

Abstract
The apical junctional complex (AJC), composed of tight and adherens junctions, maintains epithelial barrier function. Since cigarette smoking and chronic obstructive pulmonary disease (COPD), the major smoking-induced disease, are associated with increased lung epithelial permeability, we hypothesized that smoking alters the transcriptional program regulating airway epithelial AJC integrity. Transcriptome analysis revealed global down-regulation of physiological AJC gene expression in the airway epithelium of healthy smokers (n = 59) compared to nonsmokers (n = 53) in association with changes in canonical epithelial differentiation pathways such as PTEN signaling accompanied by induction of cancer-related AJC components. The overall expression of AJC-related genes was further decreased in COPD smokers (n = 23). Exposure of airway epithelial cells to cigarette smoke extract in vitro resulted in down-regulation of several AJC genes paralleled by decreased transepithelial resistance. Thus, cigarette smoking induces transcriptional reprogramming of airway epithelial AJC architecture from its physiological pattern necessary for barrier function toward a disease-associated molecular phenotype.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-010-0500-x) contains supplementary material, which is available to authorized users.
Keywords: Tight junctions, Adherens junctions, Airway epithelium, Epithelial polarity, Cigarette smoking, Transcriptional regulation, Chronic obstructive pulmonary disease
Introduction
The apical junctional complex (AJC), a distinct multicomponent structure localized to the apex of the lateral membrane compartment of polarized epithelial cells, includes two kinds of intercellular junctions, tight junctions (TJ) and adherens junctions (AJ), a unique array of transmembrane proteins connected to an extensive network of cytoplasmic scaffold and signaling proteins [1, 2]. The TJ, the most apical AJC structure, constitute a semipermeable barrier for solutes and ions and physically separate the apical and basolateral membranes [3, 4]. Just below the TJ are the AJ which mediate cell–cell adhesion [5]. Detailed studies have defined the molecular composition of AJC [2], the posttranslational events governing AJC assembly in polarized epithelia [6–8], and the transcriptional changes associated with AJC maturation during epithelial differentiation in vitro [9]. However, little is known about the global gene expression program encoding AJC components and AJC-associated molecular pathways and transcriptional networks (the “AJC gene expression architecture”) in human epithelial tissues in vivo in health and under conditions characterized by an altered epithelial barrier function.
The airway epithelium constitutes an essential tissue barrier protecting the lung from inhaled environmental challenges [10, 11]. Of these, cigarette smoke is a major risk factor for chronic obstructive pulmonary disease (COPD) and lung cancer [12–14]. Smoking can substantially compromise lung epithelial barrier function leading to increased epithelial permeability [15, 16]. These observations have been commonly interpreted as a result of smoking-induced damage to the junctional structure [17, 18] due to direct cytotoxic effects of cigarette smoke on the lung epithelial cells [19, 20]. In the present study, we hypothesized that cigarette smoking might also have a more targeted effect on the airway epithelial barrier by modifying the AJC gene expression architecture in the small airway epithelium (SAE), the primary site of cigarette smoking-associated changes in the lung [21], and that this altered AJC-related gene expression contributes to a COPD-relevant molecular phenotype.
Materials and methods
Study population
A total of 135 subjects were included in this study including 53 healthy nonsmokers, 59 healthy smokers, and 23 COPD smokers (see Supplementary Table 1, and the inclusion and exclusion criteria).
SAE sampling, cDNA preparation and microarray processing
SAE cells (10th–12th order bronchi) were collected by fiberoptic bronchoscopy by brushing and processed for microarray analysis as described in the Supplementary Methods. Total RNA was extracted using a modification of the TRIzol method (Invitrogen, Carlsbad, CA) and processed to generate cDNA. Genome-wide gene expression analysis was performed using HG-U133 Plus 2.0 array (Affymetrix, Santa Clara, CA) according to Affymetrix protocols, hardware and software. Overall microarray quality was verified by the criteria: (1) 3′/5′ ratio for GAPDH ≤3, and (2) scaling factor ≤10.0 [66]. The captured image data from the HG-U133 Plus 2.0 arrays was processed using the MAS5 algorithm. The data were normalized using GeneSpring version 7.3.1 (Agilent technologies, Palo Alto, CA). See Supplementary Methods for further details. The raw data are available at the Gene Expression Omnibus (GEO) site (http://www.ncbi.nlm.nih.gov/geo/), accession number for this dataset is GSE20257.
Characterization of the apical junctional complex gene expression
A total of 69 AJC-related genes were selected based on the literature (Table 1). Using GeneSpring 7.3.1 software, genes were considered “expressed” if detected (P call of “Present”) in ≥20% of subjects in each study group. Differentially expressed genes between two groups were identified by the criteria: (1) P call ≥20% of samples in any of the groups; and (2) P < 0.05 between the groups with a Benjamini-Hochberg correction. A gene was defined as “smoking-responsive” if its expression was significantly different in healthy smokers compared to healthy nonsmokers. Unsupervised hierarchical clustering (with standard correlation as similarity measure and the complete linkage clustering algorithm) and principal component analysis (PCA) were carried out using GeneSpring software.
Table 1.
Changes in the AJC gene expression in the SAE of healthy smokers and smokers with COPD

Color codes for significantly differentially expressed genes (vs. healthy nonsmokers); red = significantly up-regulated genes (vs. healthy nonsmokers); blue = significantly down-regulated genes (vs. healthy nonsmokers)
NS, healthy nonsmokers; S, healthy smokers; COPD, COPD smokers; N.D., not detected (P call < 20% in both study groups); N.S., not significant
aFor genes with more than one probe set, the probe set with the best P call and/or (for significant genes) the lowest P value (vs. healthy nonsmokers) is included
AJC gene expression index
The SAE “AJC gene expression index” (AJC index, IAJC) was determined as a global measure of the coordinated smoking-induced changes in the SAE AJC gene expression by integrating information on expression levels of all smoking-responsive genes encoding physiological AJC components. For genes represented by more than one probe set, the probe set with the lowest P value was used. Expression values were log2 transformed. For each gene, a mean and standard deviation were calculated from the values in healthy nonsmokers, and the “normal range” was defined as within two standard deviations (SD) of the mean, in the direction of the smoking-induced change. IAJC for each subject was defined as the percentage of AJC genes with expression levels outside the normal range, using the formula:
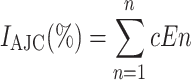 |
where E1 has a value of 1 if the expression level for gene 1 is >2 SD below that of healthy nonsmokers or a value of 0 if the expression level is ≤2 SD below that of healthy nonsmokers, E2 is the index for gene 2, etc., and the constant c (=100 divided by the number of smoking-responsive AJC genes, n) normalizes the index to the percent of AJC genes that are outside of the range of healthy nonsmokers. The statistical significance of differences in IAJC between the groups was determined using the Wilcoxon signed-ranks test.
