

Abstract
Recent advances in fluorescence localization microscopy have made it possible to image chemically fixed and living cells at 20 nm lateral resolution. We apply this methodology to simultaneously record receptor organization and dynamics on the ventral surface of live RBL-2H3 mast cells undergoing antigen-mediated signaling. Cross-linking of IgE bound to FcεRI by multivalent antigen initiates mast cell activation, which leads to inflammatory responses physiologically. We quantify receptor organization and dynamics as cells are stimulated at room temperature (22°C). Within 2 min of antigen addition, receptor diffusion coefficients decrease by an order of magnitude, and single-particle trajectories are confined. Within 5 min of antigen addition, receptors organize into clusters containing ∼100 receptors with average radii of ∼70 nm. By comparing simultaneous measurements of clustering and mobility, we determine that there are two distinct stages of receptor clustering. In the first stage, which precedes stimulated Ca2+ mobilization, receptors slow dramatically but are not tightly clustered. In the second stage, receptors are tightly packed and confined. We find that stimulation-dependent changes in both receptor clustering and mobility can be reversed by displacing multivalent antigen with monovalent ligands, and that these changes can be modulated through enrichment or reduction in cellular cholesterol levels.
Introduction
Mast cell activation results in secretion of chemical mediators of inflammation from intracellular granules as part of the adaptive immune response, which is responsible for the symptoms of allergy. The first steps of this process occur at the plasma membrane, where antigen-specific immunoglobulin E (IgE) bound to its receptor, FcεRI, is cross-linked by soluble multivalent antigen (1,2). Cross-linking of IgE-FcεRI complexes causes signal initiation by Lyn kinase phosphorylation of immunoreceptor tyrosine-based activation motifs on FcεRI β and γ2 subunits. The resulting tyrosine phosphorylation leads to Ca2+ mobilization and cellular degranulation (3). Oligomerization has readily observable effects on the spatial distribution and diffusion behavior of IgE-FcεRI. Receptors are uniformly distributed and mobile on the membrane before activation. Stimulation with antigen causes clustering of IgE-FcεRI into punctate aggregates on the cell surface and a marked decrease in receptor mobility (4,5).
Cross-linked IgE-FcεRI puncta can be visualized with conventional fluorescence microscopy. However, quantitative measurements of receptor cluster formation require subdiffraction-limited spatial resolution, comparable to the dimensions of clusters. Previous work has used a variety of experimental approaches to characterize the dynamic, antigen-induced, nanoscale reorganization of FcεRI to understand this initiation step in signaling. Receptor distributions at high spatial resolution have been studied with electron microscopy using immunogold labeling of IgE-FcεRI (6–8). In addition, dynamics of IgE-FcεRI during the time course of activation have been captured by fluorescence photobleaching recovery (FPR) (5,9,10) and fluorescence correlation spectroscopy (FCS) (11). Single-particle tracking (SPT) of individual receptors labeled with fluorescent-dye- or quantum-dot-conjugated IgE has characterized the motion of single proteins (12–14). To date, these approaches have either achieved high-resolution spatial measurements in chemically fixed systems or have measured receptor mobility in live cells using nonimaging methods or imaging without nanoscale spatial resolution.
Advances in fluorescence microscopy now enable subdiffraction imaging using photoconvertible fluorescent dyes. Superresolution techniques, including (direct) stochastic optical reconstruction microscopy (STORM/dSTORM) (15,16) and (fluorescence) photoactivation localization microscopy (PALM/FPALM) (17,18), have been used in fixed cells to quantify membrane protein distributions and clustering in other systems (19–24). Superresolution techniques in live cells capture high-resolution maps of protein distributions (25,26) in addition to diffusion information from single-molecule trajectories (27–29). Further, the use of localization microscopy for single-particle tracking methods provides improved number statistics compared to traditional single-particle tracking because the photoconversion process allows for sampling of an ensemble of receptors over the course of a single live-cell measurement. This work applies superresolution fluorescence localization microscopy, exploiting its capabilities for both high-resolution imaging and single-molecule recording of receptor diffusion. With this technique, we monitor the kinetics of clustering and mobility changes of IgE receptors in live rat basophil leukemia (RBL)-2H3 mast cells undergoing a stimulated immune response. We do this both by quantifying average properties of receptors and by examining the behavior of single molecules. In addition, we explore how receptor mobility and diffusion are altered by perturbations, including reversal of receptor cross-linking with monovalent hapten and modulation of the cholesterol content of cell membranes.
Results and Discussion
Redistribution of IgE-FcεRI upon stimulation in live cells
Through superresolution imaging of living cells, we simultaneously observe nanometer-scale receptor organization and dynamics in real time. Fig. 1 A shows a representative live RBL cell that is imaged before and after the addition of multivalent antigen at room temperature under buffer conditions that both support superresolution imaging and preserve downstream functional responses (Fig. S1 in the Supporting Material). We chose to image at room temperature rather than at 37°C because key signaling stages occur at the lower temperature, although at a slower rate. These include receptor phosphorylation, Ca2+mobilization, and endocytosis (11,30). Cells were sensitized by incubation with IgE antibodies specific for dinitrophenyl (DNP) and stimulated with the multivalent antigen DNP-bovine serum albumin (DNP-BSA). IgE-FcεRI complexes were fluorescently labeled by sensitizing with IgE directly conjugated to an Alexa Fluor 647 (AF647) and imaged as described in Materials and Methods in the Supporting Material. Live cell images are produced by following single-molecule trajectories in raw images, then reconstructing time-averaged images using only the first localized position in each trajectory. Each reconstructed image in Fig. 1 A is compiled from 2000 raw image frames acquired over 68 s of imaging time at 31 frames/s. The relatively short imaging time produces a reconstructed image that is inherently undersampled; only a fraction (estimated to be between 30% and 60%) of individual IgE proteins are represented in each image. Despite this limitation, images clearly indicate that receptors are nearly randomly organized in unstimulated cells and become more clustered in response to cross-linking by multivalent antigen.
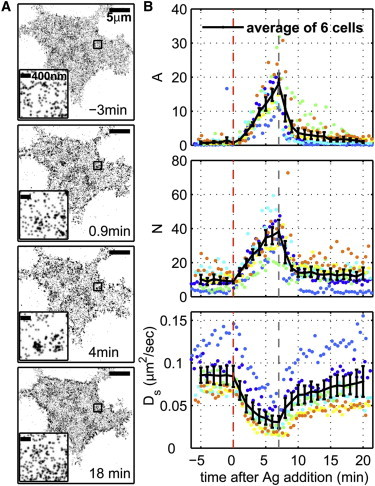
Figure 1.
