence of the sample at 340 nm to that at 380 nm; Rmin and Rmax represent the ratios of fluorescence at the same wavelengths in the presence of 0 and saturating Ca2+, respectively; and Sf2/Sb2 is the ratio of fluorescence of Fura-2 at 380 nm in 0 Ca2+ to that in saturating Ca2+. In the experiments with CHO-α1AL−1 cells, 5 nM prazosin was present throughout the measurement to mask the coexisting α1A-adrenoceptor phenotype and to selectively record the α1L response (Nishimune et al., 2010). The basal level of [Ca2+]i before stimulation was approximately 100 nM in each cell line, and the net increases in [Ca2+]i evoked by the drugs tested were normalized to the maximal response to noradrenaline recorded in cells cultured simultaneously.
Rat tissue isolation
Male and female Wistar rats (10–15 weeks of age, Charles River Japan, Inc., Yokohama, Japan) were used in the present study, which was conducted according to the Guidelines for Animal Experiments of the University of Fukui (accredited by the Ministry of Education, Culture, Sports, Science and Technology, Japan). Rats were anaesthetized with isoflurane and killed by cervical dislocation. The prostate, urinary bladder, urethra, and tail artery were rapidly isolated and immersed in modified Krebs-Henseleit solution containing (in mM): NaCl, 112.0; KCl, 5.4; MgCl2, 1.2; CaCl2, 2.0; NaH2PO4, 1.2; NaHCO3, 25.5; and d-(+)-glucose, 11.5 (pH 7.4). The solution was oxygenated with a mixture of 95% O2 and 5% CO2 and was kept on ice. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Human samples
The study with human samples was performed after obtaining full informed consent according to the guidelines of the Ethics Committee of the University of Fukui. Human urinary bladders were obtained from four male and three female patients (age range: 53–77 years) with urinary bladder cancer. Human mesenteric arteries were obtained from six male and three female patients (age range: 49–75 years) with colon cancer.
Functional studies with isolated tissues
Rat urethra and urinary bladder necks and human urinary bladder necks were transversely cut. Rat prostate was separated into lobes. Human mesenteric arteries (0.5–0.8 mm outer diameter) and rat tail arteries (0.4–0.8 mm outer diameter) were cut into 2 mm long ring preparations. The strips or ring preparations were placed in organ baths containing modified Krebs-Henseleit solution at 37°C. After equilibration for 2 h, noradrenaline was cumulatively applied twice with a 2 h interval, and the isometric tension changes were recorded through a force displacement transducer. Two hours after recording the second response to noradrenaline in each strip, the preparation was exposed to the test drugs, and the evoked response was normalized to the maximum contraction produced upon the second exposure to noradrenaline in the same preparation. In parallel with the test drug, noradrenaline was applied to other preparations and the third response to noradrenaline was used as a time control. Desipramine (0.3 μM), deoxycorticosterone acetate (5 μM) and propranolol (1 μM) were added to inhibit neural and extraneural uptake of noradrenaline, and to block β-adrenoceptors. In the experiments with human mesenteric arteries and rat tail arteries, Nω-nitro-l-arginine methyl ester (100 μM) was also added to inhibit nitric oxide release from endothelial cells. In the experiments with rat tail arteries, urinary bladders and urethras, rauwolscine (0.1 μM), was added to block α2-adrenoceptors (Lachnit et al., 1997; I. Muramatsu, unpubl. obs.). All of these blockers were added for at least 40 min before and during every contractile response. To identify the α1-adrenoceptor subtype, prazosin (1 or 10 nM), silodosin (0.3–3 nM) or BMY7378 (10–20 nM) was added 40 min before and during the third response to noradrenaline or Ro 115–1240, and the responses in the presence of antagonist were compared with the time control that was evoked simultaneously in the absence of antagonists.
Data analysis
The [Ca2+]i responses to the test drugs were normalized to the maximal response to noradrenaline (a standard drug) obtained from cells cultured at the same time, and the maximal effect of the test drug was expressed as the intrinsic activity. The potency (pEC50) of each drug was estimated from the concentration–response curve, and the relative potency of each test drug was calculated as the ratio between pEC50 values for the test drug and noradrenaline. The contractile response to each test drug in the isolated tissue strips was also normalized to the maximal contraction evoked by the second application of noradrenaline in the same strip, and the potency (pEC50) and intrinsic activity were calculated. The antagonist affinity (pKB) was determined for a single concentration of the antagonist using the concentration-ratio method (Furchgott, 1972). In this case, the concentration-ratio for noradrenaline was generally measured at 50% of the second response to noradrenaline in each preparation, whereas the ratio for Ro 115–1240 was measured at 30% of the second response to noradrenaline (Figure 4C and 4F). The procedure proposed by van Rossum was also applied to insurmountable antagonism (van Rossum, 1963). Data are presented as means ± SEM of the number of experiments (n) and were statistically analysed using Student's t-test.
Figure 4.
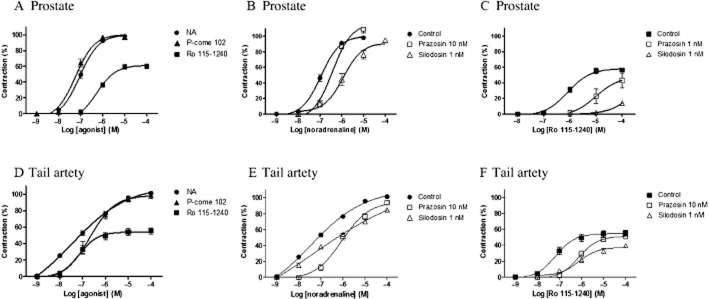
Concentration–response curves for noradrenaline (NA), Ro 115–1240, and P-come 102 in the prostate (A, B, C) and tail artery (D, E, F) of rats. The responses in the tail artery were obtained in the presence of 10 nM BMY7378. (A, D) Concentration-dependent contractions induced by three drugs in the prostate and tail artery. (B, E) Effects of prazosin (10 nM) or silodosin (1 nM) on concentration–response curve for noradrenaline. (C, F) Effects of prazosin (1 or 10 nM) and silodosin (1 nM) on the response to Ro 115–1240. Mean ± SEM of five to seven experiments. NA, not applicable.
