

Abstract
A wide range of invasive pathological outcomes originate from the loss of epithelial phenotype and involve either loss of function or downregulation of transmembrane adhesive receptor complexes, including Ecadherin (Ecad) and binding partners β-catenin and α-catenin at adherens junctions. Cellular pathways regulating wild-type β-catenin level, or direct mutations in β-catenin that affect the turnover of the protein have been shown to contribute to cancer development, through induction of uncontrolled proliferation of transformed tumor cells, particularly in colon cancer. Using single-molecule force spectroscopy, we show that depletion of β-catenin or the prominent cancer-related S45 deletion mutation in β-catenin present in human colon cancers both weaken tumor intercellular Ecad/Ecad bond strength and diminishes the capacity of specific extracellular matrix proteins—including collagen I, collagen IV, and laminin V—to modulate intercellular Ecad/Ecad bond strength through α-catenin and the kinase activity of glycogen synthase kinase 3 (GSK-3β). Thus, in addition to regulating tumor cell proliferation, cancer-related mutations in β-catenin can influence tumor progression by weakening the adhesion of tumor cells to one another through reduced individual Ecad/Ecad bond strength and cellular adhesion to specific components of the extracellular matrix and the basement membrane.
Introduction
During developmental processes, such as gastrulation, and adult pathology, such as cancer metastasis, individual epithelial/carcinoma cells modulate their mutual interactions in response to the extracellular environmental cues in a temporal and spatial manner. For example, in the course of cancer progression from an ordered epithelial layer to invading carcinoma cells deadhere from adjacent normal cells and other tumor cells to actively invade through the underlying basement membrane, travel through stromal spaces by forming short-term adhesions with stromal extracellular matrix (ECM) proteins, and intravasate between endothelial cells covering blood vessels and lymphatics to distribute to and colonize distant organs. Of critical importance in this metastatic cascade is the nature of the interactions among invading tumor cells and between these tumor cells and the other cells and proteins within the ECM that they encounter on the way.
The biochemical and biophysical stimuli offered by a tumor microenvironment can favor migrating cells to invade the stromal space as single cells or collectively (1,2) as small clusters by forming dynamic associations, which, at the end of the metastatic cascade subsequently mature at colonized sites. It is therefore likely that the cells sense the microenvironment and modulate their intercellular adhesion accordingly.
A wide range of invasive pathological outcomes originate from the loss of epithelial phenotype (3–5) and involve either loss of function or downregulation of transmembrane adhesive receptor complexes (6,7). For example, the complex comprising Ecadherin (Ecad) (8,9) and its binding partners β-catenin and α-catenin (10,11) localizes at adherens junctions and modulates the strength of intercellular adhesion in monolayers of normal cells and during cancerous processes, such as metastasis. Cellular pathways regulating the wild-type (WT) β-catenin level, or direct mutations in β-catenin that affect the turnover of the protein have been shown to contribute to cancer development, through induction of uncontrolled proliferation of transformed tumor cells particularly in colon cancer (12,13). However, direct measurements of alteration in cell-cell adhesion force due to cancer-related mutations in β-catenin have not been explored (14).
Here, we show that a cancer-related mutation in β-catenin present in human colon cancers weakens tumor cell intercellular Ecad/Ecad bond strength and the capacity of tumor environmental ECM proteins to modulate intercellular Ecad/Ecad bond strength. Thus, in addition to regulating tumor cell proliferation, cancer-related mutations in β-catenin can influence tumor progression by weakening tumor cell adhesion to one another and their microenvironment. Furthermore, we show that the activity of known β-catenin-related kinase glycogen synthase kinase 3 (GSK3β) regulates the strength of β-catenin-mediated Ecad/Ecad bonds in a wide range of cell lines.
Materials and Methods
Cell culture
McCoy’s 10A modified medium (GIBCO) was supplemented with 10% fetal bovine serum (ATCC) and 1% penicillin/streptomycin (Sigma). For regular culture, cells were plated to 75% confluence in T25 flasks, trypsinized for 2 min at 37°C and 5% CO2, resuspended in suspension medium, and replated in T25 flasks at 1:10 dilution. HBSS buffer was used for all washing steps. For preparing dishes for single-molecule force spectroscopy, ∼10,000 cells were plated into 60 mm culture dishes (either uncoated or precoated with an ECM molecule) and incubated for up to 2 days to form small colonies (4–10 cells). Immediately before the experiments, cells were switched to prewarmed serum-free medium supplemented with HEPES to maintain pH.
Single molecule spectroscopy
Preparing atomic force microscopy (AFM) cantilevers
MSNL-AUHW cantilevers (Veeco, NY) with a nominal force constant of 10 pN/nm were used for all measurements. Cantilevers were gently washed for 1 min each in 70% ethanol, 10% HCl cleaning buffer, ultrapure water, and 95% ethanol. After drying on a clean surface, the cantilevers were placed gently for 5 min in a dish containing acetone. The cantilevers were then either incubated in phosphate buffered saline (PBS) until ready for experiment or further treated for coating the surface with ECM molecules. For certain experiments, to let the cells adhere properly to the surface, 0.5 mg/ml streptavidin was used to coat the cantilevers.
Coating culture dishes with ECM
Polystyrene culture dishes (Corning, Lowell, MA) were used for coating various ECM substrates. Briefly, the dishes were washed with ethanol overnight and allowed to dry in a stream of nitrogen. The dishes were immediately coated with a solution of specific ECM. 1 ml of 50 μg/ml of human Collagen IV (Millipore, Billerica, MA), 1 ml of 50 μg/ml of Engelbreth-Holm-Swarm MatriGel, and 1 ml of 50 μg/ml of human laminin V (Abcam, Cambridge, MA) solution were used to coat the dishes. For collagen-I, precoated dishes were used (Corning).
Biotinylation of cells for dwell experiments
For experiments requiring a longer contact between cells, biotin was used to allow the cells to bind strongly to the cantilever, following standard protocol (16). Cells were washed thrice with ice-cold PBS and spun down at 1400 rpm for 4 min. Sulfo-NHS-LC-biotin (Thermo-Fisher) was used at a final concentration of 0.5 mg/ml to incubate the cells at 4°C, for 30 min. The biotin-labeled cells were washed thrice with ice-cold PBS, supplemented with glycine to quench the residual biotin, and used immediately for loading onto AFM cantilevers.
Loading live cells onto AFM cantilevers
An Eppendorf Transjector 5246 (Eppendorf N.A., Hauppauge, NY) was used to place cells on an AFM cantilever. A cell culture dish containing medium with 2% HEPES added was placed on a TE300 microscope with a 10× Plan-Fluor objective (N.A. 0.3) and a hand-pulled microneedle was introduced in solution, just over the cantilever. Up to 1000 cells were introduced into the solution near to the end of the cantilever. After the cells settled down at the base of the dish, the microneedle was used to pick up a few cells, which were gently placed on the cantilever, while ensuring that the cantilever was not strained. After the cells were attached, the microneedle was removed from the solution, an incubation chamber was placed over the dish, maintaining the cells at 37°C and 5% CO2 until they were spread. The dish was then placed in a water-heated incubator until the time of experiment.