Functional analysis of gene expression data
Genome-wide correlation analysis was performed to identify smoking-responsive gene probe sets having gene expression patterns similar to AJC genes down-regulated by smoking (“AJC-associated genes”), i.e., genes negatively correlated with the SAE IAJC in healthy smokers using the Pearson correlation coefficient (r) ≤ −0.5 as a screening cut-off criterion. To determine canonical molecular pathways enriched within the AJC-associated gene dataset, the derived gene list was subjected to Ingenuity Pathway Analysis (http://www.ingenuity.com). Criteria for pathway enrichment were: (1) the significance of association between the canonical pathway and the dataset (P < 0.05, Fischer’s exact test), and (2) the degree of enrichment (>1% unique genes from the input dataset belonging to the pathway). Functional categories enriched in the gene datasets were determined by gene ontology (GO) analysis using GeneSpring software and ProfCom analytic tool [67]. For selected gene sets, Spearman’s rank correlation coefficient (ρ) was determined using StatView (SAS Institute, Cary, NC). A smoking-sensitive network of AJC-associated protein–protein interactions was constructed using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins; http://string-db.org) database [31], applying a gene list containing smoking-responsive AJC genes and genes in the top AJC-associated molecular pathways as a combined input dataset using the Markov clustering algorithm or, for recently discovered interactions, based on literature data.
AJC transcriptional network
The core AJC network genes were identified based on: (1) P call ≥20%, (2) significantly downregulated in the SAE of healthy smokers versus healthy nonsmokers (P < 0.05, Benjamini-Hochberg correction), (3) correlation with IAJC (r ≤ −0.6; ρ ≤ −0.7, P < 0.0001), and (4) correlation with two or more known AJC genes (ρ ≥ 0.7, P < 0.05). The intrinsic AJC network genes (nodes) were identified based on the criteria: (1) well-characterized gene encoding a defined AJC component (Table 1), (2) P call ≥20%, (3) significantly downregulated in the SAE of healthy smokers versus healthy nonsmokers (P < 0.05 with Benjamini-Hochberg correction), (4) correlation with IAJC (r ≤ −0.5; ρ ≤ −0.6, P < 0.0001), (5) correlation with three or more core AJC network genes (ρ ≥ 0.7, P < 0.05), and (6) correlation with two or more known AJC genes (ρ ≥ 0.6, P < 0.05). AJC network genes were clustered into subgroups based on unsupervised hierarchical clustering.
Human airway epithelial cell cultures and in vitro studies
The human airway epithelial cell line 16HBE14o− was chosen because these cells can differentiate in a three-dimensional culture system acquiring essential characteristics of the normal airway epithelium, including functional TJ and AJ [68]. The cells were grown in Eagle’s minimum essential medium supplemented with 2 mM l-glutamine (Gibco, Carlsbad, CA), 10% fetal calf serum, 100 μg/ml streptomycin, and 100 U/ml penicillin G, at 37°C in an incubator in an atmosphere containing 5% CO2. After reaching about 90% confluence, they were passaged and seeded (105 cells/cm2) onto Transwell permeable filter inserts (12 mm diameter, pore size 0.4 μm; Costar, Cambridge, MA) and grown at 37°C in an atmosphere containing 5% CO2 under liquid-covered conditions. The volumes of the apical (400 μl) and basolateral (1,200 μl) chambers were set to avoid a significant hydrostatic pressure gradient across the cell layers [68]. Epithelial junctional integrity was determined by measuring transepithelial electrical resistance (Millicell ERS-2 epithelial volt-ohm-meter; Millipore, Bedford, MA).
Aqueous cigarette smoke extract (CSE) was generated from the combustion of one cigarette (Marlboro Red) bubbled through 12.5 ml of culture medium [69]. This medium, defined as “100% CSE”, was adjusted to pH 7.4 and filtered through a 0.22-μm filter. Experiments were started once transepithelial resistance reached ≥700 Ω×cm2, a level corresponding to the differentiation of 16HBE14o− cells expressing functional AJC components [68]. To mimic in vivo cigarette smoking, differentiated 16HBE14o− cells were exposed to CSE from the apical surface by adding 1, 2 or 4% CSE, concentrations nontoxic to these cells [46]. Control cells were treated with culture medium only. To model chronic cigarette smoke exposure, the apical medium containing CSE remained in the upper chamber until the next day, when the resistance was measured and the medium was replaced with a fresh CSE-containing medium. The experiments were carried out for 6 days. After CSE exposure was completed, cell viability was assessed by trypan blue exclusion and RNA extracted and processed for TaqMan RT-PCR analysis. Experiments were repeated at least three times.
TaqMan real-time PCR
TaqMan RT-PCR was performed to confirm differential expression of a subset of genes found to be differentially expressed by microarray, using the same RNA samples that were used for the microarray analysis, and to determine the effect of in vitro exposure of cultured airway epithelial cells to CSE on expression of selected AJC genes and known smoking-inducible oxidative stress-related genes. See Supplementary Methods for details.
Immunofluorescence analysis
Cytospin preparations of SAE obtained from randomly selected healthy smokers (n = 3) and healthy nonsmokers (n = 3) were processed for immunofluorescence analysis. Samples were blocked with donkey serum for 30 min (Jackson ImmunoResearch, West Grove, PA) and incubated with primary antibodies, mouse monoclonal antibodies against CLDN1 (10 μg/ml; Abnova, Taipei, Taiwan) and CDH1 (10 μg/ml; Invitrogen, Camarillo, CA) at 4°C overnight in a humid chamber, with mouse IgG2a and mouse IgG1 (both from R&D; Minneapolis, MN) isotype controls, respectively. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA). Images were captured using an Olympus IX 70 fluorescence microscope and analyzed using MetaMorph software (Universal Imaging Corporation, Downingtown, PA). Pseudocolor images were formed with red channel Cy5 fluorescence.
Results
Physiological AJC gene expression architecture in the human SAE
Microarray analysis revealed a distinct pattern of AJC gene expression in the SAE of healthy nonsmokers: 31 of 63 (49%) genes encoding known AJC components were expressed in all individuals, with considerable variability in expression levels of individual AJC genes (Fig. 1). Among the constitutively expressed genes encoding transmembrane TJ proteins (TJP), CXADR (coxsackie virus and adenovirus receptor) was most abundant followed by the claudin genes CLDN-7, -3, -8, and -1, and F11R (JAM-1; Fig. 1a). Of the 12 AJC genes encoding cytoplasmic TJ components, 11 (92%) including TJP1, TJP2, and TJP3 (zona occludens ZO-1, -2, and -3, respectively), CSDA (ZONAB) and others were constitutively expressed (Fig. 1b). Although expression levels of the genes encoding the epithelial polarity complexes were relatively low, genes encoding the major epithelial polarity complex PAR3/PAR6/aPKC and the core elements of the Crumbs (CRB3/INADL/MPP5) polarity complex were detected in the SAE of all healthy nonsmokers, whereas the Scribble complex (SCRIB/LLGL1/DLG) was detected in only a few subjects (Fig. 1c). Among the AJ genes, CDH1 (E-cadherin), displayed the highest relative expression levels followed by MLLT4 (afadin; AF-6), CTNNB1 and other catenin family genes (Fig. 1d). Consistent with the normal epithelial phenotype, CDH2 (N-cadherin), abundantly expressed in mesenchymal and neuronal tissues [5], was barely detected in the SAE of healthy nonsmokers (Fig. 1d). Four genes implicated in regulation of apical epithelial polarity, e.g. NUMB, PTEN, FOXA1 and FOXA2 [22–25] were constitutively expressed at low levels (Fig. 1e). Genes coding for AJC elements such as CLDN-2, -5, -6, -11, -14, -17, and -20, and nectin-1 (PVRL1) as well as putative AJC-regulating genes AMOT, HNF4A were not detected (Fig. 1).
