

Abstract
Stearoyl-CoA desaturase (SCD) is a key enzyme that converts saturated fatty acids (SFAs) to monounsaturated fatty acids (MUFAs) in the biosynthesis of fat. To date, two isoforms of scd gene (scd1 and scd5) have been found widely existent in most of the vertebrate animals. However, the evolutionary patterns of both isofoms and the function of scd5 are poorly understandable. Herein, we aim to characterize the evolutionary pattern of scd genes and further predict the function differentiation of scd genes. The sequences of scd genes were highly conserved among eukaryote. Phylogenetic analysis identified two duplications of scd gene early in vertebrate evolution. The relative rate ratio test, branch-specific dN/dS ratio tests, and branch-site dN/dS ratio tests all suggested that the scd genes were evolved at a similar rate. The evolution of scd genes among eukaryote was under strictly purifying selection though several sites in scd1 and scd5 were undergone a relaxed selection pressure. The variable binding sites by transcriptional factors at the 5′-UTR and by miRNAs at 3′-UTR of scd genes suggested that the regulators of scd5 may be different from that of scd1. This study promotes our understanding of the evolutionary patterns and function of SCD genes in eukaryote.
1. Introduction
Stearoyl-CoA desaturase (SCD) is an intrinsic membrane protein that binds to the endoplasmic reticulum (ER), composed of four transmembrane domains [1–3]. SCD is the rate-limiting enzyme that introduces the first cis-double bond at the delta-9 position of saturated fatty acids (SFAs) to thereby generate monounsaturated fatty acids (MUFAs) [4], which are major substrates for biosynthesis of polyunsaturated fatty acids (PUFAs) and complex lipids such as triglycerides, phospholipids, cholesterol esters, and wax esters being as energy storage, components of biological membrane, and signaling molecules. The ratio of unsaturated fatty acids to saturated fatty acids plays a vital role in cell signaling and membrane fluidity, in which imbalance of this ratio is often associated with diseases like diabetes, cardiovascular diseases, fatty liver, cancers and stresses resistance, and so forth [5].
The scd genes are universally present in living organisms. The number of scd genes varies from one to five, which are generally called scd1, scd2, scd3, scd4, and scd5 in different organisms [4, 6], but with other distinct names in invertebrates such as fat-5, fat-6, and fat-7 in Caenorhabditis elegans [7–9] and ole1 in Saccharomyces cerevisiae [10]. The yeast genome contains only ole-1 gene encoded SCD, and ole-1 mutant requires unsaturated fatty acids for growth [10]. The desaturase of C. elegans FAT-5, FAT-6, and FAT-7 displays substrate preferences, in which both FAT-6 and FAT-7 mainly desaturate stearic acid (18 : 0) and have less activity on palmitic acid (16 : 0). On the contrary, FAT-5 desaturates palmitic acid (16 : 0) but has nearly undetectable activity on stearic acid (18 : 0) [7]. The evolutionary history revealed that the scd genes in vertebrates could be distinctly classified into scd5 type [3, 6, 11] and scd1 type including its homologs scd2, scd3, and scd4 [6, 12]. The divergence of scd1 and scd5 genes occurred early in vertebrate evolution due to the whole genome duplication (2R) [6]. However, the scd genes may have distinct fates after gene duplication event. It is unknown whether one scd evolved faster and acquired new function more rapidly than the other, and whether the selective patterns on both scd genes were similarly changed following the duplication.
Interestingly, though the enzymes of scd genes display similar delta-9 desaturation activity [4], the expression pattern of scd1 and scd5 is variable that scd1 is ubiquitous, but scd5 is mainly in the brain and pancreas even in different species [3, 6, 11], implying that the regulation of scd1 and scd5 expression and biological function may be distinct. The promoter region of scd1 contains many consensus binding sites for numerous transcription factors, for example, SREBP1, LXR, PPARα, C/EBP-α, NF-1, NF-Y, and Sp1 [13]. However, it is unclear whether scd5 contains similar or completely different consensus binding sites with scd1. Meanwhile, it is completely unknown that the 3′-UTR of scd1 and scd5 that may also contain similar or different target sites of microRNAs regulating their expression.
Therefore, to address the above questions, we compared the sequence characteristics of scd paralogs and then reconstructed the phylogenetic trees of scd genes in eukaryote species to determine the evolutionary history of scd genes. We used the relative rate ratio test, branch-specific dN/dS ratio tests, and branch-site dN/dS ratio tests to analyze the evolutionary forces after gene duplication. Furthermore, we characterized the binding sites by transcript factors in the 5′-UTR and the target sites by microRNAs in the 3′-UTR of scd1 and scd5 genes to investigate the regulation mechanisms of both scd genes.
2. Material and Methods
2.1. SCD Homologs BLAST, Sequence Alignment, and Phylogenetic Analysis
SCD homologs were retrieved by key word “Stearoyl-CoA desaturase” from NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) and Ensemble genome database (http://asia.ensembl.org/index.html). In addition, the sequences of human SCD proteins were used to blast available genomes from NCBI GenBank and Ensemble database. Eventually, 73 scd nucleotide sequences from 39 representative eukaryote species were retrieved (see Table S1 in the Supplementart Material available online at http://dx.doi.org/10.1155/2013/856521). Sequence alignment of 73 scd nucleotides was performed with MegAlign implemented in DNAStar 6.0 software package (DNASTAR, Madison, USA) and then was confirmed visually by BioEdit 7.0.9 [14]. The ambiguous regions of alignment were discarded and eventually 720 nucleotide bases were obtained.
Phylogenetic tree was reconstructed based on the full alignment of 73 sequences by using Maximum Likelihood (ML) analysis in PHYML [15] and approximately Maximum Likelihood (ML) analysis in FastTree 2.1.3 [16]. The yeast scd ortholog, ole1, was used as the outgroup to root the tree. For ML analysis, supports for nodes among branches were evaluated using nonparametric bootstrapping [17] with 1000 bootstrap replications. For FastTree 2 analysis, a heuristics search strategy was employed with an estimated rate of evolution for each site (the “CAT” approximation), minimum-evolution subtree-pruning regrafting (SPRs), and maximum-likelihood nearest-neighbor interchanges (NNIs). The local support values were provided based on the Shimodaira-Hasegawa (SH) test [18, 19].