Generation of force-displacement curves
A pico-Newton sensitive MFP-1D (Asylum Research, Santa Barbara, CA) was used to conduct single-molecule force spectroscopy measurements, as described previously (16–21). Briefly, a cantilever with 1–2 cells attached to it was introduced into a holder. Bending of the cantilever was measured by tracking the reflection of a laser beam from the surface of the cantilever. Postexperiment calibration was also conducted to ensure that the cantilever did not change its bending rigidity during the experiment. The thermal resonance method was used to calibrate the force constant of the cantilever (15) and the photodiode sensitivity was calibrated by impinging the cantilever on a hard substrate and measuring the slope of the corresponding force-displacement curve. Immediately before the experiment, plated cells were incubated in warm serum-free medium supplemented with 2% HEPES. Cells were brought in contact for either 1 or 300 ms, and retracted at a rate of 10 μm/s and the force-distance curves were recorded. No more than 30% force-displacement curves yielded a bond rupture event, exhibited in the form of a spike and the frequency of occurrence of single, double, triple, or more than triple bond rupture events was found to follow Poisson distribution, as expected for bond formation over small timescales. In order that the cells did not strain too much and the statistical consistency was maintained, only 30 contacts per cell-pair were allowed before moving to another cell and the force of impingement was controlled so that the cells did not press each other beyond what is physiologically insignificant, in the 50–800 pN range. The integrity of the cells on the cantilever tip was confirmed both before and after the experiments were over. Up to four cell-loaded cantilever tips were used for each experiment to invoke sufficient statistical significance in our results. To negate the hydrodynamic drag on the cantilever during both descent and ascent phase, we zeroed the zero-force slope of the force-displacement curve to obtain the correct loading rate and the rupture force for each bond rupture event. Because continuous cycling of the piezo through the descent-ascent cycle sometimes leads to hysteresis, we ensured that the location of the reflected laser spot on the diode did not deviate from the initial value by >10% (16–21).
Analysis of single-molecule measurements
For each force curve exhibiting a bond rupture, only the last bond rupture was used for computation of the mean bond strength. This ensured that any possible intercadherin cooperativity arising as a result of Ecad cluster formation at the contact site did not affect our measurement of Ecad/Ecad bond strength because the last Ecad/Ecad bond ruptured is free of any cooperative influence. The mean bond strength was obtained for each condition and used for comparison. To ensure that the rupture event was indeed a single bond rupture event, the number of instances of a force-curve with none, one, two, three, or more than three bonds were counted and found to follow a Poisson distribution, thereby ensuring that most of the events registered were single bond rupture events. Analysis of individual waves was done on the IGOR 4.09 platform (Wavemetrics, Lake Oswego, OR) (16–21).
Specificity of measurements
To test that our results indeed represent Ecad-induced adhesion, we carried out control experiments using antihuman-Ecad function blocking antibodies (HECD-1, Zymed Labs) at a concentration of 200 μg/ml, for which the cells were allowed to remain incubated at 37°C and 5% CO2 for 30 min before the experiment. Alternatively, chelating agent EDTA was used to block Ecad-induced adhesion and the corresponding frequency of binding measured to ensure that bond ruptures observed in the absence of EDTA were indeed caused by the presence of Ecad/Ecad bonds between adjoining cells (16–21).
Depletion of targeting genes with lentiviral-mediated shRNA
The shRNA sequences targeting the specific genes were selected: mh-β-catenin shRNA 2501 GGTGCTGACTATCCAGTTGA; mh-a-catenin shRNA 531 GCTGAAAGTTGTGGAAGAT; mh-GSK3β 1979 GGAAGCTTGTGCACATTCA; mh-GSK3β 2193 GACCGTGGACAGACCAATA. The lentiviral vectors containing shRNA cassettes were constructed, HCT-derived cells were transduced as described (22).
Coimmunoprecipitation (CoIP), GST-beads pulldown, and cell fractionation
Subconfluent culture of HCT116-derived cells in 2X p150 culture dishes were washed with ice-cold 1XPBS twice, cells were directly lysed in the dishes with 1 ml of CoIP buffer (20 mM HEPES pH 7.5, 120 mM NaCl, 5 mM NaF, 1 mM sodium orthovanadate, 0.5 mM EDTA, 0.1% NP-40, 1 mM DTT, protease inhibitor cocktail from Sigma), collected with cell lifter, and spun for 10 min at 12,000 rpm, 4°C. Protein concentrations were determined. 2 mg of cell lysate was used for each CoIP or pulldown. For CoIP, 2 μg of mouse monoclonal antibody against β-catenin or mouse IgG were charged to washed protein G sepharose beads. For pulldown, 2 μg of GST or GST-Ecad-internal cellular domain fusion protein (plasmid kindly provided by Dr. Cara Gottardi) were charged to glutathione-agrose beads (both were 20 μl of 1:1 v/v beads slurry washed with CoIP buffer) at 4°C for 1 h, washed with CoIP buffer three times, incubated with cell lysate for 1 h on a rocker at 4°C, and washed with CoIP buffer five times. 20 μl of sodium dodecyl sulfate-sample loading buffer was added to each set of washed beads, vortexed, and boiled for 5 min.
Western blots were performed as indicated. For subcellular fractionation, medium for cells in 2X p150 tissue culture dishes was removed and cells washed with 30 ml of ice-cold H2O twice. Immediately after removal of the cold H2O, the plate was tilted for a few seconds to drain all residue liquid. 0.5 ml of cold hypotonic buffer (20 mM HEPES pH 7.5, 5 mM NaF, 1 mM sodium orthovanadate, 0.5 mM EDTA, 1 mM DTT, protease inhibitor cocktail from Sigma) was added; thereafter, cells were collected with cell lifter (Fisher), transferred to an Eppendorf tube and passed through a 25 gauge needle with a 1-ml syringe 30 times to break up the cells. Cell nucleus and mitochondria fractions were removed by spinning at 8000 × g for 10 min at 4°C, the supernatant was transferred to ultracentrifuge tubes, and spun at 100,000 × g for 1 h at 4-deg in XL90 Beckmann Ultracentrifuge. The supernatant was collected as the cytosolic fraction, whereas the pellet was dissolved in 0.5 ml of cell lysis buffer (hypotonic buffer plus 150 mM NaCl and 0.5% NP-40) for 2 h on ice, vortexed occasionally, and the mixture was transferred to an Eppendorf tube. After spinning briefly, the supernatant was collected as the cell membrane fraction. Protein concentration was determined. For Western blot, 20 μg of proteins were loaded into each lane.
Statistical analysis
Single bond rupture forces were analyzed and compared as mean ± SEM. Statistical significance was tested following the Michelin grade scale, using the one-way analysis of variance (ANOVA) nonparametric test with Bonferroni analysis for multiple comparisons and plotted using commercially available GraphPad Prism software (GraphPad, San Diego, CA). Values of p < 0.05 were considered significant. For each condition other than function blocking anti-Ecad antibody control, no less than 140 clean bond rupture events were analyzed, with a minimum N = 3 independent experiments.
Results
The adhesion strength of single intercellular Ecad/Ecad bonds is weakened for cells expressing a cancer-related β-catenin S45 deletion mutant
We first tested the hypothesis that cancer-related mutations in β-catenin, which are associated with enhanced tumor growth, can also weaken the strength of interaction between Ecad molecules on adjoining tumor cells. To do so we turned to the colon cancer cell line HCT116, which is heterozygous for the CTNNB1 gene—these cells express WT β-catenin and β-catenin containing an S45 deletion (Δ45). From these parental cells (WT/Δ45), WT/− and −/Δ45 cells were generated through homologous recombination to delete the S45− or WT β-catenin alleles, respectively (23).