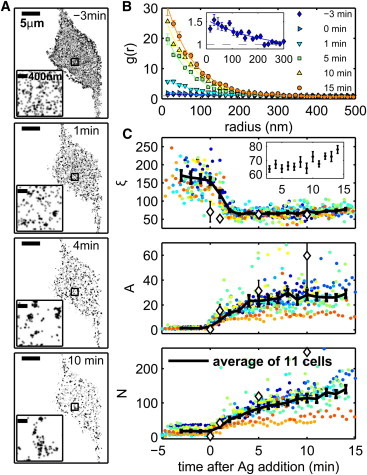
Quantitative superresolution localization microscopy imaging of IgE-FcεRI redistribution after antigen addition in live cells. (A) Reconstructed superresolution fluorescence localization images of an AF647-IgE-labeled living cell at various times in the stimulation sequence, where antigen (DNP-BSA, 1 μg/mL) is added at 0 min. Each image is reconstructed from 68 s of acquired data, as described in Materials and Methods (see Supporting Material). A movie showing complete time-lapse imaging of this cell is supplied in the Supporting Material. (Insets) Magnified images of the square regions outlined in black. (B) Autocorrelation functions, g(r), are calculated from reconstructed single-molecule centers acquired over 16 s, as described in Materials and Methods in the Supporting Material (solid symbols) and are fit to single exponentials (Eq. 1; solid lines). (Inset) The correlation function from data recorded 3 min before antigen stimulation on an expanded scale. (C) Correlation function parameters from 11 live-cell experiments, distinguished by different colors: the correlation length, ξ (upper), the correlation amplitude, A (middle), and the average number of correlated proteins, N (lower). Solid black lines indicate averages over 11 cells, and error bars represent the mean ± SE. Fit parameters extracted from two-color fixed-cell experiments are reproduced from Fig. S2, D and E and plotted for comparison in C as open black diamonds. (Inset) Average ξ for time points between 3 and 15 min after antigen addition on an expanded y-axis scale. To see this figure in color, go online.
We utilize the spatial pair correlation function as a function of radius, g(r), to quantify clustering of IgE-FcεRI complexes in reconstructed images. Pair autocorrelation functions measure the normalized probability of finding a second localized fluorophore at a given distance, r, from the average localized fluorophore. These functions are tabulated as described previously (31) and summarized in Materials and Methods in the Supporting Material. For the resulting curves, a value of 1 indicates that receptors are randomly organized. Values of >1 indicate that receptors are clustered, and the range in r over which g(r) > 1 is a measure of cluster size. The g(r) curves shown in Fig. 1 B were tabulated from images reconstructed using 500 frames of raw image data acquired over 16 s. In agreement with visual observations, autocorrelation functions generated from time-resolved images show that receptors are nearly randomly distributed before antigen addition, with g(r) ∼ 1 at all radii, and become dramatically more densely clustered after stimulation. Correlation functions measured in live cells are in good quantitative agreement with those observed in cells chemically fixed at specific time points after stimulation (Fig. S2). Although reconstructed images of live cells are undersampled compared to fixed-cell images, as long as undersampling is random, its effects alone will not change the correlation function beyond decreasing the signal/noise ratio (31).
Measured autocorrelation functions are fit to a single exponential to extract information on average cluster size and density according to the equation
| (1) |
for r > 20 nm, where A is the amplitude of correlations, which is proportional to the increased density of receptors in clusters, and ξ is the correlation length, which is approximately the average cluster radius. The average number of correlated proteins (N), or the number of correlated proteins within the average cluster, is the summation of the measured g(r) over r times the average surface density of receptors, defined by the equation
| (2) |
where we assume that the overall average surface density of receptors (ρave) is 200/μm2 (31,32). When curves are well fit to the single-exponential form given in Eq. 1 in the limit of small Δr, this sum over values of r from zero to infinity can also be written as N = ρave4πAξ2. In practice, we evaluate Eq. 2 for radii between 0 and 300 nm, with Δr = 15 nm.
This quantitative analysis, averaged over 11 cells, and a summary of extracted fit parameters is shown in Fig. 1 C. We observe dramatic redistribution of receptors into clusters after addition of multivalent antigen with weak, long-range correlations in unstimulated cells, and strong, shorter-range correlations after antigen stimulation, consistent with previous reports in live cells (33). In unstimulated cells, we observe correlations with very low values of A and N and large values of ξ. This is further illustrated by the correlation function for unstimulated cells plotted in the inset of Fig. 1 B. ξ extends to ∼200 nm in unstimulated live cells, whereas we observed ξ ≈ 80 nm in chemically fixed cells (Fig. S2). The larger ξ observed in live-cell images could arise from overcounting single molecules that are lost by our tracking algorithm, lateral motion of any correlated structures observed during data collection, or, possibly, the fact that live cells were imaged at room temperature whereas chemically fixed cells were incubated at 37°C.
We observe time-dependent increases in A and N during the first 5 min after antigen addition. After this time, the correlation amplitude, A, remains constant, the average number of correlated proteins, N, continues to increase at a slower rate, and the correlation length, ξ, slowly increases (Fig. 1 C upper, inset). The average ξ decreases within 3 min of antigen addition to ∼70 nm, in good agreement with ξ in stimulated fixed cells (Fig. 1 C, diamonds). The continuous decrease in ξ soon after antigen addition likely indicates the increasing presence of small and dense clusters in a background of larger more diffuse structure, as suggested by the image reconstructed from data acquired 1 min after antigen addition in Fig. 1 A, although we do not attempt to resolve two distinct components in g(r).
Our choice to quantify single-color live-cell images using autocorrelations relies on the assumption that live-cell superresolution images are not greatly affected by artifacts associated with overcounting single receptors. We expect this to be the case because individual fluorescently labeled receptors will typically diffuse over distances much larger than correlated structures with dimensions of several hundred nanometers or less during the time that a fluorescent molecule labeling an individual receptor remains in the dark state. This assumption may not be valid in stimulated cells, where receptors may become confined within densely cross-linked clusters, and the same receptor may be counted multiple times within a single cluster. If there is a contribution to correlation functions from overcounting, our reported results would lead to an overestimate of A. However, the values of A we observe from autocorrelation functions in live cells are systematically lower than values of A extracted from cross-correlation functions in fixed cells (Fig. 1 C, black diamonds), which are not affected by overcounting artifacts. This supports the assumption that overcounting does not affect correlation functions throughout the time course of imaging.
Our results using chemically fixed cells (Fig. S2) are consistent with and complementary to our live cell measurements in several regards. Agreement of A and N between fixed and live cells at early stimulation time points indicates that clustering occurs to approximately the same extent, although at a somewhat slower rate, in cells stimulated at room temperature compared to those stimulated at 37°C. Further, results from fixed-cell experiments demonstrate that receptors are not clustered before stimulation. This result is important in the context of our live-cell experiments, because small and highly mobile clusters that diffuse much farther than their size over a typical image acquisition time would not be detected by our live-cell quantification methods.
Mobility of IgE-FcεRI in live-cell measurements
The majority of individual fluorophores remain in a fluorescent state for multiple sequential frames, and we track these probes to form trajectories from localizations of the protein in time and space (see Materials and Methods in the Supporting Material). Visual inspection of trajectories obtained from 16 s of acquired data in unstimulated and stimulated cells suggests that IgE-FcεRI diffusion is relatively unconstrained before stimulation and that mobility decreases significantly after antigen addition (Fig. 2 A). Trajectories are quantified by tabulating the mean-square displacement (MSD) as a function of time interval (τ). Several representative curves are calculated by averaging MSD(τ) values over all trajectories acquired within 16 s, as shown in Fig. 2 B. The magnitude of the average MSD(τ) decreases after stimulation, indicating reduced receptor mobility. The representative data shown in Fig. 2, A and B, are acquired from the same cell shown in Fig. 1, A and B.
Figure 2.