Fig. 1.

Expression of AJC-related genes in the SAE of healthy nonsmokers. The ordinate represents P calls (percent subjects in each group expressing a given gene; yellow bars) and normalized log-transformed expression levels (means ± SD) of each gene (red genes with relatively high expression, ≥20; blue genes with relatively low expression, <20)
Smoking-dependent derangement of AJC gene expression architecture in healthy SAE
Genome-wide analysis revealed that, whereas 19% of all genes (6,038 of 32,436) detected in the SAE were affected significantly by smoking (Fig. 2a, left panel), the impact of smoking on the AJC transcriptional program was more profound, with 71% of the detected AJC genes (39 of 55) differentially expressed in healthy smokers versus nonsmokers (Fig. 2a, right panel). Remarkably, whereas at the genome-wide level, smoking was associated with upregulation of a considerable number of genes (Fig. 2a, left panel) that were mostly related to oxidative stress and xenobiotic metabolism (Fig. 2b), there was almost a uniform suppression of the physiological AJC gene expression program, with 37 of 39 (95%) smoking-responsive AJC genes significantly downregulated in healthy smokers compared to nonsmokers (Fig. 2a, right panel; b).
Fig. 2.

Smoking alters SAE AJC gene expression. a Volcano plot comparing the normalized expression of all gene probe sets (left panel) or AJC-encoding genes (right panel) in the SAE of healthy smokers versus healthy nonsmokers; y-axis, negative log of P value; x-axis, log2-transformed fold-change; red dots significant differentially expressed probe sets, grey dots not significant probe sets. b Differentially expressed physiological AJC-encoding genes (blue bars) and other genes belonging to various functional categories (black bars) in the SAE of healthy smokers versus healthy nonsmokers; y-axis, log2-transformed fold-change. c Diagram of the AJC architecture; significantly up- and downregulated in the SAE of healthy smokers are depicted as red boxes and blue boxes, respectively; yellow boxes indicate genes with no significant change; edges indicate known interactions between individual AJC proteins based on biochemical studies [1–7, 70]; the shapes of the nodes indicate different classes of AJC molecules determined based on the NCBI Entrez Gene database. d Immunofluorescence analysis of differential expression of CLDN1 and CDH1 proteins (red) in the cytospin preparations of SAE of healthy smokers versus healthy nonsmokers. Nuclei stained with DAPI (blue); scale bars 10 μm. More examples are shown in Supplementary Fig. 2
All AJC categories were broadly affected in healthy smokers, including 8 of 25 genes (32%) encoding transmembrane TJPs, 11 of 12 genes (92%) encoding cytoplasmic TJ, 6 of 12 polarity complex genes (50%) including all three members of the PAR3/PAR6/aPKC polarity complex, and 9 of 14 AJ genes (64%), suggesting a net effect of smoking on the physiological SAE AJC program (Fig. 2c, Table 1). From the functional perspective, smoking was associated with downregulation of the entire molecular AJC machinery, including the proteins that physically constitute cell–cell junctions, receptors for extracellular ligands, adaptor proteins, kinases, phosphatases and transcription factors (Fig. 2c). In contrast to the physiological AJC genes, expression of the TJ gene CLDN10 and the AJ gene CDH2, both associated with epithelial carcinogenesis [26–28], was significantly upregulated in the SAE of healthy smokers (Table 1).
Differential expression of several AJC genes in healthy smokers versus nonsmokers was confirmed by TaqMan RT-PCR (Supplementary Fig. 1). The expression of CLDN1 and CDH1, the two major components of TJ and AJ, respectively, was markedly decreased in the smokers’ SAE cells at the protein level (Fig. 2d; Supplementary Fig. 2). Together, these findings suggest that the physiological AJC molecular architecture in the human SAE is broadly deranged by cigarette smoking in vivo.
Dysregulation of the SAE AJC transcriptional program in COPD smokers
Microarray analysis revealed that 18 of 37 AJC-encoding genes (49%) suppressed in the SAE of healthy smokers were further downregulated in smokers with COPD (Table 1). Also the putative AJC-regulating genes PTEN, FOXA1 and FOXA2 demonstrated progressive smoking-dependent downregulation in COPD smokers (Table 1). By contrast, CLDN10 and CDH2, the two disease-associated AJC genes induced in the SAE of healthy smokers, were further upregulated in COPD smokers (Table 1) along with CLDN7, a TJ gene, implicated in epithelial carcinogenesis [29, 30]. (See Supplementary Fig. 4 for examples of AJC genes dysregulated in the SAE of COPD smokers.)
Based on the expression of all AJC genes detected in the SAE, unsupervised clustering analysis effectively segregated nonsmokers from both healthy and COPD smokers, with the COPD smokers forming a group at the pole opposite to that of nonsmokers in the clustering map (Fig. 3a). Healthy smokers were distributed throughout the map with subsets having either nonsmoker-like or COPD-like molecular patterns (Fig. 3a). PCA revealed that the groups could be separated from each other more effectively on the basis of the SAE expression of AJC genes (Fig. 3b, right panel) than on basis of the expression of all genes in the SAE transcriptome (Fig. 3b, left panel). Remarkably, the first principal component increased from 12.2% in the transcriptome-wide analysis to 28.5% in the AJC gene-restricted analysis (Fig. 3b), indicating that variability in AJC gene expression made a substantial contribution to genome-wide differences between the study groups. Similar to the cluster analysis, PCA revealed a progressive shift in AJC gene expression from healthy nonsmokers to smokers with COPD, whereas such a trend was not prominent at the genome-wide scale (Fig. 3b). This trend was not due to differences in gene number between the genome-wide and the 54-gene AJC-restricted datasets, since PCA on randomly selected datasets with the same number of genes as the AJC-restricted dataset did not reveal group-specific variability between the subjects except for the dataset composed of 54 randomly selected oxidative stress-related genes (Supplementary Fig. 3).
Fig. 3.