The tensile strength of individual Ecad/Ecad bonds formed between living cells was measured using single-molecule force spectroscopy (Fig. 1 A). A cell adherent to a cantilever was placed into contact with a cell placed on a substrate for a controlled duration, the upper cell was then retracted at a controlled speed and the strength of individual bonds formed during the time of cell-cell contact measured, as described previously (19). This method allows us to study individual Ecad/Ecad bonds formed during an extremely short duration of cell-cell contact (1 ms), without the confounding effects of an ill-defined number of molecules at the intercellular interface, as in long-term cell-cell adhesion force measurement assays such as the dual-cell micropipette assay (24). By keeping the cell-cell contact force to below half a nano-Newton and allowing bond formation to last for only 1 ms, the assay ensures the formation of very few (typically less than five) bonds during the duration of contact. During retraction of the cantilever, the distance between cells was recorded as a function of time until complete deadhesion marked by an abrupt decrease in force (red arrows, Fig. 1 B). A key advantage of this single-molecule technique is that it is largely insensitive to the levels of expression of adhesion molecules on cell surfaces (16–21), because simultaneous control over the force of impingement and the duration of contact allows for individual bond-rupture events to be registered as separate events on the force-displacement curve (Fig. 1 B), irrespective of the number of cell-surface receptors. To test that single-molecule force spectroscopy measured specific adhesive interactions between Ecad molecules, the plated cells were incubated with either a function-blocking anti-Ecad antibody (see Fig. 1 D) or EGTA (Fig. S1 in the Supporting Material) to deplete calcium in the growth medium. In both experiments, the frequency of binding between cells decreased drastically, confirming that the rupture events that appeared on force-displacement curves (red arrows, Fig. 1 B) were indeed due to the rupture of Ecad/Ecad bonds. Moreover, the occurrence of bonds formed each time cells were juxtaposed followed Poison statistics (Fig. 1 C), as expected for single-molecule force spectroscopy measurements (25) and further confirms that the force-spectroscopy measurements were dependent on the statistical probability of formation of none, one, two, three, or more than three Ecad/Ecad bonds between apposing cells, over the short duration of contact. Finally, it was ensured that the force of impingement and the duration of contact were chosen such that the frequency of binding did not exceed ∼30% (Fig. 1 D)—thereby ensuring that >80% of rupture events could be attributed to rupture of single intercellular bonds—as compared to multiple bond ruptures occurring simultaneously (16–21,25). Together these controls ensured that the values of adhesion forces reported below are specifically the result of single Ecad/Ecad bonds formed during adhesion between adjoining cells. Only one type of bond-rupture event was evaluated because we did not observe multiple peaks in the distribution of bond rupture forces.(Fig. S2).
Figure 1.
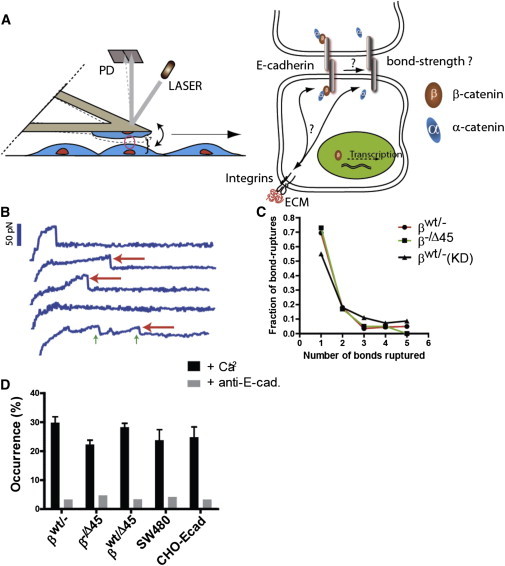
The regulation of Ecad-mediated intercellular adhesion at single-molecule resolution. (A) Schematic of the methodology used to measure Ecad/Ecad bond strength between adjoining live cells at single-molecule resolution. An AFM loaded with living cells is brought into contact with cells plated on uncoated or ECM-coated dishes. The cartoon on the right depicts the central question investigated in this work: how do changes in intracellular β-catenin and changes in extracellular ECM affect intercellular adhesion via modulation of single Ecad/Ecad bond strength. (B) Representative force-displacement curves obtained during the controlled retraction of the AFM cantilever at a speed of 20 μm/s. Red arrows mark bond rupture events; the height of the fall corresponds to the bond strength expressed in pico-Newton. Green arrows mark multiple rupture events. Only the last bond rupture was counted toward the determination of mean bond strength. (C) Distribution of occurrence of single and multiple rupture events followed Poisson statistics, shown for HCT116 cells expressing only WT (βWT/−), only ΔS45 β-catenin (β−/Δ45), and with β-catenin depleted (βWT/−(KD)). (D) The frequency of bond rupture with and without function-blocking antibody, together showing that the recorded rupture events predominantly reflect the rupture of Ecad/Ecad bonds. All error bars designate SEM.
Analysis of the distribution of bond strengths obtained by these single-molecule measurements showed that shRNAi-mediated depletion of β-catenin in WT/− cells (denoted βWT/−(KD) cells) resulted in weaker Ecad/Ecad bonds (i.e., a significantly smaller mean tensile force was required to break individual bonds between these cells) than those formed between cells expressing WT β-catenin, as expected (Fig. 2 A). Surprisingly, cells expressing only the S45 mutant β-catenin (denoted β−/Δ45 cells) formed significantly weak Ecad/Ecad bonds (Fig. 2 A).
Figure 2.
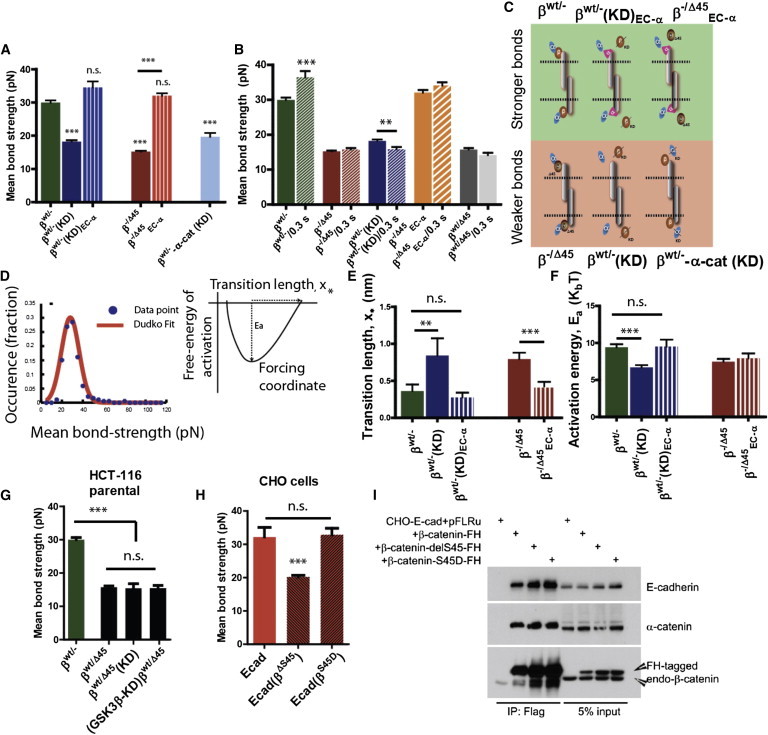
β-catenin modulates the strength of single intercellular Ecad/Ecad bonds. (A) Strength of Ecad/Ecad bonds is significantly weakened in HCT116 cells expressing only a mutant β-catenin with S45 deleted (β−/Δ45, plain red) and in HCT116 cells wherein β-catenin is knocked down (βWT/−(KD), plain blue) as compared to cells expressing only WT β-catenin (βWT/−, plain green). Chimeric fusion of α-catenin to Ecad is sufficient to rescue the strength of single Ecad/Ecad bonds, (βWT/−(KD)EC-α, threaded blue), and (β−/Δ45EC-α). Loss of α-catenin leads to significantly weaker bonds (βWT/−-α-cat(KD, light blue). Results depict one-way ANOVA variance analysis using Bonferroni’s multiple comparison test, with ∗∗∗ implying α = 0.05 (95% confidence intervals). n > 140 for each case; number of independent experiments = 3 or more. (B) Strengthening of single Ecad/Ecad bonds over time depends on the state of β-catenin. For cells expressing WT β-catenin (βWT/−, green bars), Ecad/Ecad bonds strengthened over 300 ms, whereas for cells depleted of β-catenin (blue bars), Ecad/Ecad bonds weakened overt 300 ms (p = 0.0064; n = minimum 140 cells, one-way ANOVA analysis using Bonferroni’s multiple-comparison test with α = 0.05). Cells expressing Ecad-α-catenin chimera (β−/Δ45EC-α, orange bars), cells expressing mutant β-catenin (β−/Δ45, red bars), and parental HCT116 cells did not exhibit any time-dependent behavior. (C) Illustration showing the state of β-catenin when stronger or weaker E-cad/Ecad bonds are formed. (D) Thermodynamic analysis of rupture-force distribution based on the fit of bond strength distribution by the Hummer-Dudko model (left panel (20),) give rise to computation of transition length (x∗) and activation energy (Ea). Fit shows that (E) transition length increases and (F) free energy of activation decrease significantly for mutated or depleted β-catenin. ∗∗∗ Designates p < 0.001, unpaired Student’s t-test. A minimum of 140 ruptures were analyzed for each case, with N > 3. (G) In parental HCT116 colon-carcinoma cells, heterozygous expression of both WT β-catenin and mutated β-catenin leads to weakened Ecad/Ecad bonds between adjoining cells (p = 0.0072; n >140 cells each, one-way ANOVA analysis using Bonferroni’s multiple-comparison test with α = 0.05). Genetic depletion (shRNAi) of both β-catenin and known β-catenin kinase GSK3β has the same effect. n.s: not significant. (H) In CHO cells exogenously expressing Ecad, expression of S45 deletion-mutated β-catenin leads to the formation of weaker Ecad/Ecad bonds (p = 0.0093; n >140 cells each, one-way ANOVA analysis using Bonferroni’s multiple-comparison test with α = 0.05), whereas expression of phosphomimetic S > D mutated β-catenin induces no significant (n.s.) weakening of Ecad/Ecad bonds. Immunoprecipitation results (I) exhibit no significant change in Ecad/β-catenin binding affinity in CHO cells expressing ΔS45 mutant β-catenin. All error bars designate SEM.