To evaluate the evolutionary conservation of the SCD1 and SCD5, the amino acid sequences of SCD1 and SCD5 of 11 model organisms including human, rhesus monkey, mouse, rat, tree shrew, zebrafish, Drosophila melanogaster, and C. elegans were retrieved and then aligned using Muscle (http://www.ebi.ac.uk/Tools/msa/muscle/), followed by manual adjustment with BioEdit 7.0.9 [14]. Additionally, a Neighbouring-Joining (NJ) tree was reconstructed with the amino acid sequences of SCDs from human, rhesus monkey, mouse, rat, tree shrew, and C. elegans by MEGA 4.0 [20] using amino acid p-distance model. Support for nodes among branches was evaluated using nonparametric bootstrapping [17] with 1000 bootstrap replications.
2.2. Regulation Prediction in 3′-UTR and 5′-UTR of scd Genes
Searching for the transcription factor-binding sites (TFBS) in the 5′-UTR of scd genes was carried out based on the positive effectors of transcription in the promoter region of scd1 from human, mouse, and chicken [13]. The length of 5′-UTR for this analysis was about 2500 bp upstream of the translational start sites of scd5 gene. The TFBSs were estimated by Match 1.0 with the TRANSFAC database v. 6.0 and Promo with TRANSFAC database v. 8.3 [21, 22]. The cut-off parameters were set as 0.75 for the core similarity and 0.85 for matrix similarity in Match 1.0 analysis. In Promo analysis, the species of factor and site were only constrained to animals. MultiSearchSite was used to search for binding sites sharing 15% maximum matrix dissimilarity rate in the promoter sequences of human, rhesus monkey, tree shrew, and chicken.
The microRNA targets sites in the 3′-UTR region of scd genes were predicted by using TargetScan release 6.2 (http://www.targetscan.org/). The lengths of the 3′-UTR region of scd1 and scd5 genes were about 4000 bp and 1790 bp, respectively. Only the broadly conserved sites for miRNA families among vertebrates were considered in this study. The predicted miRNAs were then introduced to the miR2Disease Base (http://www.mir2disease.org/) to establish the relationship between miRNAs and human diseases.
2.3. Relative Rate Test
The substitution rates of the scd genes were compared among different paralogs inferred from the phylogenetic tree using the relative rate test implemented in RRTree [23]. Three phylogroups were defined as vertebrate scd1, vertebrate scd5, and invertebrate scd gene. The yeast ole1 gene was used as outgroup.
2.4. Selective Pattern Analysis
The ratio of synonymous substitution to nonsynonymous substitution (ω = dN/dS) is a good indicator to estimate the evolutionary selective pressure of protein-coding regions. The ratio of ω = 1, <1, and >1 indicates a neutral selection, a purifying selection, and positive selection, respectively. The ω ratios between pairwise sequences were estimated following the method of Yang and Nielsen [24].
The codon-substitution models were implemented using CODEML in PAML package [25]. All models fixed the transition/transversion rate and codon usage biases (F3×4). To determine the evolutionary selective patterns of two scd genes, the branch-specific model was applied to the data, which assumed that the foreground clade had different ratios from the background clade [26]. In model B, scd1 and scd5 genes were set as the foreground clade. In model C, scd1, scd5, and the invertebrate SCD homologs were set as three clades. In addition, we also determined the sites evolving under positive selection in a specific clade with the branch-site model that allows variation in ω across individual codons on a specific lineage [27]. We applied the modified branch-site model A (test 1 and test 2) [27], which permits variation of the ω ratio both among sites and lineages. The likelihood ratio tests (LRTs) were constructed to compare the fit to the data of two nested models. The significant difference between two models was evaluated by calculating twice the log-likelihood difference, and followed an χ 2 distribution with the number degree of freedom equal to the difference in the number of free parameters.
3. Results
3.1. The Sequence Characteristics of SCD Orthologs
In human, the size of scd1 gene is about 17 kb and 170 kb for scd5 gene. Though the remarkably different sizes of two scd genes, the full lengths of both scd encoded proteins are very close that SCD1 has 359 aa and SCD5 330 aa (Figure 1). To determine the conservation of SCD orthologs, we first investigated the sequence characteristics of SCD proteins. Comparison of the SCD amino acid sequences from several animal organisms revealed that the three histidine motifs HRLWSH, HRAHH, and HNYHH that exist in human SCD are also highly conserved in all alignments (Figure 1). But, the three histidine motifs also display minor changes in some organisms. For example, HRLWAH exists in C. elegans FAT-5 and Drospholia SCD genes; HNFHH in C. elegans FAT-6 and FAT-7 (Figure 1). The four transmembrane hydrophobic domains marked underline are also conserved in all alignments (Figure 1). Then, we investigated the sizes and order of exons of scd genes in several representative eukaryote organisms (Figure 2). Most of the scd1 genes (e.g., chicken, human, etc.) are consisted of 6 exons. However, some vertebrate scd1 genes only have 5 exons, like platypus and zebrafish. All of the scd5 genes are consisted of 5 exons. Very interestingly, except the exon 1, the sizes and order of other exons (exon 2 (131), exon 3 (206), exon 4 (233), and exon 5 (191)) of scd5 genes were not only separately equal but also very similar to the sizes and order of the third to sixth exons of scd1 genes (exon 3 (131), exon 4 (206), exon 5 (233) and exon 6 (200)) in eukaryote organisms (Figures 2(a) and 2(b)).
Figure 1.