Smoking-dependent changes in the SAE AJC gene expression in COPD. a Unsupervised hierarchical clustering of healthy nonsmokers (green lines), healthy smokers (orange lines) and COPD smokers (blue lines) based on the SAE expression of AJC-encoding genes. Genes expressed above the average are represented in red, below average in blue, and average in white. b PCA of healthy nonsmokers (green circles), healthy smokers (orange circles) and COPD smokers (blue circles) on all expressed gene probe sets (left panel) and on AJC-encoding gene probe sets (right panel). The percentage contributions of the first three principal components are indicated. c Distribution (percent of subjects/group; ordinate) of healthy nonsmokers (green), healthy smokers (orange) and COPD smokers (blue) based on the SAE AJC index (IAJC; percent of abnormally expressed AJC-encoding genes; abscissa). The inserted box-plot shows the IAJC distribution in each group: the bottom and top of each box represent the 25th and 75th percentile (the lower and upper quartiles, respectively), and the band near the middle of each box is the 50th percentile (the median); the ends of the whiskers represent the 10th percentile and the 90th percentile; points lying outside the whiskers are considered outliers; P values were determined using the Wilcoxon signed-ranks test. d Abnormal expression frequency (ordinate; percent of subjects/group having expression of a given AJC-encoding gene below the normal range)
SAE AJC index as a measure of smoking-dependent dysregulation of the SAE AJC transcriptional program
The SAE AJC gene expression index (IAJC) was developed as a global measure of the coordinated smoking-dependent changes in the physiological SAE AJC gene expression by integrating information on expression levels of all smoking-responsive genes that encode AJC components. Consistent with the trends revealed by the clustering analysis and PCA, the average IAJC progressively increased in a smoking-dependent manner from healthy smokers to COPD smokers (P < 0.04, Fig. 3c). This analysis also revealed the contribution of individual AJC genes to the progressive smoking-dependent abnormality in the global SAE AJC gene expression architecture (Fig. 3d). Notably, the IAJC-based analysis revealed a higher magnitude of intergroup variability, suggesting that it can capture more information related to differential expression of AJC genes compared to traditional fold-change-based analysis.
Molecular pathways associated with smoking-induced changes in the SAE AJC gene expression architecture
To determine whether downregulation of the physiological AJC transcriptional program in the SAE of smokers is associated with smoking-induced changes in the canonical molecular pathways, we first performed a genome-wide screening for smoking-responsive genes negatively correlated (r < −0.5) with the IAJC, i.e., having an expression pattern similar to AJC genes in the SAE of healthy smokers. The derived gene dataset (“AJC-associated genes”) included 992 probe sets corresponding to 825 unique annotated genes (Supplementary Table 3), indicating that suppression of the AJC transcriptional program has transcriptional coherence with at least 15% of other smoking-responsive genes. Among the top AJC-associated genes was peroxiredoxin 5 (r < −0.66; ρ < −0.53, P < 0.0001), an antioxidant gene that has been suggested to protect the airway epithelium from smoking-induced injury [19]. Ingenuity pathway analysis revealed that the AJC-associated gene dataset (796 mapped genes) is enriched in elements of canonical molecular pathways implicated in regulation of epithelial differentiation, with the PTEN pathway showing the strongest association (P < 10−4; Fig. 4a). Other enriched pathways included protein kinase A signaling, insulin receptor signaling, PI3K/AKT signaling (all P < 10−3), Rac and CDC42 signaling pathways (both P < 0.005, Fig. 4a). Expression of the founding member of the PTEN pathway, the tumor suppressor PTEN, significantly correlated with overall AJC gene expression (Fig. 4b).
Fig. 4.

Smoking-sensitive AJC-associated molecular pathways. a IPA-based canonical pathways enriched among AJC-associated genes (smoking-sensitive genes negatively correlating with the IAJC with r < −0.5). The ordinate indicates the degree of enrichment (percent of AJC-associated genes belonging to the pathway; orange boxes) and shows −log P values of enrichment. The axonal guidance pathway (enrichment 3.3%; P < 0.01) was excluded as being specific to neuronal signaling. b Spearman correlation (ρ) analysis of the PTEN gene normalized expression in the SAE of healthy smokers with the IAJC. c STRING 8.2-based model of relationships between smoking-responsive physiological AJC-encoding genes and selected AJC-related genes correlating with the IAJC with r < −0.5. Each circle corresponds to an individual gene with the size proportional to the degree of correlation (r) with the IAJC, and the color corresponding to the clusters in which the genes were grouped based on known protein–protein interactions (Markov clustering); the form of the lines reflects the confidence of the association from thick (most confident) to dashed (least confident)
To construct a model of smoking-sensitive molecular interactions between individual AJC-encoding genes and associated molecular pathways, the set of AJC-encoding genes, PTEN pathway member genes and putative AJC-regulating genes correlating with the IAJC (r ≤ −0.5; ρ ≤ −0.5; P < 0.0001) were subjected to STRING network analysis. STRING is a web-based analytical tool that accumulates information on functional protein–protein associations based on the knowledge of physical interactions between individual proteins including their interaction in curated biological pathways complemented by interactions that are predicted computationally using a set of prediction algorithms [31]. This analysis revealed that, based on known and predicted protein–protein interactions, a number of smoking-sensitive AJC-encoding genes encode proteins that interact with each other and are directly or indirectly linked to PTEN (Fig. 4c), implying that AJC-associated protein–protein interactions are reprogrammed by smoking at the transcriptional level.
SAE smoking-sensitive AJC transcriptional network
A uniform downregulation of the physiological AJC gene expression pattern in the SAE of smokers suggests that transcriptional mechanisms determining the expression of individual AJC genes in the SAE are suppressed by smoking in a coordinated manner. This assumption was supported by the GO analysis, which indicated that the biological functions “Nucleic acid metabolism” (38% of dataset genes; P < 10−7) and “Transcription” (27% of dataset genes; P < 10−5) dominated among the functional GO categories significantly enriched in the AJC-associated gene dataset (Fig. 5a). Based on the concept that high gene expression correlation may provide unbiased information on possible gene regulatory networks independent of prior knowledge about the physical interaction between the gene products [32, 33], and since IAJC provides a cumulative measure of AJC gene expression, we performed a genome-wide coexpression analysis that identified a subset of 21 smoking-responsive genes (“core AJC network genes”; Fig. 5b), whose expression paralleled changes in the global AJC transcriptional program. This was evident as a strong negative correlation of their expression with the IAJC (ρ ≤ −0.7; P < 0.0001) and a strong positive correlation with at least two AJC-encoding genes (ρ > 0.7; P < 0.0001) in the SAE of healthy smokers (Supplementary Table 4). Of note, 15 of 21 core AJC network genes (71%) encoded nuclear proteins, consistent with a significant enrichment of the categories “RNA binding” (P < 10−5), “Nucleus” (P < 10−5), and “Transcription” (P < 0.001) in the 21-gene core AJC network dataset (Supplementary Table 5). The core AJC network included genes encoding well-characterized transcriptional regulators such as CRSP3, MATR3, NUCKS, and PSIP1, as well as genes related to cytokine signaling and tissue morphogenesis, such as IL6ST and PKD2, respectively (Fig. 5b; Supplementary Table 4).
Fig. 5.