The absence or loss of function of α-catenin has been shown to correlate with weaker Ecad/Ecad bonds (28,29). Because α-catenin mediates intercellular adhesion through its binding to β-catenin, which is bound to the cytoplasmic tail of Ecad (the Ecad adhesion complex) (28–31), it is not clear if these results were due to an α-catenin-independent role played by cancer-related mutant β-catenin or to a loss of α-catenin binding to the Ecad adhesion complex. To distinguish between these two scenarios, we expressed an Ecad-α-catenin chimera protein in cells with β-catenin shRNAi depleted (denoted βWT/−(KD)EC-α) and cells expressing only mutant β-catenin (denoted β−/Δ45EC-α). Measurements of Ecad/Ecad bond strengths indicated that fusion of α-catenin to the extracellular and transmembrane domain of Ecad was sufficient to rescue Ecad/Ecad bond strength as compared to Ecad/Ecad bonds formed in mutated or shRNAi depleted β-catenin backgrounds (Fig. 2 A). Moreover, genetic depletion of α-catenin in cells expressing WT β-catenin (βWT/−-α-cat(KD)) significantly weakened Ecad/Ecad bonds. Finally, analysis of bond rupture force distribution using the Hummer-Dudko model (27) showed that for cells expressing either mutated β-catenin or shRNAi-depleted of β-catenin, the characteristic bond-transition length was significantly larger than that measured for cells expressing either WT β-catenin or Ecad-α-catenin chimera (Fig. 2 E). Likewise, the free energy of activation decreased for cells with weaker Ecad/Ecad bonds (Fig. 2 F). These results are reminiscent of the Ecad/Ecad bond characteristics exhibited by cells shRNAi-depleted of α-catenin or expressing mutated α-catenin. These results are consistent with the model that β-catenin mediates intercellular adhesion through its binding partner α-catenin.
To test whether our results were limited to the two variants of HCT116 cells examined so far, we used the following two approaches: First, we measured the native Ecad/Ecad bond strength of parental HCT116 cells (βWT/Δ45) expressing both WT and ΔS45-mutated β-catenin. We found that parental cells formed significantly weaker Ecad/Ecad bonds as compared to (βWT/−) cells (Fig. 2 G). Second, the genetic depletion of β-catenin in these cells did not exhibit any change in the already weakened Ecad/Ecad bond strength. Together, these results showed that β-catenin mutations involved in colon carcinoma not only affect proliferation, but also affect Ecad/Ecad bond strength and that S45 deletion mutation in the amino terminal of β-catenin acts in a dominant negative manner.
Following another approach, we expressed Ecad in Chinese Hamster Ovary (CHO) which lack endogeneous Ecad—and expressed Ecad and either endogenous WT β-catenin or a mix of endogenous WT and exogenous S45 deleted β-catenin (Fig. 2 I). We found that CHO cells expressing endogenous WT β-catenin exhibited Ecad/Ecad bonds that were significantly stronger compared to CHO cells coexpressing WT β-catenin and ΔS45-mutated β-catenin (Fig. 2 H). Thus, the expression of ΔS45-mutated β-catenin alone weakens Ecad/Ecad bonds in a cell-type independent manner.
It is known that the serine residue 45 on the amino-terminal of β-catenin (S45) is one among four key residues that are highly conserved across species and are the consensus GSK3 phosphorylation sites. Because mutations in these S/T residues are frequent in human colon cancer, we determined if the deletion of serine 45 residue in parental colon-carcinoma cells βWT/Δ45 cells was responsible for weakening of intercellular Ecad/Ecad bonds. Upon genetic depletion of GSK3β in parental HCT116 cells (Fig. 2 I), we observed that although the strength of Ecad/Ecad bonds was significantly smaller than that for βWT/−cells, it was similar to the Ecad/Ecad bond strength of parental HCT116 cells (Fig. 2 H).
Furthermore, to test if the phosphorylation state of β-catenin at S45 indeed mediated Ecad/Ecad bond strength, we induced phosphomimetic S/D mutation in β-catenin expressed in CHO cells. We found that the phosphomimetic mutation (ser to asp) at residue 45 was alone sufficient to rescue the Ecad/Ecad bond strength to a level similar to that exhibited by CHO cells expressing only WT β-catenin. This is in consonance with our earlier result that showed that S45 residue was necessary to modulate Ecad/Ecad bond strength.
In summary, even as the presence of mutant S45 in β-catenin in colon cancer cells correlates with increased cell proliferation (22,32), our results suggest that this cancer-related mutation also plays an additional critical role in cancer progression—that of weakening individual intercellular Ecad/Ecad bonds between tumor cells.
β-catenin mediates strengthening of single Ecad/Ecad bond for increasing cell-cell contact time
The absence of α-catenin in the adhesion complex leads to weakening of Ecad/Ecad bonds over short contact durations, as opposed to the strengthening exhibited by Ecad/Ecad bonds formed when α-catenin is present. Because binding of α-catenin to Ecad is contingent on the presence of β-catenin, we allowed cells to interact for an increased time of contact and measured the corresponding strength of Ecad/Ecad bonds for cells expressing WT β-catenin (βWT/−), cells depleted of β-catenin (βWT/−(KD)), and cells expressing S45 mutant β-catenin (β−/Δ45) (Fig. 2 B). Ecad/Ecad bonds between cells expressing WT β-catenin (βWT/−) exhibited rapid strengthening over just 300 ms, whereas Ecad/Ecad bonds formed between cells expressing S45 mutant β-catenin (β−/Δ45) exhibited no significant change over the same time of contact (Fig. 2 B). The depletion of β-catenin in cells expressing WT β-catenin (βWT/−(KD)) weakened Ecad/Ecad bonds (p = 0.0064, n ∼120) (Fig. 2 B). Moreover, upon juxtaposing cells expressing Ecad-α-catenin chimera and S45-mutated β-catenin (β−/Δ45EC-α), no weakening of Ecad/Ecad bonds was observed over just 300 ms.
Together these results indicated that individual Ecad/Ecad bonds strengthen during nascent intercellular adhesion, a strengthening process mediated by β-catenin and abrogated in tumor cells expressing mutant β-catenin associated with cancer development and progression, or in cells depleted of β-catenin (summarized in Fig. 2 C). This result provides further evidence that β-catenin-mediated intercellular adhesion by acting to recruit α-catenin to the Ecad/β-catenin complex.