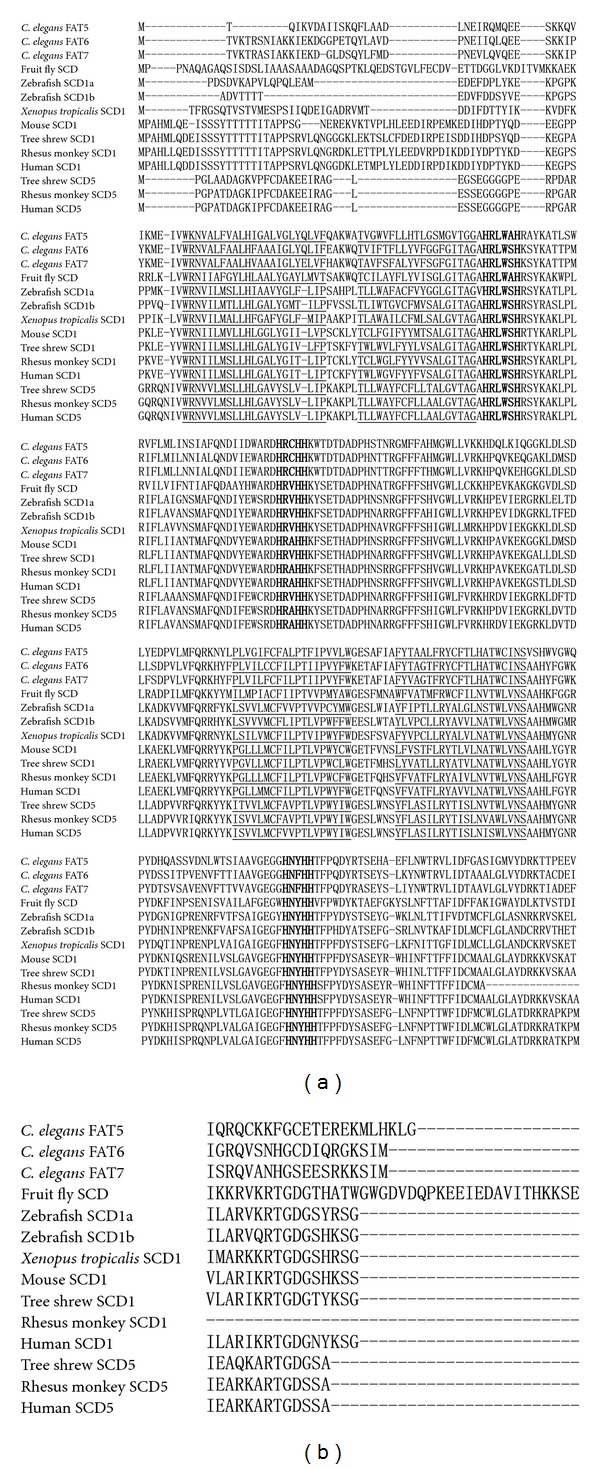
Alignment of inferred SCD protein sequences from 8 model animals. The three histidine motifs are in bold, and the four transmembrane hydrophobic domains were marked underline.
Figure 2.
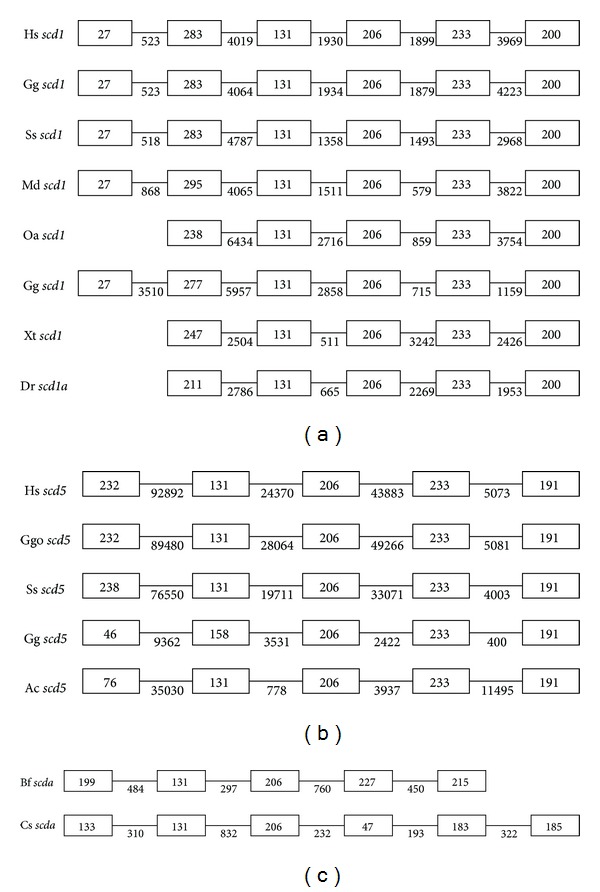
The exons size changes of scd genes. (a) Exon size changes of scd1 gene in vertebrates. (b) Exon size changes of scd5 gene in vertebrates. (c) Exon size changes of scd genes in invertebrates. Numbers in box represent the sizes of exons and numbers under bars represent the sizes of introns. Hs, Homo sapiens; Ggo, Gorilla gorilla; Ss, Sus scrofa; Md, Monodelphis domestica; Oa, Ornithorhynchus anatinus; Gg, Gallus gallus; Xt, Xenopus tropicalis; Dr, Danio rerio; Ac, Anolis carolinensis; Bf, Branchiostoma floridae; Cs, Ciona savignyi.
3.2. Phylogenetic Inference of scd Gene Lineages
The phylogenetic tree of scd genes based on the 73 nucleotide sequences from 39 species is shown in Figure 3(a) (TreeBASE Accession URL http://purl.org/phylo/treebase/phylows/study/TB2:S14739). The scd orthologs of invertebrate species are placed at the base of the tree using scd ortholog yeast ole1 as outgroup. The C. elegans fat-5, fat-6, and fat-7 are placed at the most basal position of the tree. In addition, the scd1a, scd1b,and scd1c from Ciona savignyi and amphioxus Branchiostoma floridae are just located out of the vertebrate lineages. Intriguingly, the scd genes in vertebrates are split into two lineages with strong support (support value = 99%) according to the scd gene classification, suggesting that independent duplication events occurred in vertebrates after separation from invertebrates during evolution. In teleost fish, two scd1 paralogs were also diverged into two independent clades with high support, but the scd5 gene was lost. This evolutionary pattern might suggest that the teleost fish scd experienced an ancient gene duplication event [12] or the genome duplication [6].
Figure 3.