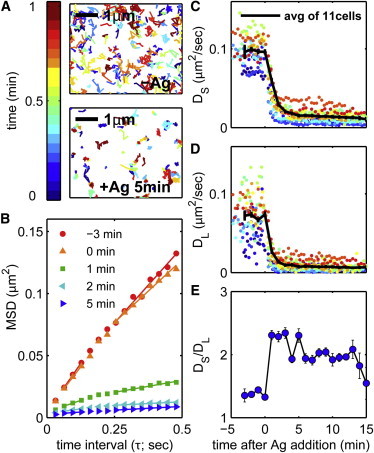
Antigen stimulation leads to slower and more confined diffusion of IgE-FcεRI receptor complexes. (A) Single-molecule trajectories of IgE-FcεRI complexes on the surface of cells under TIRF illumination before (−Ag) and after (+Ag) stimulation with 1 μg/mL DNP-BSA for 5 min. Tracks shown are accumulated over 1 min, only tracks observed for five or more frames (0.16 s) are displayed, and coloring from blue to red indicates the relative time at which a single probe was observed within the 1 min time frame. (B) MSD curves are generated by averaging over all tracks observed within a 500 frame (16 s) time period at the times during antigen stimulation indicated, as described in Materials and Methods in the Supporting Material. MSD curves are fit to Eqs. 3A and 3B to extract the short- and long-time diffusion coefficients DS and DL, respectively. (C and D) Summary of DS (C) and DL (D) extracted from MSD curves tabulated from single-molecule trajectories acquired over 500 frames (∼20 s and variable from cell to cell) for 11 distinct cells. Error bars represent the mean ± SE of the 11 live-cell experiments. (E) Confinement as a function of stimulation time, as measured by DS/DL from the same 11 live-cell experiments. Error bars represent the mean ± SE. To see this figure in color, go online.
In most cases, we find that the slopes of MSD(τ) are not linear, as expected for free diffusion, but instead are deflected to lower values at long τ, indicating that receptors are confined. We quantify both diffusion and confinement of IgE-FcεRI complexes as a function of stimulation time by fitting MSD(τ) to obtain both short- and long-time diffusion coefficients, DS and DL, respectively, which are obtained by fitting distinct time ranges of MSD(τ) curves to extract linear slopes. DS is obtained by fitting the equation:
| (3A) |
where τ2-4 indicates the second, third, and fourth time intervals of the MSD(τ) curve, typically corresponding to roughly 50–100 ms, and CS is the y-intercept of the fit that accounts for the finite localization precision of the single-molecule data. DL is obtained by fitting the MSD curve at time intervals between 250 and 500 ms to the analogous equation,
| (3B) |
Best-fit lines to Eqs. 3A and 3B, whose slopes are proportional to the values of DS and DL, are shown for the representative MSD(τ) curves in Fig. 2 B. In addition, we quantify confinement by taking a ratio of these values, DS/DL. In the examples shown in Fig. 2 B, DS/DL > 1, indicating that receptors are confined, and this ratio increases after antigen addition.
DS, DL, and their ratio for IgE-FcεRI in cells undergoing signaling responses are shown in Fig. 2, C–E, for the same 11 cells characterized in Fig. 1. Both DS and DL dramatically decrease within 5 min of antigen addition. After 5 min of stimulation, DS decreases from 0.1 to 0.02 μm2/s and DL decreases from 0.075 to 0.01 μm2/s. We also observe changes in confinement with stimulation time, as measured by DS/DL, which rapidly increases after stimulation before decreasing slightly at later stimulation time points.
Our measured values of DS versus stimulation time are in agreement with similar diffusion-coefficient parameters measured previously using SPT (10,12,13,33), FCS (11), and FPR (5,9), ranging from 0.03 to 0.26 μm2/s before stimulation and from 0.01 to 0.16 μm2/s after stimulation with a multivalent antigen. Previous measurements of receptor diffusion using SPT approaches similar to ours (10,12,13,33) agree best with our observed values, where here reported values range from 0.07 to 0.1 μm2/s before stimulation and from 0.01 to 0.05 μm2/s after stimulation. Our measurements of receptor confinement are also consistent with the observation of restricted or compartmentalized diffusion in previous SPT studies of FcεRI (10,12,33). In one of these past studies, the diffusion compartments of IgE-FcεRI were reported to shift to smaller sizes upon antigen addition, accompanied by a decrease in the diffusion coefficient for movement between compartments (33). That result is consistent with the antigen-induced increase in receptor confinement measured in this study.
Correlating receptor mobility with receptor clustering
The data presented in Figs. 1 and 2 allow for direct comparison between changes in IgE-FcεRI receptor diffusion versus spatial distribution. Taken together, the results indicate that IgE-FcεRI receptor complexes have decreased mobility (DS and DL) and increased confinement (DS/DL) that plateaus within 1–2 min after antigen addition. In contrast, the density of receptor clusters (A) increases more slowly, with the amplitude of correlations plateauing after ∼5 min (Fig. 1 C).
To explore more directly the relationship between cluster properties and receptor mobility, we plot in Fig. 3 A the average short-time receptor diffusion coefficient, DS, versus the average number of correlated proteins, N, for the stimulation time course averaged from 11 live-cell experiments (average DS and N as a function of time are shown independently in Figs. 2 C and 1 C, respectively). Interestingly, this representation suggests two distinct regimes of receptor mobility and clustering. In the first regime, DS decreases dramatically without a large corresponding change in N. In the second regime, receptors become increasingly clustered, without a large corresponding decrease in DS. The crossover between regimes occurs for N between 20 and 30 and for DS between 0.035 and 0.02 μm2/s, which correspond to stimulation times between 1 and 1:45 min, respectively, after antigen addition.
Figure 3.
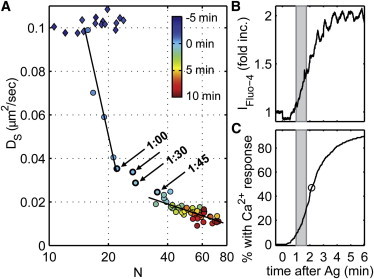
Average receptor diffusion displays two different phases of dependence on the number of proteins in the average cluster. (A) Average DS (as in Fig. 2C) is shown as a function of average N from the same live-cell experiments (as in Fig. 1C). Each point corresponds to values of Ds and N at a given time before (diamonds) and after (circles) stimulation averaged over the 11 cells imaged, and data from individual cells are binned every 15 s to facilitate averaging. Time after the addition of antigen is indicated by the color bar. Antigen (1 μg/ml) was added after the cells were imaged for 5 min. The solid black lines represent linear fits of points between 0 and 1 min and between 1:45 and 15 min after antigen stimulation, weighted by the inverse of the mean ± SE in DS and N for each point. Points spanning these two regimes are indicated with arrows and labeled with the time after antigen addition. (B) Average intensity of the cytoplasmic Ca2+ indicator Fluo-4 over a population of cells imaged as described in Materials and Methods in the Supporting Material. The increase in Fluo-4 intensity after antigen stimulation indicates the onset of Ca2+ mobilization. The time period coinciding with the timing of the transition from the first regime to the second in A is highlighted by the shaded region. (C) The cumulative distribution of cells exhibiting an initial Ca2+ response indicates that the majority (>90%) of cells have initial Ca2+ responses between 1 and 4 min after antigen stimulation. The time point when 50% of the responding cells have exhibited a Ca2+ response is indicated by the open circle. To see this figure in color, go online.
Interestingly, the beginning of the crossover between the two regimes shown in Fig. 3 A roughly coincides with the onset of Ca2+signaling in RBL-2H3 cells imaged using the Ca2+-sensitive dye Fluo-4 under nearly identical stimulation conditions (Fig. 3, B and C). Fluo-4 is loaded into sensitized RBL cells, and the fluorescence intensity is monitored across a field of several hundred cells as a function of stimulation time, as described in Materials and Methods (see Supporting Material). Soon after antigen addition, Fluo-4 intensity averaged over the population of cells begins to rapidly increase, with the bulk of the increase coming between 1 and 4 min (Fig. 3 B). There is a large cell-to-cell heterogeneity in the timing of the onset of the Ca2+ response, as indicated in the cumulative distribution shown in Fig. 3 C, with some cells initiating a response ∼1 min after antigen addition under these imaging conditions. Approximately 50% of cells have experienced a Ca2+ response within 2:15 min. Keeping in mind this large heterogeneity and the fact that we sample only a limited number of single cells in superresolution experiments, we use the Ca2+ mobilization measurement as a rough indicator of the commencement of cellular signaling.