Smoking-sensitive AJC-associated transcriptional network in the SAE. a Biological process-related GO categories significantly enriched in the dataset of AJC-associated genes (smoking-responsive; correlating with the IAJC with r < −0.5); x-axis, enrichments (percent of AJC-associated genes in the particular GO category); y-axis shows –log P values of enrichment. The dashed lines indicate the arbitrarily defined area of significant enrichment (enrichment >10%; P < 0.05). b The core AJC network genes and their coexpression-based “interactions” with the intrinsic AJC network genes were identified based on their correlation with the IAJC and each other as described in “Materials and methods”. Each circle represents an individual gene with the size differences proportional to inter-network interaction intensity, and the color corresponding to the clusters based on the unsupervised hierarchical clustering; the form of the lines reflects the strength of the correlation between the two genes (see Supplementary Table 4). c Spearman rank correlation (ρ) analysis of the PTEN gene-normalized expression and the mean AJC network gene expression in the SAE of healthy smokers. d Mean normalized expression levels of AJC network genes in the study groups; the data presented are the means±SD for each group and the P values indicate the significance of the differences between the groups
Overall AJC network gene expression in the SAE of healthy smokers strongly correlated with expression of PTEN (ρ = 0.77, P < 0.0001; Fig. 5c), indicating a link between the AJC transcriptional network and AJC-associated signaling pathways sensitive to smoking. The overall expression of the core AJC transcriptional network genes was further decreased in smokers with COPD (P < 0.0003 vs. healthy smokers; Fig. 5d), in agreement with progressive smoking-dependent suppression of the SAE AJC gene expression program in COPD (Fig. 3c).
A subset of AJC-encoding genes (TJP1, MPP5, CDH1, MPP7, DLG1, and MPDZ) exhibited a particularly high degree of coexpression with the core AJC network elements (Fig. 5b). Among these “intrinsic” AJC network genes, TJP1 and MPP5 exhibited the highest degree of interaction intensity, being strongly coexpressed (ρ > 0.7; P < 0.0001) with 18 (86%) and 16 (76%) of core AJC network genes, respectively (Fig. 5b; Supplementary Table 4). Based on the cluster analysis, individual AJC-encoding genes within the network displayed a preferential interaction with specific core network elements (Fig. 5b), suggesting heterogeneity of the regulatory pathways within the network.
Cigarette smoke modulates airway epithelial AJC in vitro
To assess whether smoking can induce changes in the AJC transcriptional program and physiological properties independent of inflammatory cells or the direct effect of cytotoxicity on the airway epithelial barrier, we exposed the apical surface of differentiated 16HBE14o− human airway epithelial cells to CSE daily at nontoxic concentrations (Fig. 6a). After 6 days of exposure, the expression of genes encoding AJC components CLDN1, CLDN8, CGN, and CDH1 was significantly downregulated (Fig. 6b). All these genes were also among the AJC genes significantly downregulated in the SAE of smokers in vivo (Table 1; Supplementary Fig. 4). Downregulation of these AJC genes was accompanied by CSE-induced suppression of genes encoding PTEN and transcription factor FOXO3A (Fig. 6b), a downstream member of the PTEN pathway [34, 35]. By contrast, expression of two smoking-inducible oxidative stress-related genes CYP1A1, CYP1B1 and NQO1 [36], was upregulated (Fig. 6b), indicating that the observed effect of CSE on AJC genes was not due to the general CSE-induced transcriptional suppression. This downregulation of physiological AJC gene expression by noncytotoxic CSE exposure in vitro was accompanied by a significant decrease in transepithelial electrical resistance (Fig. 6c), a measure of the epithelial junctional barrier integrity.
Fig. 6.

Effect of the CSE on the airway epithelial AJC gene expression and barrier integrity in vitro. a Viability of differentiated 16HBE14o− cells apically stimulated with 1, 2, or 4% CSE for 6 days as described in “Materials and methods” compared to unstimulated cells (n = 3 in each group); NS nonsignificant difference (P > 0.05) for all three comparisons. b Expression of AJC genes (CLDN1, CLDN8, CGN, CDH1), PTEN pathway genes (PTEN, FOXO3), and oxidative stress-related genes (CYP1A1, CYP1B1, NQO1) in 16HBE14o− cells stimulated with 2% CSE for 6 days as described in “Materials and methods” compared to unstimulated cells determined by TaqMan PCR; y-axis, normalized expression levels; P values indicate the significance of differences between the groups; n = 3 in each group; c Transepithelial resistance of differentiated 16HBE14o− cell monolayers (expressed as percent of initial level) was measured during 6 days of stimulation with 2% CSE, as described in “Materials and methods” compared to control (unstimulated) cells; P values indicate the significance of differences between the groups; n = 3 in each group
Discussion
Apical–basal polarity, a hallmark of differentiated epithelia, is manifested by an asymmetric distribution of specialized protein domains along the plasma membrane [7]. Localized to the apical membrane compartments of neighboring epithelial cells, the AJC is essential for epithelial barrier integrity and function, with the TJ governing the transport of solutes and ions across epithelia, and the AJ mediating cell–cell adhesion [1–4]. AJC protein complexes also function as signaling platforms that regulate gene expression, cell proliferation and differentiation [37, 38] and provide a tumor-suppressing mechanism [39, 40]. All of these functions are altered in the airway epithelium of smokers, which is leakier, hyperproliferative, abnormally differentiated and more susceptible to malignant transformation [16, 21, 41, 42] compared to that of nonsmokers. The present study suggests that one of the mechanisms central to these airway epithelial barrier abnormalities in smokers is the dysregulation of the physiological AJC transcriptional program in the airway epithelium, an alteration that progresses during the development of smoking-induced lung disease.
Physiological SAE AJC gene expression architecture
AJC barrier integrity is determined by its unique molecular composition, which varies from one anatomic location to another [4]. Under physiological conditions, the SAE appears to be leakier than the epithelium of proximal airways, indicative of a distinct SAE AJC molecular program that determines a balance between physiological permeability necessary for constitutive metabolic processes, and function as a barrier limiting diffusion of solutes, ions, and preventing penetration of inhaled microbes [1, 43, 44]. Although expression of several AJC proteins in the airway epithelium has been analyzed in detail previously [44], the entire AJC molecular composition of the SAE in vivo under physiological conditions has not been previously assessed. Microarray analysis of the present study revealed that 49% of genes encoding known AJC components are expressed in the SAE of healthy nonsmokers, with considerable variability in expression levels of individual AJC genes.
Transcriptional suppression of physiological AJC program by smoking
Cigarette smoke increases airway epithelial permeability in vivo and in vitro [15–19, 45, 46]. Since smoking is associated with dramatic gene expression changes in the airway epithelium [36], we hypothesized that chronic exposure to cigarette smoke modulates the transcriptional program necessary for the maintenance of AJC in the SAE. Of the AJC-encoding genes physiologically expressed in the SAE, 67% were found to be significantly downregulated in healthy smokers. All categories of AJC-encoding genes were broadly affected, with the genes encoding cytoplasmic TJ components being almost uniformly (92%) downregulated. This was accompanied by downregulation of putative regulators of epithelial polarity (e.g., PTEN, FOXA1, and FOXA2), suggesting that some of them might represent smoking-sensitive regulators of AJC genes in the airway epithelium. Impressively, smoking-induced differences in AJC gene expression had a higher impact on the variability between smokers and nonsmokers than the global SAE transcriptome changes induced by smoking.