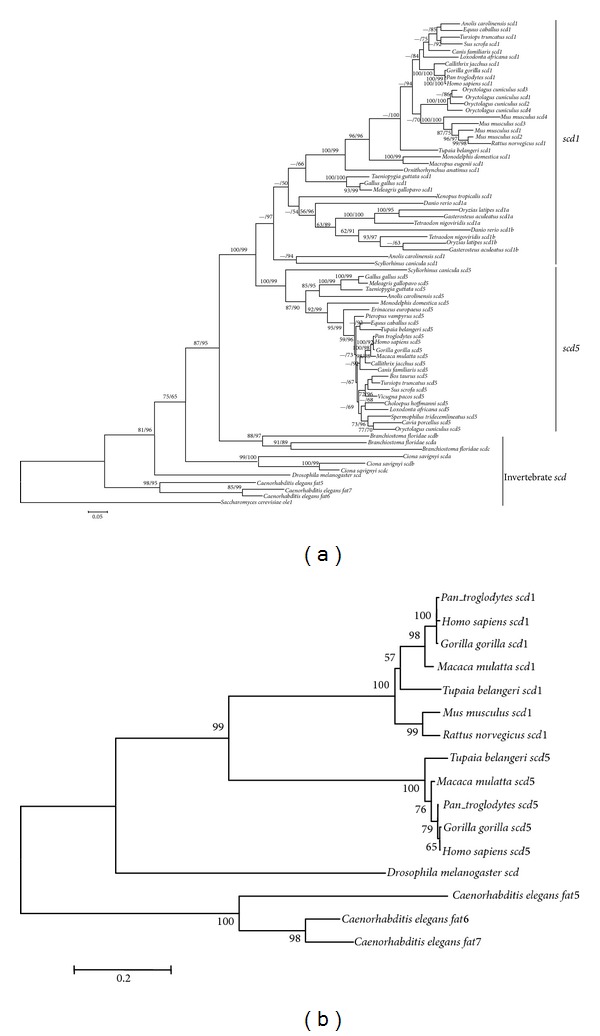
Phylogenetic trees of eukaryote scd isoforms. (a) Phylogenetic trees based on the nucleotide sequence data. The numbers on nodes indicated the support values, the former number was calculated using PHYML, and the latter number was calculated by FastTree 2.1.3. If bootstrap values were less than 50%, they were defaulted. Trees were rooted by yeast ole1 gene. (b) Phylogenetic trees based on the amino acid sequences of 9 model animals with MEGA 4.0. The numbers on nodes indicated the support values. If bootstrap values were less than 50%, they were defaulted. Trees were rooted by C. elegans SCD paralogs.
3.3. Evolutionary Rates and Selective Pattern in scd Genes
To determine whether the paralogs of scd evolve at the similar rates, the relative rate analysis was performed among scd gene and in which the invertebrate scd genes, vertebrate scd1 and scd5 genes were separated into 3 groups using the yeast ole1 as outgroup. The analysis revealed that the scd genes were evolved at the similar evolutionary rate (P < 0.05).
To address the selective constraint pattern within scd genes, the ratios of nonsynonymous (dn) to synonymous (ds) were estimated between two sequences. The analysis suggested that nearly almost pairwise comparisons of scd genes had a ω < 1, indicating a strong purifying selection. Intriguingly, the pairwise comparisons among scd1 genes of human, gorilla, and chimpanzee had a ω = ∞, which might result from that the nonsynonymous substitution occurred while the synonymous substitution did not in scd1 sequences probably because of the very close relationships among these three species.
The selective pattern of scd genes was further performed using the condon-based maximum likelihood analysis (Table 1). In this analysis, the yeast ole1 was excluded. The estimated one ratio of ω 0 (0.08684) over all sites and branches from the scd genes was substantially smaller than 1, suggesting a strong purifying selection (Table 1). In the branch-specific models, Model B assumes scd1 gene and scd5 gene as the foreground clades, respectively. In this model the estimated ω value was 0.09207 for scd1 gene and 0.07951 for the background clades. The estimated ω value was 0.06146 for scd5 gene and 0.09735 for the background clades. The LRT test suggested that the two-ratio model was not fit for the data better than the one-ratio model for scd1 gene (P > 0.05) but fit better for scd5 gene (P < 0.001). Under Model C, ω estimates for scd1, scd5, and invertebrate scd gene were 0.06140, 0.09198, and 0.11788, respectively. The LRT test indicated that Model C was significantly better fit for the data than did the one ratio model (M0) (P < 0.001).
Table 1.
Selective patterns of scd genes estimated in CODEML.
| Model | ln L | Parameters estimates | 2ΔL | Positively selected sites |
|---|---|---|---|---|
| Branch-specific models | ||||
| M0 | −20340.727636 | ω = 0.08684 | ||
| Model B | ||||
| Scd1 two ratio | −20339.045042 | ω 0 = 0.09207, ω 1 = 0.07951 | 3.365188 | |
| Scd5 two ratio | −20318.455800 | ω 0 = 0.06146, ω 1 = 0.09735 | 44.542672### | |
| Model C Three ratio |
−20315.506084 |
ω
scd1 = 0.06140 ω scd5 = 0.09198 ω invertebrate scd = 0.11788 |
50.443104### | |
|
| ||||
| Branch-site models | ||||
| Scd1 | ||||
| Model A1 | −20079.883939 |
ω
0 = 0.06891, ω
1=1, ω
2 = 1 P 0 = 0.85305, P 1 = 0.04177 |
||
| M1a | −20178.290653 |
ω
0 = 0.07950, ω
1 = 1 P 0 = 0.91928, P 1 = 0.08072 |
196.813428### | |
| Model A | −20079.883939 |
ω
0 = 0.06891, ω
1 = 1, ω
2 = 1 P 0 = 0.85305, P 1 = 0.04177 |
0 | 108L**, 109F**, 201A**, 206S, 212K**, 247Y**, 254A, 255I*, 276K**, 289V*, 315P, 330Y, 339A |
| Scd5 | ||||
| Model A1 | −20129.099054 |
ω
0 = 0.07500, ω
1 = 1, ω
2 = 1 P 0 = 0.88996, P 1 = 0.05968 |
||
| M1a | −20168.513281 |
ω
0 = 0.07951, ω
1 = 1 P 0 = 0.91900, P 1 = 0.08100 |
78.828454### | |
| Model A | −20129.099054 |
ω
0 = 0.07500, ω
1 = 1, ω
2 = 1 P 0 = 0.88996, P 1 = 0.05968 |
0 | 157A**, 194P**, 215M**, 223P, 230I, 338A** |
### P < 0.001; ##0.001 < P < 0.01;
**P > 0.99; *P > 0.95.
In addition, we determined the amino acid sites under positive selection at SCD1 and SCD5 clades on the phylogeny using the branch-site model. In this model, the SCD1 and SCD5 clades were assumed as the foreground clades, respectively. As seen in Table 1, the results of test 1 analysis designated several amino acid sites under the relaxed selection (P < 0.001) in both the scd1 and scd5 genes. However, none of the LRT test for scd genes was significant in test 2 analysis, indicating that the null hypothesis of the test 2 could not be rejected in both of the scd genes, and none of the two scd genes was underrelaxed selective constraint or under positive selection. Thus, we did not find any evidence for positive selection in both of the scd genes under these analyses.