The results reported in Fig. 3 indicate that the initial, rapid decrease in DS of IgE-FcεRI complexes is a consequence of interactions that precede Ca2+ mobilization, whereas the accumulation of receptors into densely packed clusters represents receptors after the onset of Ca2+ mobilization. This suggests that early signaling events leading to the Ca2+ response do not require that receptors be densely clustered or fully immobilized. This interpretation of our results is consistent with observations from previous studies that small IgE-FcεRI clusters that retain mobility can elicit a degranulation response (13), and that there is a high level of receptor tyrosine phosphorylation within the first few minutes of antigen stimulation, both at 37° and 15°C (34). In the latter study (34), receptor tyrosine phosphorylation at 15°C occurs on a timescale similar to that of the Ca2+ response that we measure at room temperature, as we would expect, since tyrosine phosphorylation precedes Ca2+ mobilization in the IgE receptor signaling cascade. The same study also demonstrated that exposure of DNP haptens on the surface of DNP-BSA is transient, and that antigen binding is dominated by cross-linking of receptors after a minute of exposure to antigen (34). This is also consistent with the idea that the immobilization and Ca2+ responses we observe at early stimulation times occur concurrently with the formation of small clusters, and that receptors become more heavily cross-linked at later times.
Because diffusion of cross-linked IgE-FcεRI decreases rapidly, without a corresponding large increase in N, it likely occurs as a result of IgE-FcεRI coupling to downstream signaling partners. This could be due to receptor association with other freely diffusing membrane-anchored proteins that are not labeled in these experiments. It is possible that receptor slowing is due to slower movement between corrals defined by cortical actin, either due to mechanical occlusion of the growing signaling platforms with actin-anchored proteins (36,37), or due to increased coupling of platforms to actin-stabilized, lipid-mediated heterogeneity (38). Alternatively, the reduced mobility of receptors at early signaling stages could be a consequence of direct or indirect tethering to an immobilized component such as actin. The actin cytoskeleton has been shown to be partially responsible for the immobilization of cross-linked IgE-FcεRI (12), and it plays a role in the desensitization of receptors to antigen (39), and in internalization (40). The increasing clustering of receptors that dominates later signaling stages could be a mechanism to downregulate signaling by sequestering receptors, as occurs in B cell and T cell receptor activation (41–44). It is also possible that the observed receptor clusters represent an early stage of receptor internalization that has not progressed substantially under these conditions of stimulation time and temperature (11).
Ca2+ mobilization is used here as an approximate indication of the timing of the onset of cellular signaling, but consideration should be given to its direct comparison with superresolution measurements. Clustering and immobilization of IgE receptors on the ventral surface of cells could begin more slowly than Ca2+ mobilization because the Ca2+ response originates from receptors on the dorsal surface of cells, which are expected to be more accessible to antigen. This may lead to a time delay between the initiation of the Ca2+ response and our observation via superresolution measurements of immobilization and clustering. If this is the case, receptors on the dorsal surface may be somewhat more clustered at the onset of the Ca2+ response, but this would not change our conclusion that the first phase of clustering and immobilization occurs before Ca2+ mobilization, and the second phase occurs predominantly afterward. Ca2+ mobilization itself may be considered a downstream measure of the onset of signaling. Previous work has shown that receptor phosphorylation by Lyn kinase proceeds much more rapidly than Ca2+ mobilization (35), and thus, Ca2+ mobilization represents membrane interactions that initiate a broader cellular response and occur subsequent to the initiation of signaling at the level of single receptors.
Single-molecule analysis of receptor diffusion and clustering
Early and late signaling stages are also distinguished when receptors are examined as single molecules, and several representative trajectories are shown in Fig. 4 A. Single receptor trajectories in unstimulated cells traverse large areas. Soon after antigen is added, trajectories rapidly condense, and some receptors appear to sample multiple confined areas in single trajectories, lasting ∼1 s each. After a few minutes of antigen stimulation, trajectories are compact and appear highly confined. The ensemble of single-molecule trajectories is quantified by assembling histograms of DS. Fig. 4 B shows histograms assembled using 16 s of data acquired in a single cell, which are representative of histograms obtained from other cells examined. Histograms are well described as single log-normal distributions for all time points, indicating that a single population of diffusers is resolved in these measurements. Distributions of DS rapidly shift to lower values and broaden soon after antigen is added, stabilizing after 3 min of stimulation time. These distributions are broad, in part because diffusion coefficients are not well defined when obtained from short trajectories (45). To separate this effect from real heterogeneity, we compare measured distributions of DS to those obtained by simulating Brownian trajectories with 16 frame (0.5 s) track length (Fig. S4). In unstimulated cells, the width of measured DS histograms is comparable to those of simulated trajectories. In contrast, measured histograms for receptors after antigen addition are significantly broader than the simulated distributions, indicating that the membrane environment sampled by IgE-FcεRI is heterogeneous.
Figure 4.
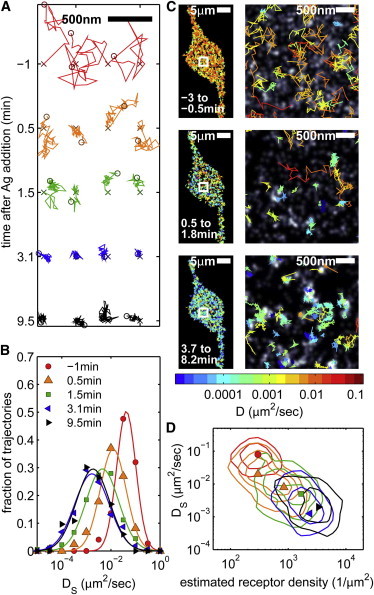
Slower and more confined diffusion of single receptors correlates with regions of high receptor density. (A) Examples of single-molecule trajectories are shown from the same cell in Fig. 1, A and B, Fig. 2, A and B, and Fig. 4C recorded before and after antigen stimulation. The tracks shown persist for at least 0.5 s for the −1 min (unstimulated) time point and for 1 s for other time points. (B) Short-time diffusion coefficients (DS) are evaluated from MSD curves tabulated from single-molecule trajectories lasting at least 0.5 s within a 16 s time period and are assembled into histograms. Histograms are normalized by the total number of tracks collected to generate each histogram. (C) Single-molecule trajectories persisting for at least 0.5 s are superimposed on a superresolution image reconstructed from unstimulated data (upper), from data acquired within 1 min of antigen addition (middle), and from data acquired after several minutes of stimulation. Track coloring indicates DS for each track on a log scale from 10−5μm2/s (blue) to 1 μm2/s (red). Images on the right are enlargements of the boxed regions in the images on the left. (D) Three-dimensional histograms of DS versus average receptor density along trajectories lasting at least 0.5 s. The average receptor density for each trajectory is determined by averaging the pixelated grayscale values from the time-averaged reconstructed image over all positions of the trajectory and then normalizing assuming ρave = 200/μm2, as described in Materials and Methods in the Supporting Material.