The data of the present study suggest a model in which the modulation of airway epithelial barrier by chronic cigarette smoking is a relatively specific molecular process involving the transcriptional reprogramming of the AJC molecular architecture. Supporting this concept, we observed that 6-day exposure of differentiated human airway epithelium to noncytotoxic concentrations of CSE in vitro resulted in downregulation of several AJC genes, and was accompanied by a reduction in transepithelial resistance. These in vitro data indicate that the effect of cigarette smoke on AJC molecular integrity in the SAE does not require the presence of inflammatory cells and their mediators previously reported to downregulate expression of AJC genes and increase airway epithelial permeability [21, 47, 48]. Downregulation of physiological AJC genes by smoking in vivo and in vitro was paralleled by decreased expression of PTEN and FOXO3A, a transcription factor in the PTEN pathway [34, 35], as well as an upregulation of oxidative stress-related genes, with a strong association between the IAJC and PRDX5, an antioxidant gene that has been suggested to limit smoking-increased airway epithelial permeability [19]. Although oxidative stress is known to induce disassembly of AJC at the protein level [49], our data suggest the possibility of a reciprocal relationship between transcriptional programs regulating AJC integrity and oxidative stress responses to smoking.
Cancer-like AJC gene expression pattern in smokers’ SAE
The broad suppression of the physiological SAE AJC transcriptional program in healthy smokers was accompanied by an upregulation of CDH2 and CLDN10, AJC genes linked to cancer-related pathological changes in epithelial tissues. Epithelial expression of CDH2, a marker of neuronal and mesenchymal cells, is a common indicator of epithelial-mesenchymal transition (EMT) [5, 50]. Other molecular changes typical of EMT, such as downregulation of CDH1, occludin, TJP1 and CXADR [51], were also detected in the smokers’ SAE, suggesting that dysregulation of SAE AJC gene expression in smokers might result from common transcriptional derangement associated with smoking-induced EMT. Consistent with this concept, nicotine, the major cigarette smoke constituent, induces EMT-like molecular and morphological changes in human lung cancer cell lines [52]. Upregulation of CDH2 and downregulation of CDH1 are the central EMT features in human lung cancer [28]. Our data provide evidence that smoking induces EMT-typical molecular changes in the healthy airway epithelium in vivo prior to clinical manifestations of lung cancer.
Another smoking-induced AJC gene was CLDN10, whose expression has been associated with increased epithelial permeability [53]. Recently, CLDN10 has been described as a marker of Clara cells in the mouse airways [54]. Given that the numbers of Clara cells are considerably reduced in the SAE of smokers [55], it is possible that smoking induces de novo expression of CLDN10 in other cell populations similar to various epithelial cancers, in which CLDN10 is overexpressed [26, 27].
Smoking-sensitive SAE AJC-associated molecular pathways and networks
Broad suppression of biologically related groups of genes suggests that central mechanisms controlling their expression, e.g. signaling pathways or transcriptional regulators, are dysregulated in a coordinated manner [32, 33, 56]. No central transcriptional mechanism regulating AJC biogenesis in the adult epithelia has been defined. The present study revealed that suppression of the physiological AJC gene expression program in the SAE of smokers is paralleled by downregulation of 15% of other smoking-responsive genes and that this AJC-associated dataset is significantly enriched with the elements of canonical molecular pathways critical for epithelial differentiation. The top enriched pathways (PTEN, PI3K/AKT, Rac- and CDC42) have been implicated in posttranslational regulation of apical epithelial polarity and AJC assembly [6, 8], but not yet linked to AJC transcriptional control. Among these pathways, the PTEN signaling pathway had the highest degree of association with smoking-induced changes in the AJC transcriptional program. The tumor suppressor PTEN is an important mediator of epithelial apical membrane identity [24]. Our data indicate an interesting possibility that cigarette smoking reprograms AJC-associated protein–protein interactions necessary for the generation of airway epithelial polarity.
Based on the concept that coexpressed genes might share common mechanisms of transcriptional regulation [33], we identified a “core AJC transcriptional network” composed of 21 smoking-responsive genes that are strongly correlating with the IAJC in the SAE of healthy smokers. One of the critical hallmarks of biologically meaningful transcriptional networks is the enrichment of certain GO categories [32, 56]. Consistent with this requirement, the core AJC transcriptional network was significantly enriched in genes encoding nuclear proteins with nucleic acid-binding activity, including the transcriptional regulators MATR3, PSIP1, NUCKS [57–59], tumor suppressor CRSP3 [60], and PKD2, a gene mediating mechanosensation in primary cilia [61]. Remarkably, MPP5, a component of the Crumbs3 polarity complex localized in the primary cilium [62], and TJP1, a TJ component capable of modulating gene expression [37], were the intrinsic AJC network components most intensively interacting with the core AJC network genes. None of identified network genes has been previously implicated in regulation of the AJC, and the network structure was different from that of the epithelial junctional complex network determined based on protein–protein interactions [63]. The overall AJC network gene expression in the healthy smokers SAE strongly correlated with PTEN expression, indicating a molecular link between the AJC transcriptional network and AJC-associated signaling pathways.
It has been previously shown that cigarette smoke, in combination with the inflammatory cytokine interleukin-1beta (IL-1β), induces AJ disassembly in endothelial cells by suppressing PTEN activity [64], indicating the possibility that cigarette smoke can utilize different molecular mechanisms to regulate AJC integrity depending on the targeted cell type and/or cellular microenvironment. Cigarette smoke is commonly associated with inflammatory responses in various tissues, including airway epithelium [65] and inflammatory cytokines, including IL-1β can induce significant derangement in the AJC structural integrity [48]. Our study extends these observations by providing evidence that smoking can induce a broad disarray in the airway epithelial AJC architecture by reprogramming transcriptional elements encoding essential components of AJC in a noninflammatory and noncytotoxic manner. It is remarkable that changes in the PTEN pathway are associated with both transcription-dependent and -independent mechanisms of AJC regulation by smoking.
In vivo gene coexpression data in our study helped identify a previously unknown relationship between the AJC complex genes, AJC transcriptional network and the PTEN pathway that exist in the human airway epithelium. Although additional mechanistic studies are necessary to determine whether PTEN is a direct regulator of the AJC transcriptional program (or vice versa), our data indicate that mechanisms that regulate the AJC transcriptional program and the PTEN signaling pathway—the two cardinal features of the epithelial apical membrane identity—are coordinated and integrated into the complex transcriptional response of the human airway epithelium to the stress of cigarette smoking in vivo.
AJC reprogramming in COPD
Another observation of the present study is that smoking-induced reprogramming of the physiological AJC architecture in the SAE progresses with the development of COPD, the major smoking-induced lung disease [13, 21, 65]. Not only the physiological AJC-encoding genes, but also the core AJC transcriptional network genes identified in healthy smokers, were progressively suppressed in the SAE of smokers with COPD, suggesting that downregulation of AJC gene expression in COPD might be a consequence of progressive dysregulation of transcriptional regulatory mechanisms that starts in smokers prior to clinical evidence of disease. Reciprocally to the suppressed physiological AJC genes, the expression of smoking-induced cancer-associated AJC components CDH2 and CLDN10 further increased in the SAE of COPD smokers, did as CLDN7, another AJC gene implicated in the promotion of epithelial carcinogenesis [29, 30]. Together, these observations suggest that the overall AJC molecular architecture in the airway epithelium in COPD is reprogrammed in a smoking-dependent manner from its normal gene expression pattern essential for maintaining an appropriate barrier function toward a cancer-like molecular phenotype.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank R.J. Kaner, A.E. Tilley, M.W. Butler, M. O’Mahony, B. Witover and B. Ferris for help with this study, and N. Mohamed for help in preparing the manuscript. These studies were supported, in part, by NIH R01 HL074326; P50 HL084936 and UL1-RR024996.