Placing the previous results on a pathological footing, we juxtaposed parental HCT116 cells simultaneously expressing both WT and S45 deleted mutant (βWT/Δ45) and observed that the mean tensile strength of the resulting Ecad/Ecad bonds was significantly lower than the Ecad/Ecad bonds formed in the presence of WT β-catenin (Fig. 2 B). Moreover, these cells formed Ecad/Ecad bonds that did not strengthen over time, in a manner similar to β-catenin depleted (βWT/−(KD) cells or β−/Δ45 cells. These results therefore suggested that βΔ45 catenin behaved in a dominant-negative inhibitory manner upon βWT catenin, as far as intercellular adhesion is concerned.
Binding affinity between Ecad and β-catenin
How then does the S45 mutant β-catenin result in weakened Ecad/Ecad bond strength between cells? One possibility could be that the S45 mutant β-catenin does not interact with the cytoplasmic tail of Ecad or α-catenin as well as WT β-catenin. As previously reported (23), mutant S45 β-catenin-containing HCT116 cells contain less total Ecad and cell surface Ecad, but the total cellular level of β-catenin and α-catenin proteins were unchanged from WT β-catenin containing HCT116 cells (Fig. 3 A). When β-catenin was immunoprecipitated, from the various HCT116 cell lines, the amount of associated Ecad was equivalent to the relative amount of Ecad detected on Western blots of input lysates from each cell type (Fig. 3 A). Furthermore, the amount of α-catenin that associated with immunoprecipitated S45 mutant β-catenin was equal to that associated with immunoprecipitated WT β-catenin. Recently, it has been suggested that an interaction between vinculin and β-catenin is critical for stabilizing Ecad at the cell surface (33). Because βWT/Δ45 and β−/Δ45 cells both have less cell surface localized and total cellular Ecad, we asked whether this could be explained by an altered interaction between vinculin and mutant β-catenin. In contrast to what was observed in breast MCF10A cells overexpressing vinculin, in HCT116 cells expressing an endogenous level of both vinculin and β-catenin, neither WT β-catenin nor S45 β-catenin coimmunoprecipitated with vinculin (Fig. 3 A), and GST-Ecad fusion proteins did not pulldown vinculin despite the presence of β-catenin (Fig. 3 B).
Figure 3.
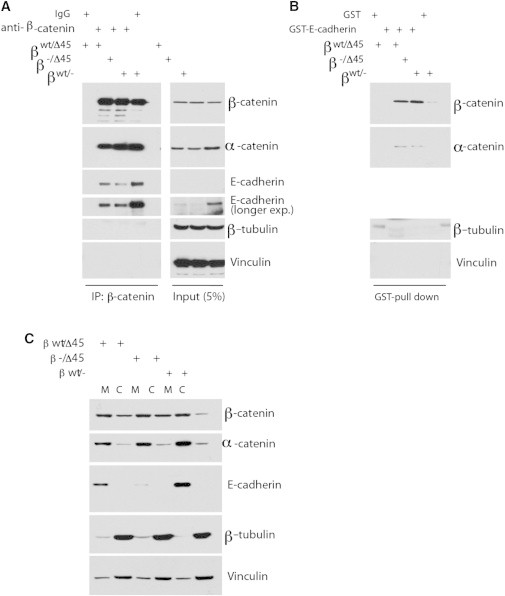
Affinity of WT and mutant β-catenin to E-cad remains unaltered. (A) β-catenin mutant binds to Ecad similarly to WT. Immunoprecipitation was performed using β-catenin antibody or mouse IgG charged protein G beads and cell lysates from HCT-derived cells as described in Methods, Western blotted as indicated. (B). Bacterially ecto-expressed E-cad cytoplasmic tail binds to β-catenin mutant. Bacterially expressed GST or GST-Ecad proteins were charged to Glutathione-agrose beads, pulldowns were performed as described and Western blotted. (C) Less cytosolic (lane C) β-catenin in WT/− than in mutant cells, as compared to membrane-bound β-catenin (lane M). Cells were fractionated and Western blotted.
In another approach we determined the amount of β-catenin and α-catenin that associated with added GST-Ecad cytoplasmic tail fusion protein, prepared and purified from bacterial lysates. S45 mutant β-catenin was readily pulled down (Fig. 3 B). The amount of WT β-catenin pulled down when GST-Ecad was added to βWT/− HCT116 cell lysates was much less (Fig. 3 B). This reflected the amount of available cytoplasmic β-catenin in each cell type as determined by Western blot analysis of membrane and cytoplasmic fractions from each cell type (Fig. 3 C). In βWT/− cells the majority of cellular β-catenin was associated with membrane Ecad, whereas in βWT/Δ45 and β−/Δ45 cells the amount of cytoplasmic β-catenin was dramatically increased (Fig. 3 C). Interestingly, and in contrast to β-catenin distribution, the amount and distribution of α-catenin between cytoplasm and membrane in the three cell types did not significantly differ (Fig. 3 C). Taken together these experiments indicated that the S45 mutant β-catenin associated with the cytoplasmic tail of Ecad and α-catenin as well as WT β-catenin. Thus, the affinity of S45 mutant β-catenin for the cytoplasmic tail of Ecad and α-catenin does not appear to be different from that of WT β-catenin.
Tumor cell Ecad/Ecad bond strength is modulated by cell adhesion to select ECM proteins and this requires WT β-catenin
In the course of their invasive migration, tumor cells encounter a host of ECM proteins—collagen IV and laminin V in the basement membrane and collagen I in the underlying ECM. Therefore, we asked whether interaction of tumor cells to these different ECM proteins could affect the adhesion strength between Ecad proteins on adjacent cells and thereby modulate tumor cell invasion and ultimate metastasis. To test this tumor cells were cultured on substrates coated with relevant ECM proteins for times much longer than integrin activation time (26, 34). A gradual weakening of Ecad/Ecad bonds between cells expressing WT β-catenin (βWT/−) occurred as the underlying substrate of cells was changed from uncoated culture dish, to Matrigel (a mixture of collagen IV and laminin V), and laminin V (Fig. 4 A). In contrast, contact of WT β-catenin-expressing cells (βWT/−) plated on collagen I and collagen IV had little influence on the strength of single Ecad/Ecad bonds compared to uncoated substrates (Fig. 4 A), at least at short times of cell-cell contact (1 ms). For cells expressing cancer-related S45 mutant β-catenin (β−/Δ45), there was no change in the mean bond strength of Ecad/Ecad bonds formed between cells exposed to any ECM protein studied (Fig. 4 A). In all the previous measurements, we ensured that the concentration of each of these ligands on the substrate was exactly the same and the substrate was prepared following identical conditions, using 1 ml of 50 μg/ml of purified protein solution in pH 7.2 PBS buffer. Moreover, altering the surface-density of ECM ligands did not change the mean Ecad/Ecad bond rupture force, thereby suggesting that our single-molecular force measurements reflect the stoichiometry-independent, asymptotic effect of ECM ligand on intercellular adhesion.
Figure 4.