We also investigated how receptor diffusion correlates with the local surface density of receptors in reconstructed images. To accomplish this, we reconstructed superresolution fluorescence images as described in Materials and Methods (see Supporting Material). Representative grayscale images for a single cell at various stimulation stages are shown in Fig. 4 C. In these images, pixel intensity is proportional to the observed receptor density, and trajectories that persist for >0.5 s are superimposed onto this image. In the unstimulated cell (Fig. 4 C, upper), individual receptors diffuse over large areas and their mobility is not visually correlated with roughly random receptor density. Soon after antigen addition (<1 min), individual receptors appear more confined, even though the spatial distribution of receptors remains largely random (Fig. 4 C, middle). At longer times, (>5 min after antigen addition), diffusing receptors are confined to regions where receptors are densely packed (Fig. 4 C, lower).
These visual observations are quantified by calculating the average pixel intensity over the length of single-molecule trajectories persisting for >0.5 s. Three-dimensional histograms are displayed as contour plots in Fig. 4 D for several stimulation times. In unstimulated cells, receptor diffusion appears relatively unconstrained, and the ensemble of single molecules experiences roughly the same local environment at the frame rates used in these experiments (∼30/s). This is not surprising given that receptors typically move hundreds of nanometers between observations, and over several square microns in a typical trajectory. Lipid-mediated and/or actin-generated obstacles to diffusion are expected to occur on smaller length-scales in unstimulated cells, and thus, single receptors are expected to sample a large number of local environments in a single trajectory. At long times, we observe a homogeneous but broad distribution in the DS versus receptor density histogram in Fig. 4 D. This is a result of both a distribution of receptor densities in puncta and individual receptors confined to sample the local environment of single puncta over their recorded trajectories.
Soon after stimulation with antigen, histograms are elongated, extending between faster-moving receptors in a low-density local environment and slower-moving receptors in a higher-density local environment. In some cases, two peaks are observed, as indicated by the orange triangles in Fig. 4 D. This elongated distribution could arise from receptors sampling a heterogeneous membrane environment. We think it is more likely that this elongated distribution arises from receptors slowly exchanging between a more mobile, less aggregated state and a less mobile, cluster-associated state over the length of the trajectory, as appears to be the case from visual inspection of single trajectories (Fig. 4 A).
Elongated distributions in Fig. 4 D are observed before the onset of Ca2+ mobilization, and transient associations of individual receptors could represent interactions that result in signaling. This also suggests that receptor aggregation is dynamic, at least in the early signaling stages. Consistent with this interpretation, it has been shown previously that readily dissociable cross-linked receptors are primarily responsible for generating downstream signaling responses (39). Previous work has also shown that initial binding of DNP-BSA to IgE is primarily monovalent, and that cross-linking occurs slowly as DNP haptens on the receptor-bound antigen subsequently become available for binding (34). This is in good agreement with our current observations that suggest transient association of receptors with receptor clusters soon after antigen addition.
Receptor clustering, immobilization, and confinement is reversible in live cells
A monovalent DNP hapten, DNP-aminocaproyl-L-tyrosine (DCT), competes with multivalent DNP-BSA for binding to anti-DNP IgE (39). The addition of an excess of DCT after antigen stimulation reverses antigen-induced cross-linking and results in the cessation of signaling (39,46,47). The representative live-cell superresolution images in Fig. 5 A show uniform distribution of AF647-IgE bound to FcεRI before antigen addition, clustered IgE-FcεRI distribution after 7 min of antigen stimulation, and uniform distribution of receptors after DCT incubation for 10 min. The average time dependence of A, N, and DS from six live-cell experiments quantifies the reversal of clustering and immobilization upon DCT exposure (Fig. 5 B). IgE-FcεRI clusters are dispersed on a timescale of several minutes, as shown by a decrease in A. Likewise, N and DS recover close to prestimulation levels within 10 min of DCT addition. These data indicate that receptor immobilization is reversible and dependent on receptor cross-linking, as has been shown previously by FPR measurements that used DCT to reverse receptor immobilization (5). This reversibility demonstrates that the densely packed and immobile receptor clusters formed within 7 min of antigen addition are not stabilized solely through interactions with other cellular components or that these interactions are insufficient to stabilize clusters in the absence of antigen cross-linking.
Figure 5.
Antigen-induced changes in receptor clustering and mobility are reversible. (A) Reconstructed images of an AF647-IgE-labeled living cell before and after stimulation and subsequent addition of DCT. Each image is reconstructed from 80 s of acquired data. (Insets) Magnifications of the boxed regions in the main image. DNP-BSA (0.1 μg/ml) was added at 0 min, and DCT (200 μM) was added at 7 min. (B) The parameters A, N, and Ds are calculated as in Figs. 1C and 2C. The average values of A, N, and Ds, indicated by black lines, for six live-cell experiments distinguished by different colors, over the time course of stimulation and DCT addition. Antigen addition is indicated by the orange dashed line at 0 min, and DCT addition is indicated by the gray dashed line at 7 min. To see this figure in color, go online.
Receptor organization and mobility in response to cholesterol perturbation in unstimulated cells
To examine the role of lipid-mediated membrane heterogeneity on early and later stages of antigen stimulation, we investigated the impact of membrane cholesterol levels on receptor organization and mobility. Receptor diffusion (DS) and clustering (N) upon cross-linking with antigen are measured for multiple live cells exposed to either methyl-β-cyclodextrin (MβCD) to reduce plasma membrane cholesterol or MβCD precomplexed with cholesterol (MβCD + chol) to enrich plasma membrane cholesterol (Figs. 6 and S5). Under our conditions, we expect an ∼20% decrease in the total cellular cholesterol content after 5 min of MβCD addition and an ∼50% decrease after 15 min (48).
Figure 6.
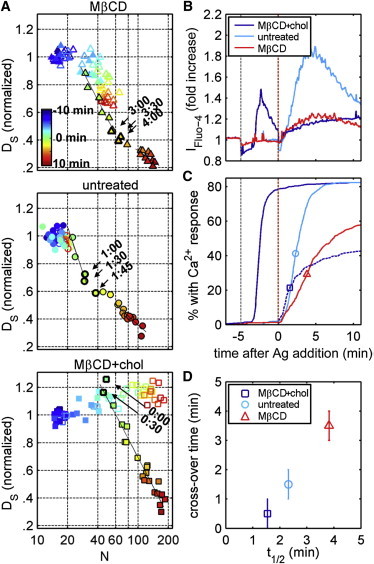
Perturbations of membrane cholesterol have corresponding effects on receptor diffusion, receptor clustering, and cellular Ca2+ responses. (A) Average DS and N from five live cells for each of six treatments—MβCD (upper), no perturbation (middle), or MβCD + chol (lower)—in the presence (solid symbols) and absence (open symbols) of antigen stimulation (0.1 μg/ml). Changing color from blue to red indicates advancement in time by 30 s for each time point from −10 to 10 min after the addition of antigen for stimulated cells or a blank addition of buffer for unstimulated cells. Cholesterol perturbations are added, where applicable, at −5 min. For stimulated time points (solid symbols outlined in black), the fit lines shown in black represent linear fits of the time points belonging to the two regimes of DS dependence on N, weighted by the inverse of the the mean ± SE in DS and N. Stimulated time points near the crossover are indicated by arrows and labeled with time after antigen addition. (B)Total fluorescence intensity is shown as a function of time for populations of cells (at least 500) loaded with Fluo-4-AM and treated with MβCD, MβCD + chol, or no perturbation. The dotted gray line at −5 min indicates the addition of MβCD or MβCD + chol, and the dotted orange line indicates the addition of antigen. (C) The cumulative distribution in time of cells with an initial Ca2+ response as monitored by Fluo-4 fluorescence. The dark blue dotted line indicates the cumulative distribution of cells treated with MβCD + chol that exhibit an additional Ca2+ response after the addition of antigen. The half-maximum times (t1/2) of cumulative curves are denoted by open symbols. (D) The estimated midpoints of the crossover times are plotted versus t1/2 for each cholesterol treatment. Error bars represent uncertainty in determining the midpoint of the crossover time by ±30 s.