Reagents
The following drugs were used: prazosin hydrochloride, A61603, BMY 7378, 5-methylurapidil and rauwolscine hydrochloride (Sigma-Aldrich Co., St. Louis, MO, USA); l-noradrenaline bitartrate and phenylephrine hydrochloride (Nacalai Tesque, Kyoto, Japan); silodosin (Kissei Pharmaceutical Co., Ltd, Matsumoto, Japan); and tamsulosin (Astellas Pharmaceutical Co., Ltd, Tokyo, Japan). Silodosin (1 mM in DMSO) was stored at −20°C and diluted with DMSO before use. NS-49, Ro 115–1240, P-come 102, MK017 and ESR1150 were synthesized by our group.
Results
Effects of various drugs on recombinant α1-adrenoceptors
CHO cells expressing different α1-adrenoceptors were stimulated with various drugs, and the changes in [Ca2+]i were monitored. Figure 2 shows the representative concentration–response curves for noradrenaline, Ro 115–1240 and P-come 102 in CHO-α1A cells and CHO-α1AL−1 cells (in the presence of 5 nM prazosin) in which α1A- or α1L-adrenoceptors were stimulated selectively (Nishimune et al., 2010). Noradrenaline evoked concentration-dependent increases in [Ca2+]i in both cell lines. The peak levels of [Ca2+]i evoked by 100 μM noradrenaline were approximately 950 nM in CHO-α1A cells and 550 nM in CHO-α1AL cells (in the presence of 5 nM prazosin), and the pEC50 values were 8.3 and 6.0 for the α1A and α1L responses respectively (Table 1). The differences in potency and efficacy for noradrenaline were considered to reflect the different densities of α1A- and α1L-adrenoceptor phenotypes expressed in these two cell lines. Ro 115–1240 also activated α1A-adrenoceptors, but the pEC50 value and the maximal response were slightly lower than those of noradrenaline (Figure 2A). In contrast, Ro 115–1240 only weakly activated the α1L-adrenoceptors (Figure 2B). Therefore, Ro 115–1240 appears to be a partial agonist that is selective for α1A-adrenoceptors. P-come 102, a newly synthesized compound, fully activated both α1A- and α1L-adrenoceptors.
Figure 2.
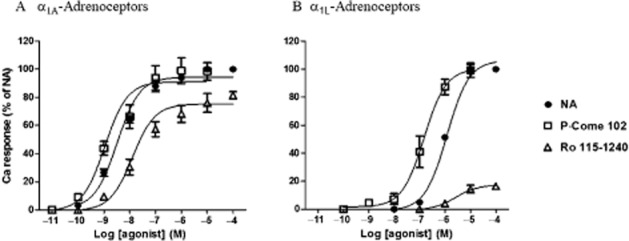
Concentration–response curves for noradrenaline (NA), Ro 115–1240, and P-come 102 at α1A- (A) and α1L-adrenoceptors (B). The increase in intracellular calcium concentration evoked by the various concentrations of agonists was normalized to the maximal response to noradrenaline in the same cells. In (B), the responses mediated through α1L-adrenoceptors were obtained after masking the concomitant α1A-adrenoceptors in the presence of 5 nM prazosin (see Methods). Mean ± SEM of four to six experiments.
Table 1.
Agonist profiles of various drugs at recombinant α1-adrenoceptors
| α1A-adrenoceptor | α1L-adrenoceptor | α1B-adrenoceptor | α1D-adrenoceptor | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Agonist | pEC50 (RP) | IA | pEC50 (RP) | IA | Selectivity (α1L/α1A) | pEC50 (RP) | IA | pEC50 (RP) | IA |
| Noradrenaline | 8.3 ± 0.07 (1.0) | 1.00 | 6.0 ± 0.06 (1.0) | 1.00 | 1.0 | 8.0 ± 0.18 | 1.00 | 8.2 ± 0.11 | 1.00 |
| Phenylephrine | 7.1 ± 0.10 (0.06) | 0.88 ± 0.02 | 5.3 ± 0.11 (0.2) | 0.91 ± 0.03 | 3.2 | 7.3 ± 0.15 (0.2) | 1.07 ± 0.07 | 6.7 ± 0.24 (0.03) | 0.62 ± 0.07 |
| P-come 102 | 8.6 ± 0.30 (2.0) | 1.00 ± 0.10 | 6.8 ± 0.16 (6.3) | 0.98 ± 0.06 | 3.2 | 0.08 ± 0.02 | 0.10 ± 0.05 | ||
| A61603 | 10.1 ± 0.19 (63.1) | 0.92 ± 0.06 | 7.3 ± 0.15 (20.0) | 1.11 ± 0.08 | 0.3 | 0.06 ± 0.00 | 0.07 ± 0.00 | ||
| Ro 115–1240 | 7.7 ± 0.11 (0.3) | 0.77 ± 0.02 | 5.6 ± 0.27 (0.4) | 0.17 ± 0.02 | 1.6 | 0.00 | 0.06 ± 0.00 | ||
| NS-49 | 7.8 ± 0.11 (0.3) | 0.81 ± 0.02 | 5.5 ± 0.11 (0.3) | 0.54 ± 0.02 | 1.0 | 0.10 ± 0.03 | 0.00 | ||
| MK017 | 10.3 ± 0.18 (100) | 0.73 ± 0.03 | 7.8 ± 0.11 (63.1) | 0.88 ± 0.04 | 0.6 | 0.16 ± 0.01 | 0.14 ± 0.06 | ||
| ESR1150 | 9.1 ± 0.08 (6.3) | 0.96 ± 0.02 | 6.5 ± 0.26 (3.2) | 0.91 ± 0.07 | 0.5 | 0.12 ± 0.01 | 0.00 | ||
RP (relative potency), potency of tested drug was compared with that of noradrenaline at the same α1-adrenoceptor subtype; IA (intrinsic activity), maximal response to tested drug relative to that of noradrenaline.