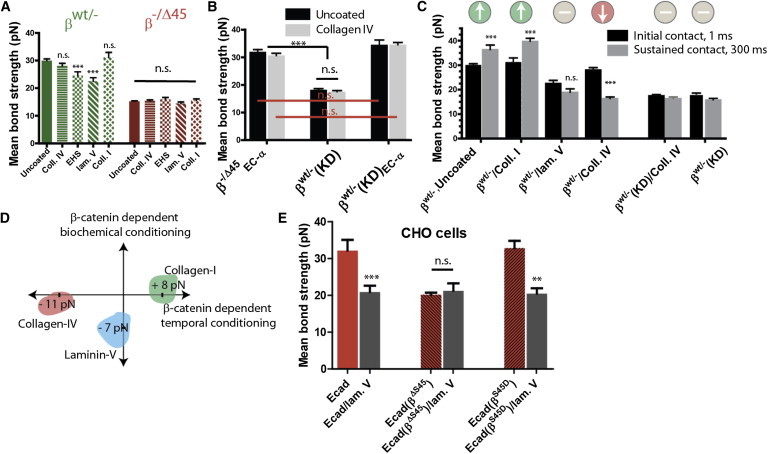
Ecad/Ecad bond strength is differentially modulated by cell adhesion to specific ECM molecules through β-catenin. (A) Ecad/Ecad bonds between HCT116 cells expressing only WT β-catenin (βWT/−) or only ΔS45-mutated β-catenin (β−/Δ45) and plated on substrates coated with different ECM ligands (Coll. IV – human collagen IV; EHS –Engelbreth-Holm-Swarm MatriGel basement membrane matrix; lam V. – human laminin V; Coll. I – rat-tail collagen I). Whereas exposure to varied ECM ligands induced change in Ecad/Ecad bond strength in (βWT/−) cells, no such response was observed for β−/Δ45cells. n = minimum 140 cells (N > 3 independent experiments) analyzed with one-way ANOVA using Bonferroni’s multiple-comparison test and α = 0.05. Unless otherwise marked, all significance is relative to (βWT/−) behavior. (B) Collagen IV does not affect single Ecad/Ecad bonds, even when α-catenin is directly fused to Ecad (βWT/−(KD)EC-α and β−/Δ45EC-α), mimicking the Ecad/Ecad bonds formed between cells with β-cateninß-catenin depleted (βWT/−(KD). n = minimum 140 cells (N > 3 independent experiments) analyzed with one-way ANOVA using Bonferroni’s multiple-comparison test and α = 0.05. (C) Response to increasing duration of cell-cell contact, to 300 ms. Cells expressing WT β-catenin (βWT/−) exhibit time-dependent weakening of Ecad/Ecad bonds when placed on collagen IV, strengthening when plated on collagen I, and no change when coated in laminin V. Cells with depleted (βWT/−(KD) do not show any time-dependent response. ∗∗∗Designates p < 0.001 under the Michelin grade scale. n = minimum 140 cells (N > 3 independent experiments) analyzed with one-way ANOVA using Bonferroni’s multiple-comparison test and α = 0.05. (D) Illustration showing the different roles played by different ECM ligands. Although Collagen IV and Collagen I induce a temporal but no sensory response, laminin V induces a sensory response, but no temporal response. Numbers depict the weakening or strengthening of Ecad/Ecad bonds. (E) Similar to βWT/− cells, CHO cells expressing either WT β-catenin or phosphomimetic S > D mutated β-catenin exhibit significant weakening of Ecad/Ecad bonds upon exposure to laminin V-coated substrate, whereas simultaneous expression of WT (endogenous) and ΔS45-mutated (exogenous) β-catenin induces no laminin V-specific response. All bars designate mean ± SEM, and n > 140 for each condition.
It was interesting that these results showed that laminin V, but not collagen IV, induced a sensory response to a change in substrate ligand, even though both of these proteins are abundantly present in the basement membrane. To examine further the insensitivity to collagen IV, we plated β−/Δ45EC-α, βWT/−(KD), and βWT/−(KD)EC-α cells on collagen IV-coated dishes and measured the corresponding Ecad/Ecad bond strength during homotypic contact of short duration (1 ms) (Fig. 4 B). In each case, collagen IV had no significant effect on the strength on Ecad/Ecad bonds. These results further confirmed our earlier finding that unlike laminin V, collagen IV had no effect on the nascent Ecad/Ecad bonds formed over <1 ms.
In contrast to cell-cell contacts of short duration, increasing the duration of cell-cell contact to 300 ms (Fig. 4 C), we found that collagen IV uniquely weakened the time-dependent change in Ecad/Ecad bond strength in WT β-catenin containing cells. WT β-catenin cells plated on laminin V showed no further weakening of change in Ecad/Ecad bonds overtime, whereas WT β-catenin cells plated on collagen I showed normal strengthening of bonds over time (Fig. 4 C). To test whether β-catenin played a role in matrix protein-mediated temporal maturation/weakening of Ecad/Ecad bonds, we plated cells depleted of β-catenin (βWT/−(KD)) on collagen IV-coated culture dishes and measured the mean Ecad/Ecad bond strength over extended duration of contact (300 ms, Fig. 4 C). We observed no temporal change in strength of already weakened Ecad/Ecad bonds (Fig. 4 C), in contrast to the temporal change in Ecad/Ecad bond strength exhibited by cells expressing WT β-catenin.
These results allowed us to divide the influence of various matrix proteins on Ecad/Ecad bond strength into two categories (Fig. 4 D): a temporal response that reflected a change in the dynamics of bond maturation and a biochemical response that determined whether the cell can sense a change in its biochemical extracellular environment. Collagen I and IV induce similar minimal level of biochemical response, as for both cases, the cells were unable to alter the strength of immediate Ecad/Ecad bond, going from uncoated to coated substrates. In contrast, laminin V induces a significant biochemical response to immediate bond formation, while not affecting the dynamics of bond maturation, as suggested by no change in time-dependent bond strength. Both collagen I and collagen IV affected the temporal maturation of bonds, but in opposite direction (Fig. 3 D). Another potentially provocative conclusion from these studies is that matrix proteins abundant in basement membrane (collagen IV, laminin V) inhibited bond strength—either immediate or temporal maturation, whereas exposure of cells to the ECM protein collagen I aided the temporal maturation of Ecad/Ecad bonds, although having no effect upon the nascent bond strength.
To further test our results in a cell-independent manner, we plated CHO cells expressing either WT β-catenin, S45 deleted β-catenin (βΔS45), or S/D phosphomimetic β-catenin (βS45D), on laminin V-coated or uncoated substrates (Fig. 4 E). We found that the presence of mutated β-catenin abrogated the substrate-dependent modulation of Ecad/Ecad bond strength, whereas the presence of WT or phosphomimetic β-catenin alone was sufficient to allow for significant substrate-dependent Ecad/Ecad bond weakening. This result again validates our earlier finding that S45 residue on β-catenin is necessary for modulation of Ecad/Ecad bond strength.
Kinase activity of GSK3β regulates the strength of Ecad/Ecad bonds but only in the presence of WT β-catenin
Previous reports have shown that S45 on β-catenin is essential for priming other downstream β-catenin phosphorylation sites (27, 35), which together induce cytoplasmic degradation of β-catenin. It is therefore possible that the state of β-catenin phosphorylation could endow the cells with a mechanism by which to control intercellular adhesion, in addition to proliferation. Similarly, several studies have shown that phosphorylation of Ecad at distinct sites may induce either an increase or a decrease in affinity for β-catenin (36–40). Therefore, phosphorylation of adhesion plaque proteins (Ecad, β-catenin, α-catenin, and to a lesser extent p120) represent a mechanism by which cells can modulate their intercellular adhesion. GSK3β is one such kinase that phosphorylates β-catenin (41) as well as Ecad (39,42,43). To quantify the effect of phosphorylation on intercellular adhesion at single-molecule level, GSK3β was shRNAi-depleted in both WT β-catenin (βWT/−) and mutant β-catenin expressing (β−/Δ45) cells and these cells were subjected to single-molecule force spectroscopy analysis (Fig. 5 A). For cells expressing WT β-catenin, when depleted of GSK3β (GSK3β-KD)βWT/−), the strength of a single intracellular Ecad bond decreased significantly, to a level similar to mutant β-catenin expressing (β−/Δ45) cells (Fig. 5 A). The strength of single Ecad/Ecad bonds in GSK3β depleted cells expressing mutant β-catenin ((GSK3β-KD) β−/Δ45) exhibited no significant change compared to mutant cells expressing GSK3β (Fig. 5 A).
Figure 5.
