

Abstract
The C-P lyase complex in bacteria catalyzes the transformation of phosphonates to orthophosphate under conditions of phosphate starvation. The first committed step in the C-P lyase catalyzed reaction is the displacement of adenine from MgATP by phosphonate substrates to yield ribose-1-phosphonate-5-triphosphate (RPnTP). In the C-P lyase complex this reaction is catalyzed by the nucleosidase PhnI, and modulated by the addition of PhnG, PhnH and PhnL. Here we describe the synthesis of Immucillin-A triphosphate, a mimic of the transition state structure for the nucleosidase reaction catalyzed by PhnI. This compound inhibits PhnI with a dissociation constant of 20 nM at pH 7.5.
Phosphonates are ubiquitous organophosphorus compounds that contain a characteristic carbon-phosphorus (C-P) bond, which is chemically inert and hydrolytically stable. These compounds represent a growing class of antibiotics (fosfomycin) herbicides (glyphosate) and antiviral therapeutics (Adefovir).1,2 The metabolism of these and other organophosphonates by the microbiome of the human gut is not well understood.
Bacteria have evolved several strategies to degrade phosphonate substrates, which include phosphonatases, C-P lyase, and a novel oxidative pathway.3,4 Of these metabolic pathways, the C-P lyase complex is the most promiscuous in terms of substrate specificity.3,4 Most phosphonates are transformed to orthophosphate under conditions of phosphate starvation by this pathway. A recent investigation has led to the successful in vitro reconstitution of the C-P lyase complex from Escherichia coli using methylphosphonate (MPn) as a model phosphonate substrate.5,6 This pathway is illustrated in Scheme 1. In E. coli there are 14 proteins encoded by the C-P lyase operon (phnC-P). The genes phnG through phnP encode for proteins that are either catalytic or form an integral part of the C-P lyase complex. Based on gene knockout studies, it was determined that proteins PhnG through PhnM form the minimal catalytic components for the C-P lyase reaction, and that proteins PhnN through PhnP perform accessory catalytic functions.3,4,7
Scheme 1. The C-P lyase pathway of E. coli.

Recent studies have identified a marine archeon Nitrosopumilus maritimus as a potential biogenic source of methylphosphonate from global ocean surfaces.8 The methylphosphonate produced from these marine organisms is degraded to methane and orthophosphate in ocean surfaces by bacteria that possess the phn operon encoding the C-P lyase pathway.8 The global methane production from ocean surfaces is substantial and amounts to approximately 4% of the total methane budget worldwide.9 Methane is 20 times more potent as a green house gas than carbon dioxide.9 Inhibitors of the C-P lyase complex have not been identified. Such compounds will be very important as high resolution structural probes of the C-P lyase complex and potentially as lead compounds in the development of new antibiotics for those bacteria that can metabolize phosphonates in phosphate limited environments.
The first committed step catalyzed by the C-P lyase complex is the synthesis of ribose-1-phosphonate-5-triphosphate (RPnTP) that occurs via the displacement of adenine from MgATP by the phosphonate co-substrate. This nucleosidase-like reaction requires the presence of four proteins from the C-P lyase complex: PhnI, PhnG, PhnH, and PhnL.5 The catalytic machinery for this transformation is most likely localized to PhnI since in the absence of PhnG, PhnH, and PhnL, this enzyme will catalyze the attack of water on the anomeric carbon of the ribose moiety of MgATP to form ribose-5-triphosphate (RTP) and adenine.5 PhnG, PhnH and PhnL are absolutely critical for the formation of RPnTP, but the precise role of these proteins is not yet clear. PhnI can utilize guanosine-5′-triphosphate (GTP) and inosine-5′-triphosphate (ITP) as substrates with near equal catalytic efficiencies as ATP. However, adenosine-5′-diphosphate (ADP) and guanosine-5′-diphosphate (GDP) are much poorer substrates for PhnI. PhnI cannot utilize adenosine, guanosine, adenosine-5′-monophosphate (AMP) or guanosine-5′-monphosphate (GMP) as substrates.5 The reaction catalyzed by PhnI in the absence of PhnG, PhnH, and PhnL is presented in Scheme 2 using ATP as the substrate.
Scheme 2.

The reaction catalyzed by PhnI in the absence of PhnGHI.
PhnI is insoluble when expressed from E. coli without an affinity tag. This enzyme was therefore cloned, expressed, and purified as an N-terminal glutathione S-transferase (GST) fusion protein.5 The addition of dithiothreitol during purification and kinetic assays was essential for measuring the catalytic activities of PhnI. The nucleosidase reaction catalyzed by PhnI with MgATP and water can be performed without the in situ cleavage of the GST protein to yield RTP. However, the formation of RPnTP, with PhnG, PhnH and PhnL from MgATP and methyl phosphonate, requires the in situ cleavage of the GST-tag. Previous studies have shown that the formation of RPnTP from the reaction of PhnI, in the presence of PhnG, PhnH and PhnL results in the formation of the α-anomer with an inversion configuration at C1 of the ribose moiety. The reaction with water is assumed to involve the same stereochemical course.