Selectivity (α1L/α1A): Selectivity of tested drug at α1L-adrenoceptor against α1A-adrenoceptor, where the affinities for noradrenaline in α1L- and α1A-responses were normalized as equal.
Mean±SEM of three to six experiments.
Noradrenaline and phenylephrine also produced concentration-dependent increases in [Ca2+]i in CHO-α1B and CHO-α1D cells. The peak level of [Ca2+]i evoked by 100 μM noradrenaline was approximately 400 nM. The affinities (pEC50) and maximal responses of various drugs at four recombinant α1-adrenoceptors are summarized in Table 1. Apart from noradrenaline and phenylephrine, all of the agonists tested (P-come 102, A61603, Ro 115–1240, NS-49, MK017 and ESR1150) were essentially inactive at α1B- and α1D-adrenoceptors. As the densities of α1A- and α1L-adrenoceptors expressed in each cell line differed (see Methods), the pEC50 values for the various drugs were normalized to that for noradrenaline in the same cell line. Then, the potencies for noradrenaline were assumed to be the same between α1A- and α1L-adrenoceptor phenotypes, and the phenotype selectivity (α1L/α1A) of each drug was estimated (Table 1). In general, large differences in selectivity were not observed between α1A- and α1L-adrenoceptor phenotypes. However, A61603, MK017 and ESR1150 showed higher potency than noradrenaline in both cell lines and showed a trend towards slightly higher selectivity for α1A-adrenoceptors than for α1L-adrenoceptors. In contrast, P-come 102 was less selective than noradrenaline for α1A-adrenoceptors (Table 1). NS-49 was less potent than noradrenaline at α1A- and α1L-adrenoceptors, and its intrinsic activity was lower at α1L-adrenoceptors (0.54) than at α1A-adrenoceptors (0.81).
Effects of noradrenaline, Ro 115–1240 and P-come 102 on rat LUT α1-adrenoceptors
As we observed distinct agonist profiles between recombinant α1A- and α1L-adrenoceptor phenotypes, we next examined the effects of representative drugs on LUT α1-adrenoceptors. Here, we used noradrenaline as a standard drug, Ro 115–1240 as an α1A-specific partial agonist, and P-come 102 as an α1A/α1L full agonist with less selectivity for α1A-adrenoceptors. Figure 3A and 3C show the concentration–contraction curves for these three drugs in male and female rat urethras. P-come 102 as well as noradrenaline showed full agonist activity, and the potency of P-come 102 was the same as or slightly higher than that of noradrenaline in both male and female rats (Table 2). In contrast, Ro 115–1240 did not induce a contraction in either male or female urethras. In the urinary bladder neck, full agonist activity was also observed in the responses to noradrenaline and P-come 102 irrespective of sex. These agonist profiles are summarized in Table 2.
Figure 3.
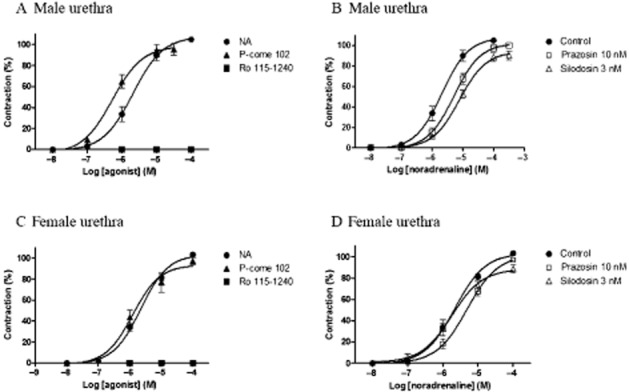
Concentration–response curves for noradrenaline (NA), Ro 115–1240, and P-come 102 in male (A, B) and female (C, D) rat urethras. Maximal contractions induced by a second application of noradrenaline were taken as 100%, and the responses to the third application of noradrenaline or tested agonists were evaluated in the same preparations. (A, C) Concentration-dependent contractions induced by three drugs in male and female rat urethras. (B, D) Effects of 10 nM prazosin and 3 nM silodosin on concentration–response curves for noradrenaline in male and female urethras. Mean ± SEM of four to seven experiments. NA, not applicable.
Table 2.
Agonist activities of noradrenaline, P-come 102 and Ro 115–1240 at native α1-adrenoceptors
| Noradrenaline | P-come 102 | Ro 115–1240 | ||||||
|---|---|---|---|---|---|---|---|---|
| Tissue | pEC50 | IA | pEC50 | IA | pEC50 | IA | ||
| Rat | ||||||||
| Urethra | Male | 5.7 ± 0.09 (5M) | 1.05 ± 0.02 | 6.3 ± 0.11* (5M) | 0.96 ± 0.06 | – (5M) | 0 | |
| Female | 5.7 ± 0.06 (5F) | 1.03 ± 0.01 | 5.9 ± 0.13 (4F) | 0.97 ± 0.04 | – (4F) | 0 | ||
| Urinary bladder neck | Male | 5.9 ± 0.04 (5M) | 0.99 ± 0.01 | 6.2 ± 0.07 (5M) | 0.96 ± 0.03 | – (4M) | 0 | |
| Female | 5.7 ± 0.06 (5F) | 1.02 ± 0.01 | 6.2 ± 0.06* (5F) | 1.04 ± 0.03 | – (4F) | 0 | ||
| Prostate | Male | 7.0 ± 0.07 (8M) | 1.0 ± 0.1 | 7.3 ± 0.07 (7M) | 1.0 ± 0.02 | 6.3 ± 0.09* (8M) | 0.60 ± 0.04* | |
| Tail artery | 7.5 ± 0.22 (5M, 5F) | 1.01 ± 0.01 | 6.9 ± 0.11 (4M,4F) | 0.99 ± 0.03 | 7.2 ± 0.13 (5M,5F) | 0.55 ± 0.04* | ||
| Human | ||||||||
| Urinary bladder neck | 5.4 ± 0.09 (4M,3F) | 0.9 ± 0.05 | 5.9 ± 0.10* (4M,3F) | 0.93 ± 0.03 | – (4M,3F) | 0 | ||
| Mesenteric artery | 6.4 ± 0.20 (6M,3F) | 1.0 ± 0.02 | 5.7 ± 0.12* (6M,3F) | 0.76 ± 0.05* | – (3M,2F) | 0 | ||
IA (intrinsic activity), maximal contraction induced by tested drug, which was compared with the maximal contraction induced by second application of noradrenaline in the same preparation. The tested response was obtained after twice application of noradrenaline in the same preparation (see Methods). The responses in rat tail artery were obtained in the presence of 10 nM BMY7378. (), number of male (M) and female (F) specimens.