In the absence of cross-linking by antigen, local receptor density changes due to variations in membrane cholesterol concentration. Fig. S5 shows representative superresolution images of AF647-IgE/FcεRI with and without antigen treatment for individual cells with MβCD or MβCD + chol. IgE-FcεRI continues to show largely random distribution in unstimulated cells with reduced cholesterol levels, but becomes tightly clustered in unstimulated cells with elevated cholesterol levels. This visual observation is quantified in Fig. S6, where DS, A, and N are plotted as a function of time and treatment with MβCD or MβCD + chol.
It is likely that other cellular processes contribute to the organization and mobility of receptors in cholesterol-loaded cells. For example, we observe robust antigen-independent activation of transient Ca2+ oscillations after cells are incubated with MβCD + chol for 2 min, and this persists until ∼5 min after MβCD + chol is added (Figs. S7 and 6, B and C). This indicates that the cellular environment changes dramatically in response to MβCD + chol in ways that may not be directly related to cholesterol’s effects on lipid-mediated membrane organization. Modulating cellular cholesterol levels also leads to changes in receptor diffusion in unstimulated cells. MβCD addition to unstimulated cells results in a time-dependent decline in DS over 15 min, and incubation with MβCD + chol leads to slight increases in DS (Fig. S6), despite the large increases in receptor clustering described above. These observations could be the result of changes in membrane surface area, changes in the surface density of immobile obstacles, or induction of solid-phase domains (49).
Receptor organization and mobility in response to cholesterol perturbation in stimulated cells
Perturbations of membrane cholesterol also affect the organization and mobility of IgE-FcεRI complexes when receptors are subsequently cross-linked with multivalent antigen (Fig. 6 A). For both cholesterol reduction and enrichment, receptor clustering increases and receptor diffusion decreases in response to antigen, qualitatively similar to trends in the absence of perturbation (Figs. 3 A and 6 A). We observe two distinct regimes in plots of DS versus N for points after antigen addition in cells pretreated with MβCD, as is also observed in cells in the absence of cholesterol modulation. The crossover between these two regimes occurs at larger values of N in cells with reduced cholesterol levels (N < 40 for untreated versus N > 60 for MβCD-treated cells (Fig. 6 A, upper)), which also corresponds to longer stimulation times at the crossover point (∼1 min for untreated vs. ∼3 min for MβCD-treated cells). For cells pretreated with MβCD + chol (Fig. 6 A, lower), only one regime is apparent in plots of DS versus N, and the N is larger before antigen addition.
Antigen-induced functional responses are also affected in cells pretreated with MβCD or MβCD + chol. Antigen-induced signaling is less effective in cells with reduced cholesterol levels when stimulated degranulation is assessed (50–53) (Fig. S8). When Ca2+ mobilization is again used as a rough measure of the onset of cellular signaling, as in Fig. 3, pretreatment of cells with MβCD results in a Ca2+ response that is both reduced in magnitude (Fig. 6 B) and delayed (Fig. 6 C) compared to untreated cells. A fraction (∼40%) of cells pretreated with MβCD fail to show Ca2+ responses within 10 min after antigen addition. We quantify the timing of antigen-induced Ca2+ mobilization by measuring the time taken for 50% of responding cells to show an initial Ca2+ response, t1/2, as indicated by the symbols on the cumulative distributions shown in Fig. 6 C. Antigen-induced signaling is attenuated in cells pretreated with MβCD + chol when they are assayed by degranulation (Fig. S8) or by measurement of Ca2+ mobilization (Fig. 6).
For the case of cholesterol enrichment, we observe an initial Ca2+response after MβCD + chol is added, as described above, followed by a second, weaker Ca2+ signal in response to antigen (Fig. 6 B). A large fraction (∼60%) of MβCD + chol-treated cells also fail to exhibit an antigen-induced Ca2+ response (Fig. 6 C), although cells that do respond do so with a minimal time lag after antigen addition. As a result, t1/2 is shorter compared to either untreated or MβCD-treated cells (Fig. 6 D). These differences in the nature of the antigen-dependent Ca2+ response may be influenced by the MβCD + chol-induced Ca2+ transient observed before antigen addition. The shape, frequency, and duration of Ca2+ oscillations are also severely affected by changes in cellular cholesterol (Fig. S7).
t1/2 is correlated with the timing of the crossover observed in plots of DS versus N for the three cholesterol treatments (Fig. 6 D). Despite the uncertainty in relating the timing of Ca2+ mobilization to superresolution measurements discussed above, we observe differences in the relative timing of the crossover and t1/2 that are both dependent on cholesterol perturbation. This observation supports our conclusion that the initial, rapid decrease in the diffusion coefficient of IgE-FcεRI receptors is a consequence of interactions that precede Ca2+ mobilization, whereas the accumulation of receptors into densely packed clusters represents receptors after the onset of signaling. For the case of cholesterol reduction, antigen-induced slowing of receptor diffusion occurs at a slower rate than in untreated or cholesterol-enriched cells (Fig. 6 A), suggesting that initial signaling steps occur over a longer time period. This is consistent with our observations of a slower Ca2+ response in MβCD-treated cells compared to untreated cells. The crossover between regimes occurs at larger values of N in MβCD treated versus untreated cells, suggesting that more receptors are needed to initiate downstream signaling events when cholesterol levels are reduced. Plots of Ds versus N for cholesterol-enriched cells indicate only a single regime, and antigen-induced Ca2+ responses occur with a minimal time lag after antigen addition.
Previous work has demonstrated the importance of membrane-lipid-mediated protein targeting for transmembrane signaling in the FcεRI cascade (54,55). Cholesterol reduction inhibits stimulated receptor phosphorylation by Lyn, and productive signaling only occurs upon the redistribution of receptors, kinases, and phosphatases via changes in the local lipid environment surrounding cross-linked receptors (53,56,57). In our previous SEM work we found that cholesterol reduction before antigen addition led to smaller IgE-FcεRI-rich clusters and reduced Lyn partitioning into receptor-rich clusters when cells were chemically fixed 1 min after antigen addition at 37°C. Although it is not possible to compare absolute numbers between these experiments due to the different labeling strategies employed, our current findings are consistent with these previous results. Specifically, we find that it takes longer for receptors to assemble into tight clusters when cells are pretreated with MβCD, so at a given time point after stimulation, we would expect receptor-rich clusters to be smaller in MβCD-pretreated cells compared to untreated cells. Our observations of a delay in Ca2+ responses in MβCD-pretreated cells relative to untreated cells are consistent with our past observations of defects in Lyn recruitment under these conditions. These findings are consistent with an inhibitory role for cholesterol reduction in signaling that has been supported by previous observations (50,52,53). An alternative explanation for the changes in diffusion versus clustering behavior and Ca2+ responses observed in MβCD- and MβCD + chol-treated cells are related to more global effects of cholesterol modulation, such as its perturbation of the actin cytoskeleton. Cholesterol reduction can disrupt cytoskeleton-membrane attachment through perturbation of plasma membrane PIP2 (58), or because actin is frequently coupled to the plasma membrane via more ordered regions (59). Therefore, the changes in receptor clustering, receptor mobility, and Ca2+ responses caused by MβCD, and by extension MβCD + chol, may be indirect results of membrane cholesterol modulation via its effects on the actin cytoskeleton.