We have synthesized an inhibitor for the metabolism of organophosphonates by construction of a compound that blocks the reaction catalyzed by PhnI, the first committed step in the C-P lyase complex. Shown in Scheme 3 is a putative transition state structure for the displacement of adenine from MgATP by water. Since the transition state of the enzymatic reaction is expected to bind more tightly to the enzyme than the substrate or products, transition state analogs can bind more tightly to the enzyme compared to ground state analogs. Thus transition state mimics can function as potent inhibitors of many classes of enzymes. Previous studies directed at enzymes catalyzing nucleosidase-type chemistry have led to the successful development of the immucillin-family of compounds, which have proven to be very potent and efficient inhibitors.10,11
Scheme 3.
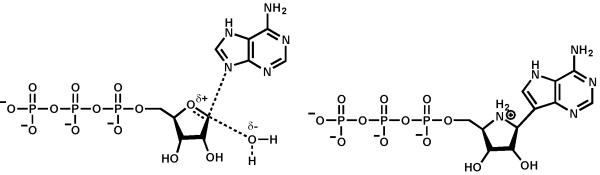
The structure of the proposed transition state for the hydrolysis of ATP by PhnI (left) and the structure of ImmA-TP (right).
The rational design of this compound family scaffold has led to successful therapeutic applications in treatment of various diseases such as malaria, bacterial infections and certain cancers.10,11 With this background, we synthesized a mimic of the anticipated transition state for the conversion of ATP to RTP and adenine. The structure of Immucillin-A triphosphate (ImmA-TP) is presented in Scheme 3. The proposed transition state of the PhnI-catalyzed reaction closely resembles the structure of ImmA-TP. The C-N bond of C-1′ of ribose and N-9 of adenine is replaced by a hydrolytically stable C-C bond from the 9-deazaadenine in ImmA-TP. The ribofuranose of ATP is replaced with a pyrrolidine moiety in ImmA-TP, closely mimicking the oxacarbenium ion generated at the transition state during the nucleosidase-type reaction catalyzed by PhnI. Lastly, the triphosphate moiety attached to the C-5′ of ribose is left intact, since the substrate profile of PhnI requires this recognition motif.
ImmA-TP was made via the enzymatic phosphorylation of Immucillin A (ImmA). ImmA (chloride salt, 25 mg; 83 μmol) was added to 2.0 mL of a solution containing 250 mM TRIS buffer, pH 8.0, 100 mM KCl, 30 mM MgCl2, and 175 mM phosphoenolpyruvate. To this reaction mixture 2.0 mg of Anopheles gambiae adenosine kinase, pyruvate kinase and myokinase were added. The phosphorylation reactions were initiated by the addition of 1.0 mM ATP, and incubated overnight at 37 °C. The reaction components were purified by reverse phase HPLC using a Luna2-C18 250 × 4.6 mm column, by elution with phosphate/tetrabutylammonium bisulfate, pH 6.0, as previously reported.12-14 Based on these conditions, ImmA-TP eluted at 16.8 min, which differed from AMP (17.3 min), ADP (18.5 min), and ATP (19.2 min). Samples were further desalted and concentrated to provide the tetrabutyl ammonium salt of the inhibitor. The compound was assessed for purity using ESI-MS (negative ion mode) with an [M-H] = 504. The compound was estimated to be more than 90% pure.
The inhibition of PhnI by ImmA-TP was analyzed using the nucleosidase activity of this enzyme in the absence of added phosphonates or other proteins needed for the biosynthesis of RPnTP. In this reaction catalyzed by PhnI, water is used to displace adenine from MgATP. Since this reaction does not require the in situ cleavage of the N-terminal GST-tag for catalytic activity, all assays where performed with the intact GST-fusion protein with PhnI.5 In typical assay, PhnI was pre-incubated with various concentrations of ImmA-TP for 45 minutes at 4 °C, after which the activity for the formation of RTP from ATP was determined. A typical assay contained 10 nM PhnI, 150 μM ATP, 150 μM Mg2+, and varying concentrations of ImmA-TP (0 - 750 nM) in 50 mM HEPES (pH 8.5 or 7.5) or CHES (pH 9.2) in a volume of 250 μL at 30 °C. The reaction rates were measured by monitoring the formation of adenine with adenine deaminase.15 The effect of ImmA-TP on the rate of the reaction catalyzed by PhnI is presented in Figure 1.
Figure 1.