Significantly different from the value for noradrenaline (P < 0.05).
–: no response.
The concentration–response curves for noradrenaline in the urethra and urinary bladder neck isolated from male and female rats were slightly inhibited by 10 nM prazosin, with estimated pKB values of 8.0–8.6 (Figure 3B and 3D for urethra, Table 3). However, BMY7378 (20 nM) had no inhibitory effect on the contractile responses to noradrenaline in the urethra and bladder neck. Silodosin (1–3 nM) suppressed the concentration–response curves for noradrenaline in male urethras (Figure 3B) and in male and female bladder necks. However, the contractile responses to noradrenaline in the female urethra were not inhibited by 3 nM silodosin (Figure 3D), suggesting a sex difference in the sensitivity to silodosin of rat urethras.
Table 3.
Antagonist profiles of the contractile responses to noradrenaline and Ro 115–1240 in various tissues
| Agonist | Antagonist (pKB) | |||||
|---|---|---|---|---|---|---|
| Tissue | Prazosin | Silodosin | BMY7378 | |||
| Rat | ||||||
| Urethra | Male | Noradrenaline | 8.2 ± 0.1 | 9.1 ± 0.0 | – | |
| Female | Noradrenaline | 8.0 ± 0.2 | – | – | ||
| Urinary bladder neck | Male | Noradrenaline | 8.6 ± 0.3 | 10.3 ± 0.4 | – | |
| Female | Noradrenaline | 8.3 ± 0.1 | 10.0 ± 0.5 | – | ||
| Prostate | Male | Noradrenaline | 8.2 ± 0.2 | 9.7 ± 0.3 | – | |
| Male | Ro 115–1240 | 9.9 ± 0.2 | 10.3 ± 0.3 | – | ||
| Tail artery | Noradrenaline | 9.4 ± 0.2 a | 9.8 ± 0.1a | 8.3 ± 0.1a | ||
| Ro 115–1240 | 9.2 ± 0.1a | 10.4 ± 0.2a | – | |||
| Human | ||||||
| Urinary bladder neck | Noradrenaline | 8.2 ± 0.1 | 9.7 ± 0.2 | – | ||
| Mesenteric artery | Noradrenaline | 9.1 ± 0.1 | 9.1 ± 0.8 | – | ||
The pKB values were estimated at a single concentration of prazosin (10 nM) or silodosin (1 or 3 nM) according to the concentration ratio method (Furchgott, 1972) or van Rossum procedure (van Rossum, 1963). The pKB values for silodosin in the response to Ro 115–1240 were estimated at 0.3 nM in rat prostate.
The pKB values for prazosin and silodosin in rat tail artery were obtained in the presence of 10 nM BMY7378, while the pKB values for BMY7378 were estimated from the ratio at 25% level of control response for noradrenaline (in the absence of BMY7378). The data obtained from both gender were combined in rat tail artery and human urinary bladder neck and mesenteric artery.
–, no inhibition by 20 nM BMY7378 or 3 nM silodosin. Mean ± SEM of four to seven experiments.
Next, the contractile responses to these three drugs were examined in the rat prostate. P-come 102 showed full agonist activity with similar potency to noradrenaline. Ro 115–1240 also induced contraction of the prostate, and the intrinsic activity of Ro 115–1240 was 0.60. The contractile response to noradrenaline in the prostate was relatively resistant to 10 nM prazosin, but was inhibited by 1 nM silodosin (Figure 4B). In contrast, the contractions evoked by Ro 115–1240 were noticeably suppressed by both prazosin (1 nM) and silodosin (1 nM; Figure 4C, Table 3).
Effects of noradrenaline, Ro 115–1240, and P-come 102 on α1-adrenoceptors in rat tail artery
The presence of functional α1A-adrenoceptors has also been reported in rat tail artery (Lachnit et al., 1997; Tanaka et al., 2004), therefore, we examined the effects of all three agonists on these arterial α1-adrenoceptors. In the presence of 10 nM BMY7378 and 100 nM rauwolscine, to mask α1D-adrenoceptors and α2-adrenoceptors, respectively, noradrenaline and P-come 102 showed full agonist activity, but Ro 115–1240 was only a partial agonist in the rat tail artery (Figure 4D). In contrast to the simple concentration–response curves for P-come 102 and Ro 115–1240, the curve for noradrenaline, produced by a wide range of noradrenaline concentrations (1 nM–100 μM), was shallow suggesting that distinct α1-adrenoceptor subtypes (probably α1A and α1B subtypes; Lachnit et al., 1997; Tanaka et al., 2004) are involved in the response to noradrenaline. The concentration–response curves for noradrenaline and Ro 115–1240 were inhibited by 10 nM prazosin and 1 nM silodosin (Figure 4E and 4F, Table 3). We observed no sex differences in the α1-adrenoceptor responses of the tail artery.