Abbreviations
- AJC
Apical junctional complex
- AJ
Adherens junctions
- CDH
Cadherin
- CGN
Cingulin
- CLDN
Claudin
- COPD
Chronic obstructive pulmonary disease
- CSE
Cigarette smoke extract
- GO
Gene ontology
- IAJC
Small airway epithelium apical junctional complex gene expression index
- PCA
Principal component analysis
- PTEN
Phosphatase and tensin homolog
- SAE
Small airway epithelium
- TJ
Tight junctions
- TJP
Tight junction protein
References
- 1.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 2.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–669. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–235. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- 4.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gumbiner BM. Regulation of cadherin adhesive activity. J Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellman I, Nelson WJ. Coordinated protein sorting, targeting and distribution in polarized cells. Nat Rev Mol Cell Biol. 2008;9:833–845. doi: 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson WJ. Remodeling epithelial cell organization: transitions between front-rear and apical-basal polarity. Cold Spring Harb Perspect Biol. 2009;1:a000513. doi: 10.1101/cshperspect.a000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samarin S, Nusrat A. Regulation of epithelial apical junctional complex by Rho family GTPases. Front Biosci. 2009;14:1129–1142. doi: 10.2741/3298. [DOI] [PubMed] [Google Scholar]
- 9.Halbleib JM, Saaf AM, Brown PO, Nelson WJ. Transcriptional modulation of genes encoding structural characteristics of differentiating enterocytes during development of a polarized epithelium in vitro. Mol Biol Cell. 2007;18:4261–4278. doi: 10.1091/mbc.E07-04-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 11.Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc. 2008;5:772–777. doi: 10.1513/pats.200805-041HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3:733–744. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]
- 13.Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel WC, Zielinski J. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–555. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- 14.Lee G, Walser TC, Dubinett SM. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Curr Opin Pulm Med. 2009;15:303–307. doi: 10.1097/MCP.0b013e32832c975a. [DOI] [PubMed] [Google Scholar]
- 15.Jones JG, Minty BD, Lawler P, Hulands G, Crawley JC, Veall N. Increased alveolar epithelial permeability in cigarette smokers. Lancet. 1980;1:66–68. doi: 10.1016/S0140-6736(80)90493-6. [DOI] [PubMed] [Google Scholar]
- 16.Knowles MR, Buntin WH, Bromberg PA, Gatzy JT, Boucher RC. Measurements of transepithelial electric potential differences in the trachea and bronchi of human subjects in vivo. Am Rev Respir Dis. 1982;126:108–112. doi: 10.1164/arrd.1982.126.1.108. [DOI] [PubMed] [Google Scholar]
- 17.Boucher RC, Johnson J, Inoue S, Hulbert W, Hogg JC. The effect of cigarette smoke on the permeability of guinea pig airways. Lab Invest. 1980;43:94–100. [PubMed] [Google Scholar]
- 18.Petecchia L, Sabatini F, Varesio L, Camoirano A, Usai C, Pezzolo A, Rossi GA. Bronchial airway epithelial cell damage following exposure to cigarette smoke includes disassembly of tight junction components mediated by the extracellular signal-regulated kinase 1/2 pathway. Chest. 2009;135:1502–1512. doi: 10.1378/chest.08-1780. [DOI] [PubMed] [Google Scholar]
- 19.Serikov VB, Leutenegger C, Krutilina R, Kropotov A, Pleskach N, Suh JH, Tomilin NV. Cigarette smoke extract inhibits expression of peroxiredoxin V and increases airway epithelial permeability. Inhal Toxicol. 2006;18:79–92. doi: 10.1080/08958370500282506. [DOI] [PubMed] [Google Scholar]
- 20.Sun W, Wu R, Last JA. Effects of exposure to environmental tobacco smoke on a human tracheobronchial epithelial cell line. Toxicology. 1995;100:163–174. doi: 10.1016/0300-483X(95)03087-V. [DOI] [PubMed] [Google Scholar]
- 21.Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- 22.Gao N, Ishii K, Mirosevich J, Kuwajima S, Oppenheimer SR, Roberts RL, Jiang M, Yu X, Shappell SB, Caprioli RM, Stoffel M, Hayward SW, Matusik RJ. Forkhead box A1 regulates prostate ductal morphogenesis and promotes epithelial cell maturation. Development. 2005;132:3431–3443. doi: 10.1242/dev.01917. [DOI] [PubMed] [Google Scholar]
- 23.Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C, Colwill K, Starostine A, Metalnikov P, Pawson T. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–548. doi: 10.1016/j.cell.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 24.Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burtscher I, Lickert H. Foxa2 regulates polarity and epithelialization in the endoderm germ layer of the mouse embryo. Development. 2009;136:1029–1038. doi: 10.1242/dev.028415. [DOI] [PubMed] [Google Scholar]
- 26.Cheung ST, Leung KL, Ip YC, Chen X, Fong DY, Ng IO, Fan ST, So S. Claudin-10 expression level is associated with recurrence of primary hepatocellular carcinoma. Clin Cancer Res. 2005;11:551–556. [PubMed] [Google Scholar]
- 27.Sugita M, Geraci M, Gao B, Powell RL, Hirsch FR, Johnson G, Lapadat R, Gabrielson E, Bremnes R, Bunn PA, Franklin WA. Combined use of oligonucleotide and tissue microarrays identifies cancer/testis antigens as biomarkers in lung carcinoma. Cancer Res. 2002;62:3971–3979. [PubMed] [Google Scholar]
- 28.Prudkin L, Liu DD, Ozburn NC, Sun M, Behrens C, Tang X, Brown KC, Bekele BN, Moran C, Wistuba II. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod Pathol. 2009;22:668–678. doi: 10.1038/modpathol.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darido C, Buchert M, Pannequin J, Bastide P, Zalzali H, Mantamadiotis T, Bourgaux JF, Garambois V, Jay P, Blache P, Joubert D, Hollande F. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res. 2008;68:4258–4268. doi: 10.1158/0008-5472.CAN-07-5805. [DOI] [PubMed] [Google Scholar]
- 30.Nubel T, Preobraschenski J, Tuncay H, Weiss T, Kuhn S, Ladwein M, Langbein L, Zoller M. Claudin-7 regulates EpCAM-mediated functions in tumor progression. Mol Cancer Res. 2009;7:285–299. doi: 10.1158/1541-7786.MCR-08-0200. [DOI] [PubMed] [Google Scholar]
- 31.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xulvi-Brunet R, Li H. Co-expression networks: graph properties and topological comparisons. Bioinformatics. 2010;26:205–214. doi: 10.1093/bioinformatics/btp632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a) Proc Natl Acad Sci U S A. 2008;105:2622–2627. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nho RS, Kahm J. beta1-Integrin-collagen interaction suppresses FoxO3a by the coordination of Akt and PP2A. J Biol Chem. 2010;285:14195–14209. doi: 10.1074/jbc.M109.052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harvey BG, Heguy A, Leopold PL, Carolan BJ, Ferris B, Crystal RG. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J Mol Med. 2007;85:39–53. doi: 10.1007/s00109-006-0103-z. [DOI] [PubMed] [Google Scholar]