3.4. The Regulation Analysis of scd Genes
Numerous transcription factors, for example, SREBP1, LXR, PPARα, C/EBP-α, NF-1, NF-Y, and Sp1, have been revealed to bind to the scd1 promotor region [13]. The consensus binding sites for the SREBP1, PPAR-α, C/EBP-α, NF-1, and NF-Y were known to mediate the insulin response, whereas the binding sites for Sp1 and AP1 were known to be the leptin response element. To determine whether these transcription factors also bind to scd5 promotor region, the transcription binding site prediction was performed by using TRANSFAC and Promo. C/EBP-α, AP1, SP1, NF-1, NF-Y, and SREBP1 were detected at the promoter region of scd5 gene of four species (Table 2). But SREBP1 was not detected in the promoter region of scd5 gene in other mammals (results not shown). Because SREBPs are weak transcriptional activators on their own, they interact with their target promoters in cooperation with additional regulators, most commonly including one or both of the transcription factors NFY and SP1 [28–30], and their binding sites were possessed a high degree of overlap [31]. We also detected the binding sites of NFY and SP1 near the binding site of SREBP1 in human. In this analysis, we detected the binding site of PPARα by Promo, but not by TRANSFAC. However, the binding site of PPARα detected in scd5 gene was different from that of in scd1 gene (Table 2). Though most of the transcription factor binding sites in scd1 gene could be detected in scd5 gene, the regulation of these transcription factors on scd5 gene still needs further experimental verification.
Table 2.
Transcription factor binding sites predicted at the 5′UTR of hscd5.
| Transcription factor | Binding sites | Position (hscd1)$ | Position (hscd5) |
|---|---|---|---|
| C/EBPα | GMAAA | −219 | −1061, −1648 |
| AP1 | TGACC | −204, −271 | −580, −643 |
| SP1 | GGCGG | −304, −314, −551 | −286, −946 |
| NF-Y | CCAAT | −458, −501 | −397, −976 |
| NF-1 | TTGGC | −459, −502 | −395 |
| SREBP1 | TCACC | −517 | −892* |
| PPARα | AAAG/GGTCA | −1186 | −579# |
| T3R | GGTCA | −2228 | −1223, −2245 |
T3R: tri-iodothyronine receptor; AP1: activator protein 1; NF-1/Y: nuclear factor 1/Y; SREBP1: sterol regulatory element binding protein; PPARα: peroxisome proliferator-activated receptor; C/EBPα: CAAT/enhancer binding protein.
$These transcription factor binding sites were from [64].
#Predicted by PROMO.
*Only found in human using TRANSFAC.
In order to compare the microRNAs regulation on scd genes, we predicted the microRNA target sites at the 3′-UTR region of scd1 and scd5 genes using TargetScan. The lengths of 3′-UTR region of scd1 and scd5 gene were about 4000 bp and 1790 bp, respectively (Figure 4). Within the 3′-UTR region of scd1 gene, 8 conserved sites of microRNA families were predicted among vertebrates and 5 conserved sites were predicted among mammals (Figure 4(a)). Among these 13 microRNA families, almost all of them were closely associated with the cancers, for example, the miR-128, Let 7, miR-206, and miR-124a linked to breast cancer [32–35], hepatocellular carcinoma [36–38], and pancreatic cancer [39, 40]. In addition, plenty of evidence has described that the scd1 acted as a potential target to prevent or treat metabolic syndrome. Among these microRNA families, several microRNAs were associated with the nonalcoholic fatty liver disease (NAFLD), type 2 diabetes, and diabetic nephropathy; for example, miR-429 and Let 7cde were closely related to NAFLD [41, 42]; miR-181a related to diabetes [43]; miR-216a related to diabetic nephropathy [44]. At the 3′-UTR region of scd5 gene, 5 conserved sites of microRNA families were predicted among vertebrates and 2 conserved sites were predicted among mammals (Figure 4(b)). All of these microRNA families were closely associated with cancers. scd5 gene was mainly expressed in brain and pancreas. Several microRNAs were associated with the neurological disorder and pancreatic cancers. miR-106a was associated with autism [45], miR-17 with glioma [46], miR-20b with schizophrenia [47]. miR-205, miR-221, miR-222, miR-17-5p, and miR-20a were associated with pancreatic cancers [39, 48–50]. Only 2 microRNAs, miR-200ab and miR-17, were linked to NAFLD [41, 42].
Figure 4.
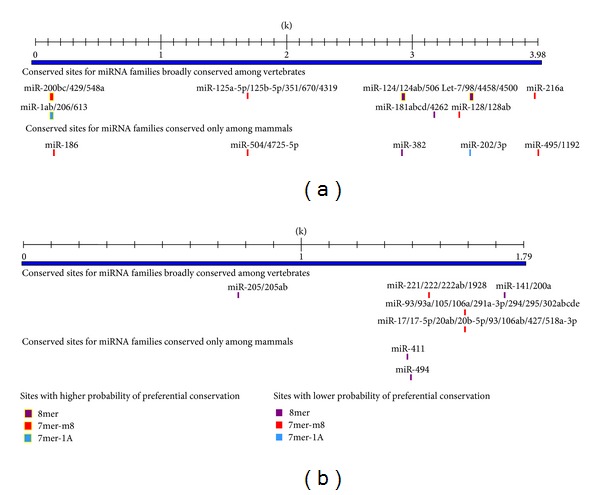
The target sites for miRNA families conserved among mammals and vertebrates at the 3′-UTR region of human scd1 gene (a) and scd5 gene (b). The sites with different probability of preferential conservation were marked in different colors. The target sites sharing among miRNAs separated by slash were marked with same color.
4. Discussion
The phylogenetic trees show that homologs of scd gene from invertebrates were all placed at the basal position of the tree, whereas the scd genes in vertebrates were diverged into two independently duplicated genes early in vertebrate evolution with strong support, in which all scd1 genes form a distinct clade and all scd5 genes clustered into another clade (Figures 3(a) and 3(b)). Our phylogenetic analysis was consistent with the previous studies by Castro et al. [6] and Lengi and Corl [11]. This pattern of duplication might be resulted from part of the two rounds of genome duplication in vertebrate ancestry [6].