Conclusion
In conclusion, we demonstrate that superresolution fluorescence localization imaging is a powerful method for quantifying the organization and mobility of immune receptors in cells undergoing stimulated responses. Simultaneous measurements of clustering and diffusion enable the resolution of two distinct temporal phases of receptor clustering and immobilization. At early times after stimulation, receptor-rich clusters increase marginally in size and receptors slow dramatically when averaged over the population of receptors. When examined as individual molecules, either as monomers or as members of clusters, single receptors appear to reversibly associate with small and slowly moving receptor clusters soon after the addition of antigen. These behaviors are observed at stimulation times preceding Ca2+ mobilization, leading us to conclude that they arise from interactions associated with initial signaling steps. At later times, receptors in clusters become increasingly dense and are largely immobile. Since these behaviors occur at times after the initial Ca2+ response, we hypothesize that receptor immobilization into densely packed clusters leads to subsequent cellular interactions related to downregulation of signaling. Before these terminating steps, dense receptor clusters are dispersed when the cross-linking antigen is displaced by a monovalent ligand. Receptor clustering and dynamics are also altered in cells with modulated cholesterol levels. Most notably, receptors cluster in cells with increased cholesterol levels even in the absence of antigen, and we observe changes in the duration of the initial phase of receptor clustering and immobilization in stimulated cells with modulated cholesterol levels. The onset of Ca2+ mobilization requires association of activated receptors with multiple proteins, and this signaling complex formation appears to occur before or during the crossover between the two regimes defined by our analysis. We observe differences in the timing of Ca2+ mobilization for cells with modulated cholesterol levels that correspond to changes in the timing of the initial phase of clustering. These findings are motivators for future work investigating the physical interactions that give rise to the observed changes in receptor organization and mobility, and how these translate into cellular functions.
Acknowledgments
The authors thank Amit Singhai for assistance with experiments and for providing reagents, and Christopher V. Kelly, Matthew B. Stone, and Benjamin B. Machta for helpful discussions.
Research was supported through grants from the National Institutes of Health to S.L.V. (R00GM087810) and B.B. and D.H. (RO1 AI018306). S.A.S. acknowledges partial support from a National Institutes of Health Molecular Biophysics Training Grant (T32GM008267).
Supporting Material
References
- 1.Siraganian R.P. Mast cell signal transduction from the high-affinity IgE receptor. Curr. Opin. Immunol. 2003;15:639–646. doi: 10.1016/j.coi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Blank U., Rivera J. The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 2004;25:266–273. doi: 10.1016/j.it.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Gilfillan A.M., Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schlessinger J., Webb W.W., Metzger H. Lateral motion and valence of Fc receptors on rat peritoneal mast cells. Nature. 1976;264:550–552. doi: 10.1038/264550a0. [DOI] [PubMed] [Google Scholar]
- 5.Menon A.K., Holowka D.A., Baird B.A. Cross-linking of receptor-bound IgE to aggregates larger than dimers leads to rapid immobilization. J. Cell Biol. 1986;102:541–550. doi: 10.1083/jcb.102.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stump R.F., Pfeiffer J.R., Oliver J.M. Mapping gold-labeled IgE receptors on mast cells by scanning electron microscopy: receptor distributions revealed by silver enhancement, backscattered electron imaging, and digital image analysis. J. Histochem. Cytochem. 1988;36:493–502. doi: 10.1177/36.5.2965720. [DOI] [PubMed] [Google Scholar]
- 7.Wilson B.S., Pfeiffer J.R., Oliver J.M. Observing FcεRI signaling from the inside of the mast cell membrane. J. Cell Biol. 2000;149:1131–1142. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veatch S.L., Chiang E.N., Baird B.A. Quantitative nanoscale analysis of IgE-FcεRI clustering and coupling to early signaling proteins. J. Phys. Chem. B. 2012;116:6923–6935. doi: 10.1021/jp300197p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menon A.K., Holowka D.A., Baird B.A. Clustering, mobility, and triggering activity of small oligomers of immunoglobulin E on rat basophilic leukemia cells. J. Cell Biol. 1986;102:534–540. doi: 10.1083/jcb.102.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feder T.J., Brust-Mascher I., Webb W.W. Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys. J. 1996;70:2767–2773. doi: 10.1016/S0006-3495(96)79846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larson D.R., Gosse J.A., Webb W.W. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J. Cell Biol. 2005;171:527–536. doi: 10.1083/jcb.200503110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews N.L., Lidke K.A., Lidke D.S. Actin restricts FcepsilonRI diffusion and facilitates antigen-induced receptor immobilization. Nat. Cell Biol. 2008;10:955–963. doi: 10.1038/ncb1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrews N.L., Pfeiffer J.R., Lidke D.S. Small, mobile FcεRI receptor aggregates are signaling competent. Immunity. 2009;31:469–479. doi: 10.1016/j.immuni.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spendier K., Lidke K.A., Thomas J.L. Single-particle tracking of immunoglobulin E receptors (FcεRI) in micron-sized clusters and receptor patches. FEBS Lett. 2012;586:416–421. doi: 10.1016/j.febslet.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rust M.J., Bates M., Zhuang X. Stochastic optical reconstruction microscopy (STORM) provides sub-diffraction-limit image resolution. Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heilemann M., van de Linde S., Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 17.Betzig E., Patterson G.H., Hess H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 18.Hess S.T., Girirajan T.P.K., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenfield D., McEvoy A.L., Liphardt J. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 2009;7:e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lillemeier B.F., Mörtelmaier M.A., Davis M.M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen D.M., Rentero C., Gaus K. PALM imaging and cluster analysis of protein heterogeneity at the cell surface. J. Biophotonics. 2010;3:446–454. doi: 10.1002/jbio.200900089. [DOI] [PubMed] [Google Scholar]
- 22.Williamson D.J., Owen D.M., Gaus K. Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events. Nat. Immunol. 2011;12:655–662. doi: 10.1038/ni.2049. [DOI] [PubMed] [Google Scholar]
