

Glial cells are historically known for their role in regulating extracellular potassium concentration. It was postulated almost 50 years ago (1) that astrocytes were important for clearing activity-induced rises in extracellular potassium and that astrocytes, via manipulation of extracellular potassium concentration, may regulate neural network activity. Subsequent work in the leech central-nervous-system preparation (2) and isolated retinal Muller cells (3) helped solidify glial ability to buffer extracellular potassium, but it was only recently shown that astrocytes can be recruited to actively regulate extracellular potassium concentration with indirect effects on neural network activity (4).
Potassium spatial buffering is the capacity of astrocytes to redistribute locally elevated extracellular potassium following neuronal activity. Evidence from acute slices and astrocytic cocultures suggests redistribution is directed to sites of low extracellular potassium, to prevent subsequent changes in membrane excitability. Due to the tortuosity and lack of extracellular space in the complex mammalian central nervous system, the bulk of this redistribution is achieved through the high selective potassium conductance of astrocytes associated with significant astrocytic gap junction coupling. The general understanding has been that astrocytic inwardly rectifying potassium channels are important for establishing baseline extracellular potassium levels and astrocytic resting membrane potentials, in addition to playing a role in minimizing the postactivity undershoot in extracellular potassium concentration. Active uptake of potassium by the Na+/K+ ATPase has been considered the major driving force behind buffering of activity-induced potassium rises (5,6).
Based on early computer modeling, astrocytes were originally suggested to regulate cerebral blood flow by K+ siphoning (7). However, subsequent experimental testing of this model in retinal explants was unable to confirm a role for potassium in neurovascular coupling (8). Despite this lack of a role for K+ in neurovascular coupling in the retina, evidence strongly supporting a role for K+ in cortical neurovascular coupling has been experimentally confirmed (9,10). In this issue, Witthoft et al. (11) incorporate and highlight the importance of astrocytic inwardly rectifying channels in a new computer model showing that these channels accelerate siphoning of potassium into the perivascular space. This helps reconcile the long delay to functional hyperemia predicted by previous models that did not consider astrocytic inward rectifying channels. Witthoft et al. (11) also address and provide a framework to integrate astrocytic IP3 and Ca2+-mediated release of epoxyeicosatrienoic acids (EETs) into the K+ siphoning hypothesis. As such, it would suggest that all Gq-coupled receptors (mGluR1, mGluR5 glutamatergic, α1-adrenergic, M1, M3, M5 muscarinic, 5-HT2 serotonergic, and H1 histaminergic receptors) on astrocytes may elicit similar effects on the vasculature.
The model proposed by Witthoft et al. (11) relies on mGluR and IP3 signaling pathways. However, this signaling pathway may not apply to adult animals lacking mGluR-mediated calcium signaling (12). Interestingly, they provide data that suggest the mGluR/IP3 signaling pathway is only important with weaker stimulations. The relatively large potassium rises used in their model were sufficient to result in very similar vasodilation responses when glutamatergic signaling was omitted. Thus, the potassium-siphoning model for neurovascular coupling appears to be a very basic, and possibly ubiquitous, mechanism for coupling dilation to large activity-induced changes in extracellular potassium. Smaller, more refined effects on the vasculature likely result from the integration of nitric oxide, EET, and cyclooxygenase pathways that are closely linked to the local tissue metabolic state (13). In fact, potassium-mediated neurovascular coupling may explain the residual cortical vasodilation seen during vibrissal stimulation under conditions where all the above-mentioned pathways are blocked pharmacologically (14). As Witthoft et al. (11) point out, posttranslational modifications of TRPV4 and BK channels may alter channel kinetics that would alter the relative contribution of this pathway. Thus, potassium contribution to vascular responses probably varies considerably depending on levels of local activity and metabolic demands. It may be reasonable to assume, however, that K+ siphoning always contributes to vasodilation under conditions of high or excessive neural activity.
Another very interesting aspect of the model proposed by Witthoft et al. (11) is the inclusion of a bidirectional communication scenario whereby vessels can also affect astrocytic stretch-activated TRPV4 channels on the foot processes via changes in vessel diameter. Stretch or strain activation of surrounding astrocytic foot processes opens TRPV4 channels, allowing calcium to depolarize the foot process. This activates BK channels and permits potassium to flood the perivascular space, leading to smooth muscle inward rectifier-mediated hyperpolarization and vasodilation. This pathway may be important for quelling vasomotion or spasticity as the authors suggested in a previous work (15), but may also have interesting implications for propagating astrocytic endfoot calcium-mediated (BK channel) dilation along the vascular tree to dilate feeding arteries/arterioles (Fig. 1). Thus, astrocyte endfeet alone may be sufficient for rapid propagation of K+-mediated dilatory signals along the vasculature.