When a gene duplication event occurs, the duplicated genes have redundant functions. The fate of the duplicated genes might be loss of function, gaining a new function, or subfunctionalization [51]. Subfunctionalization occurred when both duplicates can be stably maintained in the genome [52]. The division of gene expression after gene duplication appears to be a general form of subfunctionalization [53, 54]. In this model, after gene duplication, complementary degenerate mutations are fixed randomly underrelaxed functional constraints [55]. Previous studies suggested that both scd1 and scd5 encode the same functional delta-9 desaturase and are localized on endoplasmic reticulum (ER) membrane [3, 56]. However, the scd1 gene expressed ubiquitous, and scd5 gene expressed mainly in brain in different species [3, 6, 11]. We inferred that the evolution of scd genes might be a division of gene expression subsequent to gene duplication. This pattern was supported by the evolutionary forces behind the expression division of duplicate genes. The relative rate test suggested that the two duplicated scd genes evolved at the similar rate. The selective constraints analysis suggested that the scd1 and scd5 were both under strict purifying selection (Table 1), which was consistent with the conserved delta-9 desaturase of both scd genes. Intriguingly, in the branch-site analysis, we detected that some sites within scd1 and scd5 were underrelaxed selective pressure. These sites might be resulted from the random fixation of the complementary degenerate mutations that were underrelaxed functional constraints.
Though both of scd1 and scd5 encoded delta-9 desaturase, producing a palmitoleic acid (16:1n7) and oleic acid (18:1n9) [3, 56], they expressed diversely in the physiological process. Previous studies had proposed that scd1 were associated with a variety of diseases including cancers, type 2 diabetes, and cardiovascular disorders [13], whereas scd5 might act a potential role for maintaining the optimum levels of oleic acid in brain development and physiological activities [3, 57]. Castro et al. proposed that the major distinction between scd5 and scd1 would be at the regulatory level, in which scd1 gene expression was mainly modulated at the transcriptional level by a wide variety of hormones and nutrients, whereas scd5 was not responsive to external inputs like food sources [6]. In this study, we predicted the transcription factor binding sites at the 5′-UTR region and the miRNA target sites at the 3′-UTR region of human scd5 gene. The transcription factor binding sites detected in scd1 gene [13] could also be detected in scd5 gene. However, the SREBP1 binding site only presents in human scd5 gene, but not in other mammals, for example, rhesus monkey, pig and others. This might be that the prediction of transcription factor (TF) binding sites was based on known TF binding sites so that some new TF binding sites can not be detected. Recent studies have suggested that SREBP1 regulates the expression of scd5 in primary cultures of human skeletal muscle cells [58], or directly binding to the promoter region of scd5 in bovine [59]. In contrast, a study on human hepatocyte cell line suggested that SREBP1 only binds to the scd1 gene, but not to scd5 gene [31]. This discrepancy might be the distinct expression of scd5 gene in different species or tissues. From our prediction, we conclude that the TF binding sites predicted in scd5 gene were very similar to these of scd1 gene, suggesting that the regulators may also be similar between two scd genes. Certainly, these TF predictions need further experimental verification.
miRNAs regulation is another gene regulatory mechanism in posttranscriptional regulation. Gu et al. estimated the time of vertebrate miRNA duplication events and suggested that gene/genome duplications in the early stage of vertebrates may expand the protein-encoding genes and miRNAs simultaneously [60]. Gene duplication events, followed by subfunctionalization and neofunctionalization processes, are considered to be a major source for emergence of novel miRNA genes [61]. In this study, the lengths of 3′-UTR of scd1 and scd5 gene are about 4000 bp and 1790 bp, respectively (Figure 4). A previous study suggested that genes with longer 3′-UTRs are regulated by more distinct types of miRNAs [62]. In our analysis, 13 miRNAs targeting sites are detected in the 3′-UTR of scd1 gene, while 7 miRNAs targeting sites are detected in the 3′-UTR of scd5 gene. Additionally, the length changes of 3′-UTRs in these two scd genes might suggest a differentiation of the regulatory mechanisms. miRNAs predicted to target the 3′-UTR region of scd1 gene are associated with breast cancers, hepatocellular carcinoma, and metabolic syndromes such as diabetes, NAFLD. However, most of the miRNAs predicted to target the 3′-UTR region of scd5 gene are related to the neurogenic disease and pancreatic cancer; and only 2 microRNAs are associated with the NAFLD. This regulatory pattern might be due to the high expression of scd5 gene in brain and pancreas [3]. Additionally, a recent study has reported that the scd5 gene plays a key role in the regulation of the neuronal cell proliferation and differentiation [56]. These results might indicate that the expression of scd5 is implicated in brain development and physiological activity.
In addition, we also investigated the size and order of exons of scd genes. We found that the scd1 gene has an extra exon (exon1) compared to scd5 gene (Figure 2). The first 45 amino acids of SCD1 were highly different from those of SCD5 (Figure 1). Though there is no histidine domain and transmembrane domain exists in this part of SCD1, about 30 residues constitute a motif responsible for the rapid degradation of SCD [63]. This result indicated that the degradation of two SCD might be very different. However, due to no information on the degradation of SCD5, the evolutionary changes of regulation on both scd genes and SCD proteins still need further investigation.
5. Conclusion
In summary, this study of evolutionary pattern of scd genes showed that scd1 and scd5 genes emerged due to the duplication event as well as that they may play different roles. We also detected that the scd genes were evolved at the similar rate and were under strictly purifying selection, consistent with the conserved function of delta-9 desaturase of both SCD. Furthermore, our study revealed several potentially adaptive amino acid changes, which might be resulted from the random fixation of the complementary degenerate mutations underrelaxed functional constraints. The prediction of transcriptional factor binding sites at the 5′-UTR and miRNAs at 3′-UTR of scd genes suggested that the regulators of scd5 may be different from scd1 gene, supportting the differentiation at the regulatory levels between scd5 and scd1. These findings increase the current knowledge of evolutionary patterns and function of scd genes in eukaryote. Yet, further experimental investigations need to elucidate the regulation and function of scd genes, especially the scd5 gene.
Supplementary Material
Supplementary Table: Information of species, genes and sequence code used in this study.
Conflict of Interests
All authors declared no conflict of interests.
Authors' Contribution
Xiaoyun Wu and Xiaoju Zou contributed equally to this paper.