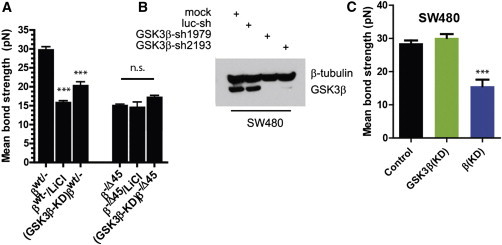
The kinase activity of GSk3β regulates the strength of Ecad/Ecad bonds but only in the presence of β-catenin. (A) Upon shRNAi depletion of GSk3β, HCT116 cells expressing WT β-catenin (GSK3β-KD)βWT/−) exhibit significantly weaker Ecad/Ecad bonds, whereas HCT116 cells expressing mutant β-catenin (GSk3β-KD)β−/Δ45) do not show this weakening. Inhibition of GSk3β (using LiCl, βWT/−/LiCl and β−/ΔS45/LiCl) also weakened individual intercellular Ecad/Ecad bonds. ∗∗∗ Designates p < 0.001, unpaired Student’s t-test. (B and C) In SW480 cells with endogenous APC mutation, genetic depletion of GSK3β (B) induces no change in the Ecad/Ecad bond strength (C), whereas depletion of endogenous WT β-catenin causes significant weakening of Ecad/Ecad bonds. All bars designate mean ± SEM, and n > 140 for each case.
In another approach testing the role of GSK3β in modulating Ecad/Ecad bond strength, we measured the strength of Ecad/Ecad bonds formed between cells expressing either WT (βWT/−) or ΔS45 mutant β-catenin pretreated with the GSK3β inhibitor LiCl (37, 44). Upon inhibition of GSK3β, the strength of single Ecad/Ecad bonds decreased significantly for βWT/− cells, whereas β−/Δ45 cells did not exhibit any change in Ecad/Ecad bond strength, confirming our results with GSK3β depleted cells (Fig. 5 A). Together, these results suggested that the presence of WT β-catenin was essential for the regulation of intercellular adhesion by intracellular cues such as GSK3β activity, that are in turn governed by extracellular cues such as basement membrane proteins and growth-factors such as the Wnt factor (43, 45).
To put our studies on a more pathologic footing, we noted that in the context of colon carcinoma, APC/Axin mutations are frequent. Because GSK3β associates with the APC/Axin complex and together, this complex targets β-catenin for ubiquitination and degradation, we asked if mutations in any component of this GSK3/APC/Axin complex will have the same effect on Ecad/Ecad bonds as genetic deletion of GSK3β. Using SW480 cells that have endogenous colon-cancer causing APC mutation, we knocked down (Fig. 5 B) either endogenous β-catenin or GSK3β. We found that whereas shRNAi depletion of β-catenin induced significant weakening of Ecad/Ecad bonds, depletion of GSK3β had no significant effect on the Ecad/Ecad bond strength (Fig. 5 C). Interestingly, it was also found that parental SW480 cells form Ecad/Ecad bonds that are as strong as Ecad/Ecad bonds formed between HCT116 cells expressing only WT β-catenin (βWT/−) but stronger than parental HCT116 cells. Therefore, it appears that although the actual strength of Ecad/Ecad bonds varies across cell types, the modulation of Ecad/Ecad bonds based on phosphorylation state of β-catenin is conserved and GSK3β is a critical kinase mediating Ecad/Ecad bond strength.
Discussion
Examination of all the conditions studied in this work (Figs. 2–4) readily reveals that single Ecad/Ecad bonds display a consistent low tensile strength basal state under conditions that induce weakened intercellular adhesion. A variety of stimuli including intracellular protein modifications, such as the presence of β-catenin and α-catenin or mutations in β-catenin, and extracellular cues in the form of changes in ECM ligands, allow the cell to undergo a molecular-level decision making process vis-à-vis intercellular adhesion: Whether to rapidly enhance or weaken the adhesion strength of single nascent Ecad/Ecad bond. For increased time of contact between cells or the type of contact of cells with various ECM molecules, cells can modulate the tensile strength of their intercellular Ecad bonds in the presence of WT β-catenin, whereas expression of ΔS45 β-catenin or the absence of WT β-catenin abrogates this response. Because cells as far apart as HCT116 cells, CHO cells and SW480 cells all exhibit a similar β-catenin-mediated modulation of single Ecad/Ecad bonds, it is likely that like Ecad β-catenin also plays a much conserved role in intercellular adhesion.
These results suggest that for intercellular inside-out signaling to function properly, the presence of WT β-catenin is essential. Apart from β-catenin, kinase activity of key proteins (e.g., GSK3β, CKI, CKII) also governs the robustness of this inside-out signaling. Because a variety of external cues can modulate the state of β-catenin-related kinases and phosphatases, it is likely that the net adhesion state of a cell is a quorum decision involving all external cues. Although in this work, only the role of substrate biochemistry is investigated, the role of soluble factors may be equally as interesting. Specifically, the role played by Wnt proteins in modulating Ecad/Ecad bond strength via GSK3β-induced β-catenin processing merits further investigation.
Extracellular biochemical cues in the form of ligand presentation can uniquely govern the final mechanical state of Ecad/Ecad bonds, whereas intracellular conditions such as the phosphorylation of adhesion-plaque proteins forms the other level of control over intercellular adhesion. Our results therefore indicate that β-catenin serves as a clutch that facilitates the transition from the low adhesion state of Ecad/Ecad bond to its strong adhesion state. Moreover, although genetic fusion of α-catenin to Ecad is enough to strengthen an intracellular Ecad/Ecad bond, the presence of WT β-catenin is essential for the cell to respond to altered ECM ligand. Such a stepped control over the strength of Ecad/Ecad bonds allows the cells to modulate their global intercellular adhesion. Such a scenario is ideal for efficient control of global intercellular adhesion—both in development as well as in cancer progression.
Our present results generate several intriguing questions in understanding intercellular adhesion modulation. The existence of a single basal level of weak Ecad/Ecad bonds and multilevel strengthening of Ecad/Ecad bonds suggests that a simple transition of the Ecad molecule from one conformation state to another is unlikely. This possibility is further strengthened by the fact that analysis of crystal structures of Ecad-β-catenin complexes has not revealed different stable conformations (46,47).
Our results show that intercellular adhesion at the level of single Ecad/Ecad bonds is weakened when colorectal cancer cells (HCT116) that express WT β-catenin were exposed to laminin V but not collagen IV, which are the major ECM components of the basement membrane. However, even though the strength of Ecad/Ecad bonds is weaker when the cells are plated on reconstituted Engelbreth-Holm-Swarm and laminin V (as compared to uncoated substrate or substrate coated with collagen I or collagen IV), it is still higher than the basal-level of adhesion. Thus, the regime of moderate intercellular Ecad/Ecad bonds represents the optimal regime of regulation of intercellular adhesion: Ecad/Ecad bonds are strong enough to maintain the integrity of the epithelial layer and weak enough to allow dynamic modulation in response to various intercellular adhesion cues. Also interesting is the fact that colon-carcinoma derived βWT/− cells (but not β−/Δ45 cells) plated on collagen I form Ecad/Ecad bonds, which are stronger than Ecad/Ecad bonds formed by cells plated on proteins that comprise the basement membrane of epithelial cells. In contrast, it is known that pancreatic carcinoma cells respond to collagen I by weakening of adherens junctions (48). Tissue-specific pathways therefore seem to govern the ultimate response of cells toward exposure to various ECM substrates.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01GM084204, RO1GM080673, and U54CA143868.
Contributor Information
Denis Wirtz, Email: wirtz@jhu.edu.
Gregory D. Longmore, Email: greglongmor@dom.wustl.edu.