Figure 1.
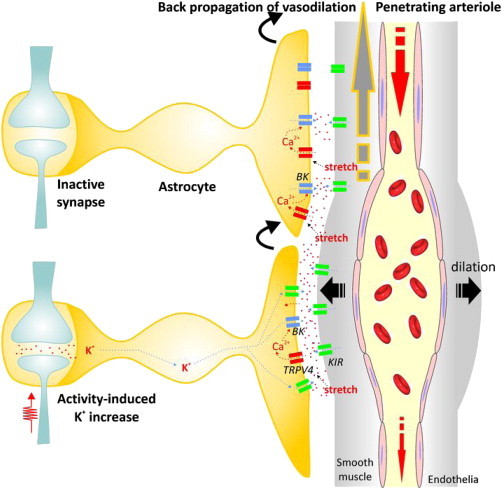
Back-propagation of vasodilation through astrocyte vascular endfeet. As outlined in Witthoft et al. (11), large-activity-induced potassium increases will result in astrocyte depolarization and siphoning of excess potassium to the gliovascular interface, where it activates inwardly rectifying potassium channels on arteriole smooth muscle, resulting in hyperpolarization and relaxation/dilation. This dilation results in stretch of glial foot processes and opening of TRPV4 channels, allowing calcium in to activate BK channels. This will further flood the perivascular space with potassium, enlarging and expanding the dilatory response. As the dilation expands, the stretch will activate TRPV4 channels on the adjacent nonstimulated astrocyte process, initiating the same processes leading to further dilation. This cascade can jump from foot process to foot process being regenerated with each jump, much like saltatory jumping of the action potential down myelinated fibers. In this way, local dilatory signals can rapidly propagate back up the vascular tree to dilate the larger supply arteries.
In summary, the model proposed by Witthoft et al. (11) integrates Gq-coupled IP3 and EET production with existing potassium homeostatic mechanisms to provide an elegant means for neurovascular coupling. Extending this model as a framework to include astrocytic nitric oxide, EET, and cyclooxygenase pathways under more modest activity scenarios firmly places astrocytes as primary conduits for neurovascular coupling.
Contributor Information
Lane K. Bekar, Email: lane.bekar@usask.ca.
Maiken Nedergaard, Email: nedergaard@urmc.rochester.edu.
References
- 1.Hertz L. Possible role of neuroglia: a potassium-mediated neuronal-neuroglial-neuronal impulse transmission system. Nature. 1965;206:1091–1094. doi: 10.1038/2061091a0. [DOI] [PubMed] [Google Scholar]
- 2.Orkand R.K., Nicholls J.G., Kuffler S.W. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 3.Newman E.A., Frambach D.A., Odette L.L. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang F., Smith N.A., Nedergaard M. Astrocytes modulate neural network activity by Ca²+-dependent uptake of extracellular K+ Sci. Signal. 2012;5:ra26. doi: 10.1126/scisignal.2002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chever O., Djukic B., Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 2010;30:15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Ambrosio R., Gordon D.S., Winn H.R. Differential role of KIR channel and Na+/K+-pump in the regulation of extracellular K+ in rat hippocampus. J. Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- 7.Paulson O.B., Newman E.A. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237:896–898. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metea M.R., Kofuji P., Newman E.A. Neurovascular coupling is not mediated by potassium siphoning from glial cells. J. Neurosci. 2007;27:2468–2471. doi: 10.1523/JNEUROSCI.3204-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filosa J.A., Bonev A.D., Nelson M.T. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat. Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 10.Girouard H., Bonev A.D., Nelson M.T. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc. Natl. Acad. Sci. USA. 2010;107:3811–3816. doi: 10.1073/pnas.0914722107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witthoft A., Filosa J.A., Karniadakis G.E. Potassium buffering in the neurovascular unit: models and sensitivity analysis. Biophys. J. 2013;105:2046–2054. doi: 10.1016/j.bpj.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun W., McConnell E., Nedergaard M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339:197–200. doi: 10.1126/science.1226740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon G.R., Choi H.B., MacVicar B.A. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X., Li C., Koehler R.C. Relative contribution of cyclooxygenases, epoxyeicosatrienoic acids, and pH to the cerebral blood flow response to vibrissal stimulation. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H1075–H1085. doi: 10.1152/ajpheart.00794.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witthoft A., Karniadakis G.E. A bidirectional model for communication in the neurovascular unit. J. Theor. Biol. 2012;311:80–93. doi: 10.1016/j.jtbi.2012.07.014. [DOI] [PubMed] [Google Scholar]