Acknowledgments
The authors thank the anonymous referees for their constructive and helpful comments and suggestions. This work was supported by the Innovative Program of the Chinese Academy of Sciences (KSCX2-EW-R-12 and KSCX2-EW-J-23) to Bin Liang, the National 863 Project of China (2012AA021801 and 2012AA022402), the National Natural Science Foundation of China (Y301261041, U1202223, 31160216, and 31171134), and the West Light Foundation to Xiaoyun Wu from the Chinese Academy of Sciences, and State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences (GREKF13-03).
References
- 1.Heinemann FS, Ozols J. Stearoyl-CoA desaturase, a short-lived protein of endoplasmic reticulum with multiple control mechanisms. Prostaglandins Leukotrienes and Essential Fatty Acids. 2003;68(2):123–133. doi: 10.1016/s0952-3278(02)00262-4. [DOI] [PubMed] [Google Scholar]
- 2.Strittmatter P, Spatz L, Corcoran D. Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(11):4565–4569. doi: 10.1073/pnas.71.11.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Yu L, Schmidt RE, et al. Characterization of HSCD5, a novel human stearoyl-CoA desaturase unique to primates. Biochemical and Biophysical Research Communications. 2005;332(3):735–742. doi: 10.1016/j.bbrc.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Paton CM, Ntambi JM. Biochemical and physiological function of stearoyl-CoA desaturase. American Journal of Physiology—Endocrinology and Metabolism. 2009;297(1):E28–E37. doi: 10.1152/ajpendo.90897.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flowers MT, Ntambi JM. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Current Opinion in Lipidology. 2008;19(3):248–256. doi: 10.1097/MOL.0b013e3282f9b54d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castro LFC, Wilson JM, Gonçalves O, Galante-Oliveira S, Rocha E, Cunha I. The evolutionary history of the stearoyl-CoA desaturase gene family in vertebrates. BMC Evolutionary Biology. 2011;11(1, article 132) doi: 10.1186/1471-2148-11-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watts JL, Browse J. A palmitoyl-CoA-specific Δ9 fatty acid desaturase from Caenorhabditis elegans. Biochemical and Biophysical Research Communications. 2000;272(1):263–269. doi: 10.1006/bbrc.2000.2772. [DOI] [PubMed] [Google Scholar]
- 8.Brock TJ, Browse J, Watts JL. Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genetics. 2006;2(7, article e108) doi: 10.1371/journal.pgen.0020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brock TJ, Browse J, Watts JL. Fatty acid desaturation and the regulation of adiposity in Caenorhabditis elegans. Genetics. 2007;176(2):865–875. doi: 10.1534/genetics.107.071860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stukey JE, McDonough VM, Martin CE. Isolation and characterization of OLE1, a gene affecting fatty acid desaturation from Saccharomyces cerevisiae. The Journal of Biological Chemistry. 1989;264(28):16537–16544. [PubMed] [Google Scholar]
- 11.Lengi AJ, Corl BA. Comparison of pig, sheep and chicken SCD5 homologs: evidence for an early gene duplication event. Comparative Biochemistry and Physiology B. 2008;150(4):440–446. doi: 10.1016/j.cbpb.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Evans H, De Tomaso T, Quail M, et al. Ancient and modern duplication events and the evolution of stearoyl-CoA desaturases in teleost fishes. Physiological Genomics. 2008;35(1):18–29. doi: 10.1152/physiolgenomics.90266.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauvoisin D, Mounier C. Hormonal and nutritional regulation of SCD1 gene expression. Biochimie. 2011;93(1):78–86. doi: 10.1016/j.biochi.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Hall TA. Bioedit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/Nt. Nucleic Acids Symposium Series. 1999;41:95–98. [Google Scholar]
- 15.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 16.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5(3) doi: 10.1371/journal.pone.0009490.e9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenstein J. Confidence limits on phylogenies: an approach using bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 18.Guindon S, Delsuc F, Dufayard J-F, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods in Molecular Biology. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 19.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 20.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 21.Messeguer X, Escudero R, Farré D, Núñez O, Martínez J, Albà MM. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics. 2002;18(2):333–334. doi: 10.1093/bioinformatics/18.2.333. [DOI] [PubMed] [Google Scholar]
- 22.Farré D, Roset R, Huerta M, et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Research. 2003;31(13):3651–3653. doi: 10.1093/nar/gkg605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson-Rechavi M, Huchon D. RRTree: relative-rate tests between groups of sequences on a phylogenetic tree. Bioinformatics. 2000;16(3):296–297. doi: 10.1093/bioinformatics/16.3.296. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution. 2000;17(1):32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z. Inference of selection from multiple species alignments. Current Opinion in Genetics and Development. 2002;12(6):688–694. doi: 10.1016/s0959-437x(02)00348-9. [DOI] [PubMed] [Google Scholar]
- 27.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology and Evolution. 2005;22(12):2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 28.Sanchez HB, Yieh L, Osborne TF. Cooperation by sterol regulatory element-binding protein and Sp1 in sterol regulation of low density lipoprotein receptor gene. The Journal of Biological Chemistry. 1995;270(3):1161–1169. doi: 10.1074/jbc.270.3.1161. [DOI] [PubMed] [Google Scholar]
- 29.Jackson SM, Ericsson J, Mantovani R, Edwards PA. Synergistic activation of transcription by nuclear factor Y and sterol regulatory element binding protein. Journal of Lipid Research. 1998;39(4):767–776. [PubMed] [Google Scholar]
- 30.Dooley KA, Millinder S, Osborne TF. Sterol regulation of 3-hydroxy-3-methylglutaryl-coenzyme A synthase gene through a direct interaction between sterol regulatory element binding protein and the trimeric CCAAT-binding factor/nuclear factor Y. The Journal of Biological Chemistry. 1998;273(3):1349–1356. doi: 10.1074/jbc.273.3.1349. [DOI] [PubMed] [Google Scholar]
- 31.Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genetics. 2008;4(7) doi: 10.1371/journal.pgen.1000133.e1000133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foekens JA, Sieuwerts AM, Smid M, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(35):13021–13026. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iorio MV, Ferracin M, Liu C-G, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Research. 2005;65(16):7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 34.Kondo N, Toyama T, Sugiura H, Fujii Y, Yamashita H. MiR-206 expression is down-regulated in estrogen receptor α-positive human breast cancer. Cancer Research. 2008;68(13):5004–5008. doi: 10.1158/0008-5472.CAN-08-0180. [DOI] [PubMed] [Google Scholar]