Supporting Material
References
- 1.Giampieri S., Manning C., Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009;11:1287–1296. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hidalgo-Carcedo C., Hooper S., Sahai E. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol. 2011;13:49–58. doi: 10.1038/ncb2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu B., Chappuis-Flament S., Leckband D. Functional analysis of the structural basis of homophilic cadherin adhesion. Biophys. J. 2003;84:4033–4042. doi: 10.1016/S0006-3495(03)75129-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou J.J., Gaemers S., Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 5.Zimrin A.B., Villeponteau B., Maciag T. Models of in vitro angiogenesis: endothelial cell differentiation on fibrin but not matrigel is transcriptionally dependent. Biochem. Biophys. Res. Commun. 1995;213:630–638. doi: 10.1006/bbrc.1995.2178. [DOI] [PubMed] [Google Scholar]
- 6.Shimoyama Y., Nagafuchi A., Hirohashi S. Cadherin dysfunction in a human cancer cell line: possible involvement of loss of α-catenin expression in reduced cell-cell adhesiveness. Cancer Res. 1992;52:5770–5774. [PubMed] [Google Scholar]
- 7.Onder T.T., Gupta P.B., Weinberg R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 8.Huang S.H., Wu J.C., Wang S.M. Distribution of the cadherin-catenin complex in normal human thyroid epithelium and a thyroid carcinoma cell line. J. Cell. Biochem. 1998;70:330–337. [PubMed] [Google Scholar]
- 9.Joo Y.E., Rew J.S., Kim S.J. Expression of e-cadherin and catenins in early gastric cancer. J. Clin. Gastroenterol. 2002;35:35–42. doi: 10.1097/00004836-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Czyzewska J., Guzińska-Ustymowicz K., Kemona A. The expression of E-cadherin-catenin complex in patients with advanced gastric cancer: role in formation of metastasis. Folia Histochem. Cytobiol. 2010;48:37–45. doi: 10.2478/v10042-010-0017-z. [DOI] [PubMed] [Google Scholar]
- 11.Davies B.R., Worsley S.D., Ponder B.A. Expression of E-cadherin, alpha-catenin and beta-catenin in normal ovarian surface epithelium and epithelial ovarian cancers. Histopathology. 1998;32:69–80. doi: 10.1046/j.1365-2559.1998.00341.x. [DOI] [PubMed] [Google Scholar]
- 12.Moon R.T., Bowerman B., Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- 13.van de Wetering M., Sancho E., Clevers H. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 14.Gottardi C.J., Gumbiner B.M. Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J. Cell Biol. 2004;167:339–349. doi: 10.1083/jcb.200402153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutter J.L., Bechhoefer J. Calibration of atomic force microscope tips. Rev. Sci. Instrum. 1993;64:1868–1873. [Google Scholar]
- 16.Hanley W.D., McCarty O., Konstantopoulos K. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 2003;278:10556–10561. doi: 10.1074/jbc.M213233200. [DOI] [PubMed] [Google Scholar]
- 17.Raman P.S., Alves C.S., Konstantopoulos K. Single-molecule binding of CD44 to fibrin versus P-selectin predicts their distinct shear-dependent interactions in cancer. J. Cell Sci. 2011;124:1903–1910. doi: 10.1242/jcs.079814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanley W.D., Wirtz D., Konstantopoulos K. Distinct kinetic and mechanical properties govern selectin-leukocyte interactions. J. Cell Sci. 2004;117:2503–2511. doi: 10.1242/jcs.01088. [DOI] [PubMed] [Google Scholar]
- 19.Dobrowsky T.M., Panorchan P., Wirtz D. Chapter 15: Live-cell single-molecule force spectroscopy. Methods Cell Biol. 2008;89:411–432. doi: 10.1016/S0091-679X(08)00615-8. [DOI] [PubMed] [Google Scholar]
- 20.Dobrowsky T.M., Zhou Y., Wirtz D. Monitoring early fusion dynamics of human immunodeficiency virus type 1 at single-molecule resolution. J. Virol. 2008;82:7022–7033. doi: 10.1128/JVI.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang M.I., Panorchan P., Wirtz D. Single-molecule analysis of human immunodeficiency virus type-1 gp120-receptor interactions in living cells. J. Virol. 2005;79:14748–14755. doi: 10.1128/JVI.79.23.14748-14755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Y., Nie L., Longmore G.D. A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics. 2010;8:238–245. doi: 10.1016/S1672-0229(10)60025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan T.A., Wang Z., Kinzler K.W. Targeted inactivation of CTNNB1 reveals unexpected effects of β-catenin mutation. Proc. Natl. Acad. Sci. USA. 2002;99:8265–8270. doi: 10.1073/pnas.082240999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu Y.S., Thomas W.A., Dufour S. Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J. Cell Biol. 2004;167:1183–1194. doi: 10.1083/jcb.200403043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans E., Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bayas M.V., Leung A., Leckband D. Lifetime measurements reveal kinetic differences between homophilic cadherin bonds. Biophys. J. 2006;90:1385–1395. doi: 10.1529/biophysj.105.069583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudko O.K., Hummer G., Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc. Natl. Acad. Sci. USA. 2008;105:15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bajpai S., Feng Y., Wirtz D. Loss of α-catenin decreases the strength of single E-cadherin bonds between human cancer cells. J. Biol. Chem. 2009;284:18252–18259. doi: 10.1074/jbc.M109.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bajpai S., Correia J., Wirtz D. alpha-Catenin mediates initial E-cadherin-dependent cell-cell recognition and subsequent bond strengthening. Proc. Natl. Acad. Sci. USA. 2008;105:18331–18336. doi: 10.1073/pnas.0806783105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drees F., Pokutta S., Weis W.I. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada S., Pokutta S., Nelson W.J. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korinek V., Barker N., Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 33.Peng X., Cuff L.E., DeMali K.A. Vinculin regulates cell-surface E-cadherin expression by binding to β-catenin. J. Cell Sci. 2010;123:567–577. doi: 10.1242/jcs.056432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miao H., Burnett E., Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 35.Liu C., Li Y., He X. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi H., Takeo Y., Fukami K. Lipid rafts and caveolin-1 are required for invadopodia formation and extracellular matrix degradation by human breast cancer cells. Cancer Res. 2009;69:8594–8602. doi: 10.1158/0008-5472.CAN-09-2305. [DOI] [PubMed] [Google Scholar]
- 37.Aberle H., Schwartz H., Kemler R. Cadherin-catenin complex: protein interactions and their implications for cadherin function. J. Cell. Biochem. 1996;61:514–523. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 38.Behrens J., Vakaet L., Birchmeier W. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J. Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lickert H., Bauer A., Stappert J. Casein kinase II phosphorylation of E-cadherin increases E-cadherin/β-catenin interaction and strengthens cell-cell adhesion. J. Biol. Chem. 2000;275:5090–5095. doi: 10.1074/jbc.275.7.5090. [DOI] [PubMed] [Google Scholar]
- 40.Serres M., Filhol O., Schmitt D. The disruption of adherens junctions is associated with a decrease of E-cadherin phosphorylation by protein kinase CK2. Exp. Cell Res. 2000;257:255–264. doi: 10.1006/excr.2000.4895. [DOI] [PubMed] [Google Scholar]
- 41.Hart M.J., de los Santos R., Polakis P. Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3 β. Curr. Biol. 1998;8:573–581. doi: 10.1016/s0960-9822(98)70226-x. [DOI] [PubMed] [Google Scholar]
- 42.Huber O., Bierkamp C., Kemler R. Cadherins and catenins in development. Curr. Opin. Cell Biol. 1996;8:685–691. doi: 10.1016/s0955-0674(96)80110-4. [DOI] [PubMed] [Google Scholar]
- 43.Huber A.H., Weis W.I. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- 44.Andrews N.A., Jones A.S., Kinsella A.R. Expression of the E-cadherin-catenin cell adhesion complex in primary squamous cell carcinomas of the head and neck and their nodal metastases. Br. J. Cancer. 1997;75:1474–1480. doi: 10.1038/bjc.1997.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McManus E.J., Sakamoto K., Alessi D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi K., Nakanishi H., Takai Y. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 1999;145:539–549. doi: 10.1083/jcb.145.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gumbiner B.M. Regulation of cadherin adhesive activity. J. Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koenig A., Mueller C., Menke A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006;66:4662–4671. doi: 10.1158/0008-5472.CAN-05-2804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.