- 37.Balda MS, Matter K. Tight junctions and the regulation of gene expression. Biochim Biophys Acta. 2009;1788:761–767. doi: 10.1016/j.bbamem.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 38.Koch S, Nusrat A. Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann N Y Acad Sci. 2009;1165:220–227. doi: 10.1111/j.1749-6632.2009.04025.x. [DOI] [PubMed] [Google Scholar]
- 39.Itoh M, Bissell MJ. The organization of tight junctions in epithelia: implications for mammary gland biology and breast tumorigenesis. J Mammary Gland Biol Neoplasia. 2003;8:449–462. doi: 10.1023/B:JOMG.0000017431.45314.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee M, Vasioukhin V. Cell polarity and cancer – cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci. 2008;121:1141–1150. doi: 10.1242/jcs.016634. [DOI] [PubMed] [Google Scholar]
- 41.Auerbach O, Stout AP, Hammond EC, Garfinkel L. Changes in bronchial epithelium in relation to cigarette smoking and in relation to lung cancer. N Engl J Med. 1961;265:253–267. doi: 10.1056/NEJM196108102650601. [DOI] [PubMed] [Google Scholar]
- 42.Wistuba II, Mao L, Gazdar AF. Smoking molecular damage in bronchial epithelium. Oncogene. 2002;21:7298–7306. doi: 10.1038/sj.onc.1205806. [DOI] [PubMed] [Google Scholar]
- 43.Ballard ST, Taylor AE. Bioelectric properties of proximal bronchiolar epithelium. Am J Physiol. 1994;267:L79–L84. doi: 10.1152/ajplung.1994.267.1.L79. [DOI] [PubMed] [Google Scholar]
- 44.Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1166–L1178. doi: 10.1152/ajplung.00182.2003. [DOI] [PubMed] [Google Scholar]
- 45.Olivera DS, Boggs SE, Beenhouwer C, Aden J, Knall C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal Toxicol. 2007;19:13–22. doi: 10.1080/08958370600985768. [DOI] [PubMed] [Google Scholar]
- 46.Gangl K, Reininger R, Bernhard D, Campana R, Pree I, Reisinger J, Kneidinger M, Kundi M, Dolznig H, Thurnher D, Valent P, Chen KW, Vrtala S, Spitzauer S, Valenta R, Niederberger V. Cigarette smoke facilitates allergen penetration across respiratory epithelium. Allergy. 2009;64:398–405. doi: 10.1111/j.1398-9995.2008.01861.x. [DOI] [PubMed] [Google Scholar]
- 47.Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, Schulzke JD. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000;113(Pt 11):2085–2090. doi: 10.1242/jcs.113.11.2085. [DOI] [PubMed] [Google Scholar]
- 48.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13:3218–3234. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci. 2008;13:7210–7226. doi: 10.2741/3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11:943–950. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, Banerjee S, Carless M, Kim E, Coppola D, Haura E, Chellappan S. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gunzel D, Stuiver M, Kausalya PJ, Haisch L, Krug SM, Rosenthal R, Meij IC, Hunziker W, Fromm M, Muller D. Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J Cell Sci. 2009;122:1507–1517. doi: 10.1242/jcs.040113. [DOI] [PubMed] [Google Scholar]
- 54.Zemke AC, Snyder JC, Brockway BL, Drake JA, Reynolds SD, Kaminski N, Stripp BR. Molecular staging of epithelial maturation using secretory cell-specific genes as markers. Am J Respir Cell Mol Biol. 2009;40:340–348. doi: 10.1165/rcmb.2007-0380OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lumsden AB, McLean A, Lamb D. Goblet and Clara cells of human distal airways: evidence for smoking induced changes in their numbers. Thorax. 1984;39:844–849. doi: 10.1136/thx.39.11.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ge H, Si Y, Roeder RG. Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 1998;17:6723–6729. doi: 10.1093/emboj/17.22.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grundt K, Haga IV, Huitfeldt HS, Ostvold AC. Identification and characterization of two putative nuclear localization signals (NLS) in the DNA-binding protein NUCKS. Biochim Biophys Acta. 2007;1773:1398–1406. doi: 10.1016/j.bbamcr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 59.Zeitz MJ, Malyavantham KS, Seifert B, Berezney R. Matrin 3: chromosomal distribution and protein interactions. J Cell Biochem. 2009;108:125–133. doi: 10.1002/jcb.22234. [DOI] [PubMed] [Google Scholar]
- 60.Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP, Welch DR. Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003;63:432–440. [PubMed] [Google Scholar]
- 61.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 62.Fan S, Hurd TW, Liu CJ, Straight SW, Weimbs T, Hurd EA, Domino SE, Margolis B. Polarity proteins control ciliogenesis via kinesin motor interactions. Curr Biol. 2004;14:1451–1461. doi: 10.1016/j.cub.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 63.Paris L, Bazzoni G. The protein interaction network of the epithelial junctional complex: a system-level analysis. Mol Biol Cell. 2008;19:5409–5421. doi: 10.1091/mbc.E08-05-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbieri SS, Ruggiero L, Tremoli E, Weksler BB. Suppressing PTEN activity by tobacco smoke plus interleukin-1beta modulates dissociation of VE-cadherin/beta-catenin complexes in endothelium. Arterioscler Thromb Vasc Biol. 2008;28:732–738. doi: 10.1161/ATVBAHA.107.159434. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 66.Raman T, O’Connor TP, Hackett NR, Wang W, Harvey BG, Attiyeh MA, Dang DT, Teater M, Crystal RG. Quality control in microarray assessment of gene expression in human airway epithelium. BMC Genomics. 2009;10:493. doi: 10.1186/1471-2164-10-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antonov AV, Schmidt T, Wang Y, Mewes HW. ProfCom: a web tool for profiling the complex functionality of gene groups identified from high-throughput data. Nucleic Acids Res. 2008;36:W347–W351. doi: 10.1093/nar/gkn239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ehrhardt C, Kneuer C, Fiegel J, Hanes J, Schaefer UF, Kim KJ, Lehr CM. Influence of apical fluid volume on the development of functional intercellular junctions in the human epithelial cell line 16HBE14o−: implications for the use of this cell line as an in vitro model for bronchial drug absorption studies. Cell Tissue Res. 2002;308:391–400. doi: 10.1007/s00441-002-0548-5. [DOI] [PubMed] [Google Scholar]
- 69.Gelbman BD, Heguy A, O’Connor TP, Zabner J, Crystal RG. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir Res. 2007;8:10. doi: 10.1186/1465-9921-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vogelmann R, Nelson WJ. Fractionation of the epithelial apical junctional complex: reassessment of protein distributions in different substructures. Mol Biol Cell. 2005;16:701–716. doi: 10.1091/mbc.E04-09-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.