- 35.Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Research. 2007;67(4):1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 36.Huang X-H, Wang Q, Chen J-S, et al. Bead-based microarray analysis of microRNA expression in hepatocellular carcinoma: MiR-338 is downregulated. Hepatology Research. 2009;39(8):786–794. doi: 10.1111/j.1872-034X.2009.00502.x. [DOI] [PubMed] [Google Scholar]
- 37.Gramantieri L, Ferracin M, Fornari F, et al. Cyclin G1 is a target of miR-122a, a MicroRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Research. 2007;67(13):6092–6099. doi: 10.1158/0008-5472.CAN-06-4607. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Y, Jia H-L, Zhou H-J, et al. Identification of metastasis-related microRNAs of hepatocellular carcinoma in hepatocellular carcinoma cell lines by quantitative real time PCR. Chinese Journal of Hepatology. 2009;17(7):526–530. [PubMed] [Google Scholar]
- 39.Volinia S, Calin GA, Liu CG, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eun JL, Gusev Y, Jiang J, et al. Expression profiling identifies microRNA signature in pancreatic cancer. International Journal of Cancer. 2007;120(5):1046–1054. doi: 10.1002/ijc.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng L, Lv G-C, Sheng J, Yang Y-D. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-α expression, a novel mechanism for the pathogenesis of NAFLD. Journal of Gastroenterology and Hepatology. 2010;25(1):156–163. doi: 10.1111/j.1440-1746.2009.05949.x. [DOI] [PubMed] [Google Scholar]
- 42.Alisi A, Da Sacco L, Bruscalupi G, et al. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Laboratory Investigation. 2011;91(2):283–293. doi: 10.1038/labinvest.2010.166. [DOI] [PubMed] [Google Scholar]
- 43.Klöting N, Berthold S, Kovacs P, et al. MicroRNA expression in human omental and subcutaneous adipose tissue. PLoS ONE. 2009;4(3) doi: 10.1371/journal.pone.0004699.e4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato M, Putta S, Wang M, et al. TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nature Cell Biology. 2009;11(7):881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abu-Elneel K, Liu T, Gazzaniga FS, et al. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics. 2008;9(3):153–161. doi: 10.1007/s10048-008-0133-5. [DOI] [PubMed] [Google Scholar]
- 46.Malzkorn B, Wolter M, Liesenberg F, et al. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathology. 2010;20(3):539–550. doi: 10.1111/j.1750-3639.2009.00328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perkins DO, Jeffries CD, Jarskog LF, et al. MicroRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biology. 2007;8(2, article R27) doi: 10.1186/gb-2007-8-2-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bloomston M, Frankel WL, Petrocca F, et al. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. The Journal of the American Medical Association. 2007;297(17):1901–1908. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 49.Park J-K, Lee EJ, Esau C, Schmittgen TD. Antisense inhibition of microRNA-21 or -221 arrests cell cycle, induces apoptosis, and sensitizes the effects of gemcitabine in pancreatic adenocarcinoma. Pancreas. 2009;38(7):e190–e199. doi: 10.1097/MPA.0b013e3181ba82e1. [DOI] [PubMed] [Google Scholar]
- 50.Greither T, Grochola LF, Udelnow A, Lautenschläger C, Würl P, Taubert H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. International Journal of Cancer. 2010;126(1):73–80. doi: 10.1002/ijc.24687. [DOI] [PubMed] [Google Scholar]
- 51.Zhang JZ. Evolution by gene duplication: an update. Trends in Ecology and Evolution. 2003;18(6):292–298. [Google Scholar]
- 52.Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388(6638):167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- 53.Wagner A. Decoupled evolution of coding region and mRNA expression patterns after gene duplication: implications for the neutralist-selectionist debate. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6579–6584. doi: 10.1073/pnas.110147097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu Z, Nicolae D, Lu HH-S, Li W-H. Rapid divergence in expression between duplicate genes inferred from microarray data. Trends in Genetics. 2002;18(12):609–613. doi: 10.1016/s0168-9525(02)02837-8. [DOI] [PubMed] [Google Scholar]
- 55.Force A, Lynch M, Pickett FB, Amores A, Yan Y-L, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151(4):1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinner DI, Kim GJ, Henderson GC, Igal RA. StearoylCoA desaturase-5: a novel regulator of neuronal cell proliferation and differentiation. PLoS One. 2012;7(6) doi: 10.1371/journal.pone.0039787.e39787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lengi AJ, Corl BA. Identification and characterization of a novel bovine stearoyl-CoA desaturase isoform with homology to human SCD5. Lipids. 2007;42(6):499–508. doi: 10.1007/s11745-007-3056-2. [DOI] [PubMed] [Google Scholar]
- 58.Rome S, Lecomte V, Meugnier E, et al. Microarray analyses of SREBP-1a and SREBP-1c target genes identify new regulatory pathways in muscle. Physiological Genomics. 2008;34(3):327–337. doi: 10.1152/physiolgenomics.90211.2008. [DOI] [PubMed] [Google Scholar]
- 59.Lengi AJ, Corl BA. Regulation of the bovine SCD5 promoter by EGR2 and SREBP1. Biochemical and Biophysical Research Communications. 2012;421(2):375–379. doi: 10.1016/j.bbrc.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 60.Gu X, Su Z, Huang Y. Simultaneous expansions of micrornas and protein-coding genes by gene/genome duplications in early vertebrates. Journal of Experimental Zoology B. 2009;312(3):164–170. doi: 10.1002/jez.b.21273. [DOI] [PubMed] [Google Scholar]
- 61.Berezikov E. Evolution of microRNA diversity and regulation in animals. Nature Reviews Genetics. 2011;12(12):846–860. doi: 10.1038/nrg3079. [DOI] [PubMed] [Google Scholar]
- 62.Cheng C, Bhardwaj N, Gerstein M. The relationship between the evolution of microRNA targets and the length of their UTRs. BMC Genomics. 2009;10, article 431 doi: 10.1186/1471-2164-10-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mziaut H, Korza G, Ozols J. The N terminus of microsomal Δ9 stearoyl-CoA desaturase contains the sequence determinant for its rapid degradation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):8883–8888. doi: 10.1073/pnas.97.16.8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang L, Ge L, Tran T, Stenn K, Prouty SM. Isolation and characterization of the human stearoyl-CoA desaturase gene promoter: requirement of a conserved CCAAT cis-element. Biochemical Journal. 2001;357, part 1:183–193. doi: 10.1042/0264-6021:3570183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table: Information of species, genes and sequence code used in this study.