Effects of noradrenaline, Ro 115–1240 and P-come 102 on α1-adrenoceptors in the urinary bladder neck and mesenteric artery of humans
Finally, we examined the effects of the three drugs on human tissues. Figure 5A and 5C show the concentration–contraction curves for noradrenaline, Ro 115–1240 and P-come 102 in urinary bladder neck and mesenteric artery tissues. P-come 102 showed full agonist activity with slightly higher potency than noradrenaline in the urinary bladder neck preparations (four male and three female patients). However, the potency and intrinsic activity of P-come 102 were lower than that of noradrenaline in the mesenteric artery (six male and three female patients; Table 2). Ro 115–1240 was inactive in both human preparations. The contractile response to noradrenaline in the urinary bladder neck was resistant to 10 nM prazosin, whereas a significant rightward shift of the concentration–contraction relationship was produced by the same concentration of prazosin in the mesenteric artery (Figure 5B and 5D). Silodosin (1 nM) caused insurmountable inhibition of the concentration–response curve for noradrenaline in the urinary bladder neck (Figure 5B) and suppressed the responses to high concentrations of noradrenaline in the mesenteric artery (Figure 5D). BMY7378 (20 nM) failed to affect the contractile response to noradrenaline in either tissue (Table 3).
Figure 5.
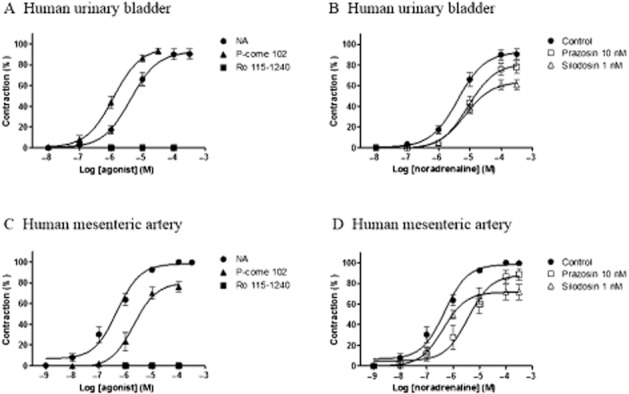
Concentration–response curves for noradrenaline (NA), Ro 115–1240, and P-come 102 in the human urinary bladder (A, B) and mesenteric artery (C, D). (A, C) Concentration-dependent contractions induced by three drugs in the urinary bladder and mesenteric artery. (B, D) Effects of prazosin (10 nM) or silodosin (1 nM) on concentration–response curve for noradrenaline. Mean ± SEM of seven experiments in the urinary bladder and nine in the mesenteric artery. NA, not applicable.
Discussion
In the LUT it has been proposed that adrenergic contractions are induced by activation of α1-adrenoceptors, but the identity of its functional phenotype is still contentious. We have hypothesized that this may be due to the expression of distinct α1-adrenoceptor phenotypes (α1A and α1L) from a single α1A-adrenoceptor gene. Therefore, in the present study, we first examined the agonist profiles of recombinant α1A- and α1L-adrenoceptor phenotypes using various drugs, A61603, NS-49, Ro 115–1240 and ESR1150 (Figure 1), which have been developed for SUI therapy (Knepper et al., 1995; Obika et al., 1995; Blue et al., 2004; Musselman et al., 2004; Matsumaru et al., 2005; Nishimune et al., 2010). Other than noradrenaline and phenylephrine, these drugs showed high specificity for α1A- and α1L-adrenoceptor phenotypes and were essentially inactive at the α1B- and α1D-adrenoceptors. In particular, A61603, MK017 and ESR1150 were more potent than noradrenaline at the α1A-adrenoceptor phenotype. P-come 102 (a newly synthesized compound) was less selective than noradrenaline for the α1A phenotype. With regard to their intrinsic activity, A61603, ESR1150 and P-come 102 showed full-agonist activity at both α1A and α1L phenotypes, whereas NS-49, Ro 115–1240 and MK017 were partial agonists at both phenotypes. From these results, most of the agonists developed to treat SUI appeared to have high activity at the α1A-adrenoceptor phenotype. In particular, Ro 115–1240 and NS-49 appeared to be specific for the α1A-adrenoceptor phenotype, because their intrinsic activity at the α1L-adrenoceptor phenotype was minimal. In contrast to these drugs, P-come 102 was a full agonist at α1A- and α1L-adrenoceptor phenotypes.
We next characterized the pharmacological properties of functional α1-adrenoceptors in LUT tissues (rat prostate, urethra, and urinary bladder neck, and human urinary bladder neck) using three selected agonists. P-come 102 showed full-agonist activity in the LUT tissues, whereas Ro 115–1240 (a partial agonist of α1A-adrenoceptor) failed to produce a contraction in the LUT tissues tested other than the rat prostate. Pharmacological analysis showed that the contractile responses to noradrenaline in LUT tissues (except female rat urethra) were relatively resistant to inhibition by prazosin (α1A, α1B and α1D-selective antagonist at ∼3 nM), highly sensitive to silodosin (α1A and α1L-selective antagonist at ∼3 nM), but insensitive to BMY7378 (α1D-selective antagonist at ∼20 nM), suggesting the predominant involvement of the α1L-adrenoceptor phenotype in the contractile responses to noradrenaline in these LUT tissues. These results conform with those found previously in human prostate and other LUT tissues of many species (Testa et al., 1993; 1997; Ford et al., 1996; Van der Graaf et al., 1997; Morishima et al., 2007; Nishimune et al., 2012). In human prostate, Ro 115–1240 was also found to be inactive, but P-come 102 as well as noradrenaline produced α1L–adrenoceptor-mediated contractions (I. Muramatsu, unpubl. obs.).
In contrast to the male rat urethra, the contractile response to noradrenaline in female rat urethra was not suppressed by 3 nM silodosin (Figure 3D). This silodosin-resistant contraction was observed in the presence of BMY7378 (α1D antagonist) and rauwolscine (α2 antagonist) and was inhibited by prazosin with a low pKB value (8.0). This atypical feature of female urethra α1-adrenoceptors is not compatible with the criteria of α1-adrenoceptor classification, and suggests the presence of another phenotype showing a unique drug-binding property. Apart from its low sensitivity to prazosin (pKB = 8.38; Taki et al., 1999), no detailed information on women urethra α1-adrenoceptors has been reported. Therefore, further analysis with human materials is urgently needed to determine the pharmacological properties of α1-adrenoceptors in human urethra.