- 23.Hsu C.-J., Baumgart T. Spatial association of signaling proteins and F-actin effects on cluster assembly analyzed via photoactivation localization microscopy in T cells. PLoS ONE. 2011;6:e23586. doi: 10.1371/journal.pone.0023586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman E., Barr V., Samelson L.E. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity. 2011;35:705–720. doi: 10.1016/j.immuni.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gunewardene M.S., Subach F.V., Hess S.T. Superresolution imaging of multiple fluorescent proteins with highly overlapping emission spectra in living cells. Biophys. J. 2011;101:1522–1528. doi: 10.1016/j.bpj.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones S.A., Shim S.-H., Zhuang X. Fast, three-dimensional super-resolution imaging of live cells. Nat. Methods. 2011;8:499–508. doi: 10.1038/nmeth.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manley S., Gillette J.M., Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- 28.Manley S., Gillette J.M., Lippincott-Schwartz J. Single-particle tracking photoactivated localization microscopy for mapping single-molecule dynamics. Methods Enzymol. 2010;475:109–120. doi: 10.1016/S0076-6879(10)75005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giannone G., Hosy E., Cognet L. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys. J. 2010;99:1303–1310. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gadi D., Wagenknecht-Wiesner A., Baird B. Sequestration of phosphoinositides by mutated MARCKS effector domain inhibits stimulated Ca2+ mobilization and degranulation in mast cells. Mol. Biol. Cell. 2011;22:4908–4917. doi: 10.1091/mbc.E11-07-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veatch S.L., Machta B.B., Baird B.A. Correlation functions quantify super-resolution images and estimate apparent clustering due to over-counting. PLoS ONE. 2012;7:e31457. doi: 10.1371/journal.pone.0031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erickson J., Goldstein B., Baird B. The effect of receptor density on the forward rate constant for binding of ligands to cell surface receptors. Biophys. J. 1987;52:657–662. doi: 10.1016/S0006-3495(87)83258-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barisas B.G., Smith S.M., Roess D.A. Compartmentalization of the Type I Fc ε receptor and MAFA on mast cell membranes. Biophys. Chem. 2007;126:209–217. doi: 10.1016/j.bpc.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 34.Xu K., Goldstein B., Baird B. Kinetics of multivalent antigen DNP-BSA binding to IgE-Fc ε RI in relationship to the stimulated tyrosine phosphorylation of Fc ε RI. J. Immunol. 1998;160:3225–3235. [PubMed] [Google Scholar]
- 35.Paolini R., Jouvin M.H., Kinet J.P. Phosphorylation and dephosphorylation of the high-affinity receptor for immunoglobulin E immediately after receptor engagement and disengagement. Nature. 1991;353:855–858. doi: 10.1038/353855a0. [DOI] [PubMed] [Google Scholar]
- 36.Ritchie K., Iino R., Kusumi A. The fence and picket structure of the plasma membrane of live cells as revealed by single molecule techniques (Review) Mol. Membr. Biol. 2003;20:13–18. doi: 10.1080/0968768021000055698. (Review) [DOI] [PubMed] [Google Scholar]
- 37.Murase K., Fujiwara T., Kusumi A. Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys. J. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Machta B.B., Papanikolaou S., Veatch S.L. Minimal model of plasma membrane heterogeneity requires coupling cortical actin to criticality. Biophys. J. 2011;100:1668–1677. doi: 10.1016/j.bpj.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierini L., Harris N.T., Baird B. Evidence supporting a role for microfilaments in regulating the coupling between poorly dissociable IgE-Fc εRI aggregates downstream signaling pathways. Biochemistry. 1997;36:7447–7456. doi: 10.1021/bi9629642. [DOI] [PubMed] [Google Scholar]
- 40.Ra C., Furuichi K., White K.N. Internalization of IgE receptors on rat basophilic leukemic cells by phorbol ester. Comparison with endocytosis induced by receptor aggregation. Eur. J. Immunol. 1989;19:1771–1777. doi: 10.1002/eji.1830191002. [DOI] [PubMed] [Google Scholar]
- 41.Liu C., Miller H., Song W. A balance of Bruton’s tyrosine kinase and SHIP activation regulates B cell receptor cluster formation by controlling actin remodeling. J. Immunol. 2011;187:230–239. doi: 10.4049/jimmunol.1100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee K.-H., Dinner A.R., Shaw A.S. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302:1218–1222. doi: 10.1126/science.1086507. [DOI] [PubMed] [Google Scholar]
- 43.Varma R., Campi G., Dustin M.L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weetall M., Holowka D., Baird B. Heterologous desensitization of the high affinity receptor for IgE (Fc ε R1) on RBL cells. J. Immunol. 1993;150:4072–4083. [PubMed] [Google Scholar]
- 45.Saxton M.J. Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 1997;72:1744–1753. doi: 10.1016/S0006-3495(97)78820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fewtrell C. Plenum Press; New York: 1985. Calcium in Biological Systems. [Google Scholar]
- 47.Seagrave J.C., Deanin G.G., Oliver J.M. DNP-phycobiliproteins, fluorescent antigens to study dynamic properties of antigen-IgE-receptor complexes on RBL-2H3 rat mast cells. Cytometry. 1987;8:287–295. doi: 10.1002/cyto.990080309. [DOI] [PubMed] [Google Scholar]
- 48.Surviladze Z., Dráberová L., Dráber P. Differential sensitivity to acute cholesterol lowering of activation mediated via the high-affinity IgE receptor and Thy-1 glycoprotein. Eur. J. Immunol. 2001;31:1–10. doi: 10.1002/1521-4141(200101)31:1<1::AID-IMMU1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 49.Nishimura S.Y., Vrljic M., Moerner W.E. Cholesterol depletion induces solid-like regions in the plasma membrane. Biophys. J. 2006;90:927–938. doi: 10.1529/biophysj.105.070524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kato N., Nakanishi M., Hirashima N. Cholesterol depletion inhibits store-operated calcium currents and exocytotic membrane fusion in RBL-2H3 cells. Biochemistry. 2003;42:11808–11814. doi: 10.1021/bi034758h. [DOI] [PubMed] [Google Scholar]
- 51.Yamashita T., Yamaguchi T., Nagasawa S. Detergent-resistant membrane domains are required for mast cell activation but dispensable for tyrosine phosphorylation upon aggregation of the high affinity receptor for IgE. J. Biochem. 2001;129:861–868. doi: 10.1093/oxfordjournals.jbchem.a002930. [DOI] [PubMed] [Google Scholar]
- 52.Silveira e Souza A.M.M., Mazucato V.M., Oliver C. The α-galactosyl derivatives of ganglioside GD(1b) are essential for the organization of lipid rafts in RBL-2H3 mast cells. Exp. Cell Res. 2008;314:2515–2528. doi: 10.1016/j.yexcr.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 53.Sheets E.D., Holowka D., Baird B. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcεRI and their association with detergent-resistant membranes. J. Cell Biol. 1999;145:877–887. doi: 10.1083/jcb.145.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silveira E Souza A.M.M., Mazucato V.M., Oliver C. Lipid rafts in mast cell biology. J. Lipids. 2011;2011:752906. doi: 10.1155/2011/752906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holowka D., Gosse J.A., Hammond A.T., Han X., Sengupta P. Lipid segregation and IgE receptor signaling: a decade of progress. Biochim. Biophys. Acta. 2005;1746:252–259. doi: 10.1016/j.bbamcr.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 56.Field K.A., Holowka D., Baird B. Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. J. Biol. Chem. 1997;272:4276–4280. doi: 10.1074/jbc.272.7.4276. [DOI] [PubMed] [Google Scholar]
- 57.Young R.M., Holowka D., Baird B. A lipid raft environment enhances Lyn kinase activity by protecting the active site tyrosine from dephosphorylation. J. Biol. Chem. 2003;278:20746–20752. doi: 10.1074/jbc.M211402200. [DOI] [PubMed] [Google Scholar]
- 58.Kwik J., Boyle S., Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. USA. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holowka D., Sheets E.D., Baird B. Interactions between FcεRI and lipid raft components are regulated by the actin cytoskeleton. J. Cell Sci. 2000;113:1009–1019. doi: 10.1242/jcs.113.6.1009. [DOI] [PubMed] [Google Scholar]
- 60.Naal R.M.Z.G., Tabb J., Baird B. In situ measurement of degranulation as a biosensor based on RBL-2H3 mast cells. Biosens. Bioelectron. 2004;20:791–796. doi: 10.1016/j.bios.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 61.Hardy R.R. 4th ed. Blackwell Scientific; Oxford, United Kingdom: 1986. Handbook of Experimental Immunology. [Google Scholar]
- 62.Jaqaman K., Loerke D., Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.