Inhibition of PhnI by ImmA-TP at various pH values: pH 9.2 (blue), pH 8.5 (green) and pH 7.5 (red).
| (1) |
The inhibition of ImmA-TP on the reaction catalyzed by PhnI was fit to the equation developed by Morrison for a tight binding reversible inhibitor (Equation 1).16 In this equation, vi is the activity in the presence of the inhibitor I, vo is the activity in the absence of the inhibitor I, I is the inhibitor concentration (varying concentrations in assay), ET is the total enzyme concentration used in the assay, and Kd is the inhibition constant. For the uninhibited reaction, the kinetic constants for the nucleosidase reaction catalyzed by PhnI with ATP as the substrate are 1.4 ± 0.04 s−1, 95 ± 12 μM and (1.3 ± 0.2) × 104 M−1s−1 for kcat, Km and kcat/Km respectively. The Kd values for ImmA-TP obtained from these studies demonstrated that the inhibitor is more potent at lower pH values. The Kd values are 84 ± 16 nM, 80 ± 11 nM and 20 ± 4 nM at pH values of 9.2, 8.5, and 7.5, respectively. It was not possible to test the efficacy of ImmA-TP at pH values below pH 7.5 due to the instability of PhnI.
The immucillin-family of compounds consists of many inhibitors of human, bacterial and parasitic nucleosidases. These include adenosine nucleosidases, purine nucleoside phosphorylases (PNP), methylthioadenosine phosphorylases (MTAP), and methylthioadenosine nucleosidases (MTAN). Immucillin-H, a first generation immucillin, is a picomolar inhibitor of the human PNP, and is under clinical investigations in treating relapsed B-cell chronic lymphocytic leukemia. Biosynthetic pathways involving SAM are regulated largely through MTAP in humans, and are prime candidates as anticancer targets, as methylation is up regulated in these systems. Immucillins have thus far proved to be very potent pico- and femto-molar inhibitors of MTAPs.10,11 Variants of the immucillin class of compounds have also proved very effective against the malarial pathogen, Plasmodium falciparum, bacterial pathogens such as Heliobacter pylori, E. coli, S. pneumoniae, and Vibrio cholerae, by disrupting purine metabolism and severing quorum sensing pathways. The immucillins studied thus far show significant binding potency to the target, with Km/Kd ratios in excess of 500,000.10,11 The apparent binding efficiency of ImmA-TP to PhnI (Km/Kd) is approximately 10,000 at pH 7.5. Structure-activity studies around ImmA-TP might help develop more potent inhibitors of PhnI and further enhance the binding efficiency of ImmA-TP to this enzyme.
In this investigation we have identified a potent in vitro inhibitor of PhnI that can be used to inhibit the cascade of reactions catalyzed by the C-P lyase complex. This compound should prove invaluable in the identification of the active site contained within PhnI and as a molecular probe for inhibiting bacterial phosphonate metabolism.
Acknowledgments
Funding Sources
This work was supported in part by the Robert A. Welch Foundation (A-840) and the NIH (GM 103917) to FMR. We thank the laboratories of Vern L. Schramm, Albert Einstein College of Medicine, Bronx, New York, and Peter C. Tyler, Callaghan Innovation, Carbohydrate Chemistry Team, New Zealand for the gift of Immucillin-A. The authors declare no conflicting financial interest.
REFERENCES
- 1.White AK, Metcalf WW. Annu. Rev Microbiol. 2007;61:379–400. doi: 10.1146/annurev.micro.61.080706.093357. [DOI] [PubMed] [Google Scholar]
- 2.Clercq ED, Holy A. Nature Rev Drug Discovery. 2005;4:928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 3.Kamat SS, Raushel FM. Curr. Opin. Chem. Biol. 2013;17:589–596. doi: 10.1016/j.cbpa.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Peck SC, van der Donk WA. Curr. Opin. Chem. Biol. 2013;17:580–588. doi: 10.1016/j.cbpa.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamat SS, Williams HJ, Raushel FM. Nature. 2011;480:570–573. doi: 10.1038/nature10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamat SS, Williams HJ, Dangott LJ, Chakrabarti M, Raushel FM. Nature. 2013;497:132–136. doi: 10.1038/nature12061. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Q, van der Donk WA. Chembiochem. 2012;13:627–629. doi: 10.1002/cbic.201200020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metcalf WW, et al. Science. 2012;337:1104–1107. doi: 10.1126/science.1219875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reeburgh WS. Chem. Rev. 2007;107:486–513. doi: 10.1021/cr050362v. [DOI] [PubMed] [Google Scholar]
- 10.Schramm VL. Annu. Rev. Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schramm VL. ACS Chem. Biol. 2013;8:71–81. doi: 10.1021/cb300631k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans GB, Furneaux RH, Gainsford GJ, Schramm VL, Tyler PC. Tetrahedron. 2000;56:3053–3062. [Google Scholar]
- 13.Cassera MB, Ho MC, Merino EF, Burgos ES, Rinaldo-Matthis A, Almo SC, Schramm VL. Biochemistry. 2011;50:1885–1893. doi: 10.1021/bi101921w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgos ES, Schramm VL. Biochemistry. 2008;47:11086–11096. doi: 10.1021/bi801198m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamat SS, et al. Biochemistry. 2011;50:1917–1927. doi: 10.1021/bi101788n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrison JF. Biochim Biophys Acta. 1969;185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]