In contrast to the LUT tissues, the rat prostate and tail artery produced a partial contraction in response to Ro 115–1240, and these contractions were potently inhibited by prazosin and silodosin. α1A–Adrenoceptor-mediated contractions have been reported in rat prostate (Yazawa and Honda, 1993; Forray et al., 1994; Chang et al., 2000) and rat tail artery previously (Lachnit et al., 1997; Tanaka et al., 2004). The present results in rat prostate and tail artery show that Ro 115–1240 can act as a selective agonist on rat α1A-adrenoceptors. As α1A- and α1L-adrenoceptor phenotypes have been demonstrated to occur concomitantly in many tissues (Hiraizumi-Hiraoka et al., 2004; Morishima et al., 2007; 2008; Muramatsu et al., 2008), the inability of Ro 115–1240 to induce a contraction in LUT tissues, other than the rat prostate, may reflect tissue-dependent differences either in the coupling efficiency to the contractile system between the α1A- and α1L-adrenoceptor phenotypes or in the density of expression of each subtype. Although the exact mechanism of these different responses needs to be explored in future studies, the present findings suggest that distinct phenotypes originating from a single receptor gene are independently activated by different agonists.
To reduce the adverse effects associated with SUI treatments, ‘uroselectivity’ (that is drugs that act selectively on LUT compared with other tissues such as vascular vessels) should be a prerequisite in the development of therapeutic agonists (Andersson and Wein, 2004). Previous studies have reported that human blood vessels are regulated through α1A- and/or α1B-adrenoceptors (Rudner et al., 1999; Michelotti et al., 2000; Murata et al., 2000). The present results with prazosin and silodosin show that α1A- and α1B-adrenoceptors coexist in human mesenteric artery and are both involved in the contractile response to noradrenaline, although the contribution of the α1B-adrenoceptors appears to be more dominant. The lower activity of P-come 102 relative to noradrenaline in human mesenteric artery and the lack of response to Ro 115–1240 may partly reflect a difference in the densities of expression of each α1-adrenoceptor subtype or the minor involvement of the α1L-adrenoceptor phenotype.
Recent studies have revealed that many GPCRs including α1A-adrenoceptors have the capacity to interact with more than one G-protein subtype as well as induce alternative signalling pathways or effector proteins, resulting in drug-dependent functional selectivity (Audet and Bouvier, 2008; Evans et al., 2010; Kenakin and Miller, 2010). In the present study, we measured [Ca2+]i and smooth muscle contraction to determine LUT function. However, if different biochemical approaches are applied, distinct agonist profiles or distinct subtype selectivities may be revealed, which may be relevant for the detection of new clinical therapies.
In summary, the present results revealed different agonist profiles between recombinant α1A- and α1L-adrenoceptor phenotypes and suggest that the α1L-adrenoceptor phenotype is predominantly involved in the contractile responses of LUT. However, we do not yet have selective or specific agonists for the α1L-adrenoceptor phenotype and future studies are needed to develop such compounds that may then provide uroselective and clinically useful drugs for SUI therapy.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by a grant from the Smoking Research Foundation of Japan, by the Organization for Life Science Advancement Programs (Research and Education Program for Life Science, Translational Research Program) and the Life Science Research Laboratory, University of Fukui.
Glossary
- A61603
N-(5-[4,5-dihydro-1H-imidazol-2-y]-2-hydroxy-5,6,7,8-tetrahydronaphtalen-1-yl)methanesulfonamide hydrobromide
- BMY7378
(8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]-8-azaspiro[4,5]decane-7,9-dione dihydrochloride
- ESR1150
4-bromo-N3-imidazolidin-2-ylidine-2,N1,N1-trimethyl-benzene-1,3-diamine hydrochloride
- LUT
lower urinary tract; MK017, 2-[(5-chloro-3-isopropyl-2-methylphenyl)methyl]-4,5-dihydro-1H-imidazole hydrochloride
- NS-49
(R)-(-)-3′-(2-amino-1-hydroxyethyl)-4′-fluoromethanesulphonanilide
- P-come 102
2-[(6-dimethylamino-3-isopropyl-2-methyphenyl)methyl]-4,5-dihydro-1H-imidazole hydrochloride
- Ro 115-1240 (Dabuzalgron, R450)
N-[6-chloro-3-[(4,5-dihydro-1H-imidazol-2yl)methoxy]-2-methylphenyl]methanesulphonamide
- RS-17053
N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-α, α-dimethyl-1 H-indole-3-ethanamine hydrochloride
- SNAP5089
2,6-dimethyl-4-(4-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate-N-[3-(4,4-diphenylpiperidin-1-yl)propyl]amide methyl ester
- SUI
stress urinary incontinence
Conflict of interest
The authors have no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE. Pharmacology of lower urinary tract smooth muscles and penile erectile tissues. Pharmacol Rev. 1993;45:253–308. [PubMed] [Google Scholar]
- Andersson KE. Alpha-adrenoceptors and benign prostatic hyperplasia: basic principles for treatment with alpha-adrenoceptor antagonists. World J Urol. 2002;19:390–396. [PubMed] [Google Scholar]
- Andersson KE, Wein AJ. Pharmacology of the lower urinary tract: basis for current and future treatments of urinary incontinence. Pharmacol Rev. 2004;56:581–631. doi: 10.1124/pr.56.4.4. [DOI] [PubMed] [Google Scholar]
- Argyle SA, McGrath JC. An α1A-/α1L-adrenoceptor mediates contraction of canine subcutaneous resistance arteries. J Pharmacol Exp Ther. 2000;295:627–633. [PubMed] [Google Scholar]
- Audet M, Bouvier M. Insights into signaling from the β2-adrenergic receptor structure. Nat Chem Biol. 2008;4:397–403. doi: 10.1038/nchembio.97. [DOI] [PubMed] [Google Scholar]
- Blue DR, Daniels DV, Gever JR, Jett MF, O'yang C, Tang HM, et al. Pharmacological characteristics of Ro 115–1240, a selective α1A/1L-adrenoceptor agonist: a potential therapy of stress urinary incontinence. BJU Int. 2004;93:162–170. doi: 10.1111/j.1464-410x.2004.04577.x. [DOI] [PubMed] [Google Scholar]
- Chang RS, Chen TB, O'Malley SS, Pettibone DJ, Disalvo J, Francis B, et al. In vitro studies on L-771, 688 (SNAP 6383), a new potent and selective α1A-adrenoceptor antagonist. Eur J Pharmacol. 2000;409:301–312. doi: 10.1016/s0014-2999(00)00854-2. [DOI] [PubMed] [Google Scholar]
- Drew GM. What do antagonists tell us about α1-adrenoceptors? Clin Sci. 1985;68(Suppl. 10):15s–19s. doi: 10.1042/cs068s015. [DOI] [PubMed] [Google Scholar]
- Evans BA, Broxton N, Merlin J, Sato M, Hutchinson DS, Christopoulos A, et al. Quantification of functional selectivity at the human α1A-adrenoceptor. Mol Pharmacol. 2010;79:298–307. doi: 10.1124/mol.110.067454. [DOI] [PubMed] [Google Scholar]
- Faure C, Pimoule C, Vallancien G, Langer SZ, Graham D. Identification of α(-adrenoceptor subtypes present in the human prostate. Life Sci. 1994;54:1595–1605. doi: 10.1016/0024-3205(94)90031-0. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Vanhoutte PM. α-Adrenoceptor subclassification in vascular smooth muscle. Trends Pharmacol Sci. 1986;7:347–349. [Google Scholar]
- Ford APDW, Arredondo NF, Blue DR, Jr, Bonhaus DW, Jasper J, Kava MS, et al. RS-17053 (N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-alpha, alpha-dimethyl-1H-indole-3-ethanamine hydrochloride), a selective α1A-adrenoceptor antagonist, displays low affinity for functional α1-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol Pharmacol. 1996;49:209–215. [PubMed] [Google Scholar]
- Forray C, Bard JA, Wetzel JM, Chiu G, Shapiro E, Tang R, et al. The α(-adrenergic receptor that mediates smooth muscle contraction in human prostate has the pharmacological properties of the cloned human α(C subtype. Mol Pharmacol. 1994;45:703–708. [PubMed] [Google Scholar]
- Furchgott RF. The classification on adrenoceptors (adrenergic receptors): an evaluation from the standpoint of receptor theory. In: Blaschko H, Muscholl E, editors. Handbuch der Experimentellen Pharmacology. Vol. 3. New York: Springer; 1972. pp. 283–335. [Google Scholar]
- Graham RM, Perez DM, Hwa J, Piascik MT. α1-Adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ Res. 1996;78:737–749. doi: 10.1161/01.res.78.5.737. [DOI] [PubMed] [Google Scholar]
- Gray KT, Short JL, Ventura S. The α1A-adrenoceptor gene is required for the α1L-adrenoceptor-mediated response in isolated preparations of the mouse prostate. Br J Pharmacol. 2008;155:103–109. doi: 10.1038/bjp.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, et al. International Union of Pharmacology. X. Recommendation for nomenclature of α1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- Hiraizumi-Hiraoka Y, Tanaka T, Yamamoto H, Suzuki F, Muramatsu I. Identification of α1L-adrenoceptor in rabbit ear artery. J Pharmacol Exp Ther. 2004;310:995–1002. doi: 10.1124/jpet.104.066985. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapenshifting protein: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J Pharmacol Exp Ther. 1995;274:97–103. [PubMed] [Google Scholar]
- Lachnit WG, Tran AM, Clarke DE, Ford AP. Pharmacological characterization of an α1A-adrenoceptor mediating contractile responses to noradrenaline in isolated caudal artery of rat. Br J Pharmacol. 1997;120:819–826. doi: 10.1038/sj.bjp.0700983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomasney JW, Cotecchia S, Lefkowitz RJ, Caron MG. Molecular biology of α-adrenergic receptors: implications for receptor classification and for structure-function relationships. Biochem Biophys Acta. 1991;1095:127–139. doi: 10.1016/0167-4889(91)90075-9. [DOI] [PubMed] [Google Scholar]
- Marshall I, Burt RP, Chapple CR. Noradrenaline contractions of human prostate mediated by α(A- (α1c-) adrenoceptor subtype. Br J Pharmacol. 1995;115:781–786. doi: 10.1111/j.1476-5381.1995.tb15001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumaru T, Sugiura R, Sakai K, Igarashi T, Kuno T. Comparison of toxicity and toxicokinetics/pharmacokinetics of an α1L-adrenoceptor agonist in rats and Rhesus monkeys. J Pharm Sci. 2005;97:273–283. doi: 10.1254/jphs.fp0040248. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Vrydag W. α1-, α2-, and β-adrenoceptors in the urinary bladder, urethra, and prostate. Br J Pharmacol. 2006;147:S88–S119. doi: 10.1038/sj.bjp.0706619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelotti GA, Price DT, Schwinn DA. α1-Adrenergic receptor regulation: basic science and clinical implications. Pharmacol Ther. 2000;88:281–309. doi: 10.1016/s0163-7258(00)00092-9. [DOI] [PubMed] [Google Scholar]
- Morishima S, Tanaka T, Yamamoto H, Suzuki F, Akino H, Yokoyama O, et al. Identification of α1L- and α1A-adrenoceptors in human prostate by tissue segment binding. J Urol. 2007;177:377–381. doi: 10.1016/j.juro.2006.08.080. [DOI] [PubMed] [Google Scholar]
- Morishima S, Suzuki F, Yoshiki H, Anisuzzaman AMS, Sathi ZS, Tanaka T, et al. Identification of the α1L-adrenoceptor in rat cerebral cortex and possible relationship between α1L- and α1A-adrenoceptors. Br J Pharmacol. 2008;153:1485–1494. doi: 10.1038/sj.bjp.0707679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu I, Ohmura T, Kigoshi S, Hashimoto S, Oshita M. Pharmacological subclassification of α1-adrenoceptors in vascular smooth muscle. Br J Pharmacol. 1990;99:197–201. doi: 10.1111/j.1476-5381.1990.tb14678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu I, Ohmura T, Hashimoto S, Oshita M. Functional subclassification of vascular α(-adrenoceptors. Pharmacol Commun. 1995;6:23–28. [Google Scholar]
- Muramatsu I, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman ASM, Tanaka T, et al. Identification of α1L-adrenoceptor in mice and its abolition by α1A-adrenoceptor gene knockout. Br J Pharmacol. 2008;155:1224–1234. doi: 10.1038/bjp.2008.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Taniguchi T, Muramatsu I. Pharmacological analysis of the novel, selective α1-adrenoceptor antagonist, KMD-3213, and its suitability as a tritiated radioligand. Br J Pharmacol. 1999;127:19–26. doi: 10.1038/sj.bjp.0702489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Taniguchi T, Takahashi M, Okada K, Akiyama K, Muramatsu I. Tissue selectivity of KMD-3213, an α1-adrenoceptor antagonist, in human prostate and vasculature. J Urol. 2000;164:578–583. [PubMed] [Google Scholar]
- Musselman DM, Ford AP, Gennevois DJ, Harvison ML, Laurent AL, Mokatrin AS, et al. A randomized crossover study to evaluate Ro 115–1240, a selective α1A/1L-adrenoceptor partial agonist, in women with stress urinary incontinence. BJU Int. 2004;93:78–83. doi: 10.1111/j.1464-410x.2004.04560.x. [DOI] [PubMed] [Google Scholar]
- Nishimune A, Suzuki F, Yoshiki H, Morishima S, Muramatsu I. Identification of cysteine-rich epidermal growth factor-like domain 1α (CRELD 1α) as a novel α1A-adrenoceptor-down-regulating protein and establishment of an α1L-adrenoceptor-expressing cell line. J Pharmacol Sci. 2010;113:169–181. doi: 10.1254/jphs.10093fp. [DOI] [PubMed] [Google Scholar]
- Nishimune A, Yoshiki H, Uwada J, Anisuzzaman AS, Umada H, Muramatsu I. Phenotype pharmacology of lower urinary tract α1-adrenoceptors. Br J Pharmacol. 2012;165:1226–1234. doi: 10.1111/j.1476-5381.2011.01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obika A, Shibata A, Horie K, Foglar R, Kimura K, Tsuijimoto G. NS-49, a novel α1a-adrenoceptor-selective agonist using recombinant human α1-adrenoceptors. Eur J Pharmacol. 1995;291:327–334. doi: 10.1016/0922-4106(95)90073-x. [DOI] [PubMed] [Google Scholar]
- Perez DM, Piascik MT, Graham RM. Solution-phase library screening for the identification of rare clones: isolation of an α1D-adrenoceptor cDNA. Mol Pharmacol. 1991;40:876–883. [PubMed] [Google Scholar]
- Price DT, Schwinn DA, Lomasney JW, Allen LF, Caron MG, Lefkowitz RJ. Identification, quantification, and localization of mRNA for three distinct alpha1 adrenergic receptor subtypes in human prostate. J Urol. 1993;150:546–551. doi: 10.1016/s0022-5347(17)35544-1. [DOI] [PubMed] [Google Scholar]
- Rudner XL, Berkowitz DE, Booth JV, Funk BL, Cozal KL, D'Amico EB, et al. Subtype specific regulation of human vascular α1-adrenergic receptors by vessel bed and age. Circulation. 1999;100:2333–2343. doi: 10.1161/01.cir.100.23.2336. [DOI] [PubMed] [Google Scholar]
- Ruffolo R, Jr, Hieble JP. Adrenoceptor pharmacology: urogenital applications. Eur Urol. 1999;36(Suppl. 1):17–22. doi: 10.1159/000052313. [DOI] [PubMed] [Google Scholar]
- Su TH, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman ASM, Tanaka T, et al. Native profiles of α1A-adrenoceptor phenotypes in rabbit prostate. Br J Pharmacol. 2008;155:906–912. doi: 10.1038/bjp.2008.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki N, Taniguchi T, Okada K, Moriyama N, Muramatsu I. Evidence for predominant mediation of α1-adrenoceptor in the tonus of entire urethra of women. J Urol. 1999;162:1829–2832. [PubMed] [Google Scholar]
- Tanaka T, Zhang L, Suzuki F, Muramatsu I. Alpha-1 adrenoceptors: evaluation of receptor subtype binding kinetics in intact arterial tissues and comparison with membrane binding. Br J Pharmacol. 2004;141:468–476. doi: 10.1038/sj.bjp.0705627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa R, Guarneri L, Ibba M, Strada G, Poggesi E, Taddei C, et al. Characterization of α1-adrenoceptor subtypes in prostate and prostatic urethra of rat, rabbit, dog, and man. Eur J Pharmacol. 1993;249:307–315. doi: 10.1016/0014-2999(93)90527-o. [DOI] [PubMed] [Google Scholar]
- Testa R, Guarneri L, Angelico P, Poggesi E, Taddei C, Sironi G, et al. Pharmacological characterization of the uroselective alpha-1 antagonist Rec 15/2739 (SB 216469): role of the alpha-1L adrenoceptor in tissue selectivity, part II. J Pharmacol Exp Ther. 1997;281:1284–1293. [PubMed] [Google Scholar]
- Van der Graaf PH, Deplanne V, Duquenne C, Angel I. Analysis of α1-adrenoceptors in rabbit lower urinary tract and mesenteric artery. Eur J Pharmacol. 1997;327:25–32. doi: 10.1016/s0014-2999(97)89674-4. [DOI] [PubMed] [Google Scholar]
- Van Rossum JM. Cumulative dose–response curves. II. Technique for the making of dose–response curves in isolated organs and the evaluation of drug parameters. Arch Int Pharmacodyn Ther. 1963;143:299–330. [PubMed] [Google Scholar]
- Yazawa H, Honda K. α1-Adrenoceptor subtype in the rat prostate is preferentially the α1A type. Jpn J Pharmacol. 1993;62:297–304. doi: 10.1254/jjp.62.297. [DOI] [PubMed] [Google Scholar]