

Abstract
In the tumor microenvironment, CD11b+Gr1+ bone marrow-derived cells are a predominant source of pro-tumorigenic factors such as matrix metalloproteinases (MMPs), but how distal tumors regulate these cells in the bone marrow is unclear. Here we addressed the hypothesis that the parathyroid hormone-related protein (PTHrP) potentiates CD11b+Gr1+ cells in the bone marrow of prostate tumor hosts. In two xenograft models of prostate cancer, levels of tumor-derived PTHrP correlated with CD11b+Gr1+ cell recruitment and microvessel density in the tumor tissue, with evidence for mediation of CD11b+Gr1+ cell-derived MMP-9 but not tumor-derived VEGF-A. CD11b+Gr1+ cells isolated from mice with PTHrP-overexpressing tumors exhibited relatively increased pro-angiogenic potential, suggesting that prostate tumor-derived PTHrP potentiates this activity of CD11b+Gr1+ cells. Administration of neutralizing PTHrP monoclonal antibody reduced CD11b+Gr1+ cells and MMP9 in the tumors. Mechanistic investigations in vivo revealed that PTHrP elevated Y418 phosphorylation levels in Src family kinases in CD11b+Gr1+ cells via osteoblast-derived IL-6 and VEGF-A, thereby upregulating MMP-9. Taken together, our results showed that prostate cancer-derived PTHrP acts in the bone marrow to potentiate CD11b+Gr1+ cells, which are recruited to tumor tissue where they contribute to tumor angiogenesis and growth.
Keywords: bone, metastasis, microenvironment, myeloid-derived suppressor cells, prostate cancer
INTRODUCTION
The tumor microenvironment provides primary tumor cells to mix with multiple types of stroma such as endothelium, fibroblasts and immune cells (1). Such heterogeneity of cell populations presents a major impediment for developing a cure for cancer. Increasing evidence supports that stromal cells in the tumor microenvironment not only occupy a significant fraction of the tumor bulk, but also play critical roles in proliferation, invasion and/or metastasis of tumor cells (2). In this regard, bone is an essential partner for tumor progression, since bone marrow serves as the supplying organ for numerous critical cells in the tumor microenvironment (3,4). However, it is unclear how tumor cells co-opt the bone and/or bone marrow to facilitate a favorable tumor microenvironment.
Among the bone marrow-derived cells, CD11b+Gr1+ cells (commonly referred to as myeloid-derived suppressor cells, MDSCs) correlate with tumor progression (5). MDSCs were originally investigated for their roles in evasion of host immune surveillance, especially via suppression of T-cell-dependent anti-tumoral immunity by production of arginase, reactive oxygen species and inducible nitric oxide synthase (6). Subsequent studies demonstrated that MDSCs are increased in tumor-bearing mice and cancer patients, and infiltrate primary tumor tissue to promote angiogenesis by secreting matrix metalloproteinases (MMPs), and also by direct incorporation into tumor endothelium (7,8). More recently, MDSCs have been shown to play key roles in recovery after radiation therapy (9,10) and anti-angiogenic therapy (11).
In parallel, multiple mechanisms have been proposed to explain the increased recruitment of MDSCs in tumor tissue. Yang et al. demonstrated that CXC chemokine ligand (CXCL)-5/CXC receptor (CXCR)-2 and stromal derived factor (SDF)-1/CXCR-4 axes recruit circulating MDSCs to tumor tissue (12). More recently, expression of a single integrin (α4β1) promotes MDSC invasion into tumors via activation of phosphatidylinositol 3-kinase (PI3K) (13). However, despite such clear evidence supporting the tumorigenic functions of MDSCs and also the potential mechanisms of recruitment to the tumor tissue, MDSCs are poorly understood regarding their regulation in the supplying organ (i.e. bone marrow) of the tumor host, and also their potential crosstalk with distant primary tumor cells.
The current study was designed to elucidate how CD11b+Gr1+ cells are regulated in the bone marrow of prostate tumor hosts, contributing to tumor growth and angiogenesis. Prostate cancer provides a unique perspective on this process because of its devastating mortality and morbidity associated with its preferential metastasis to the skeleton (14). Accordingly, prostate cancer cells secrete numerous important bone-modulating cytokines, leading to osteoblastic/osteolytic reactions that facilitate growth factor and cytokine release from bone cells and matrix (15). In particular, parathyroid hormone-related protein (PTHrP) is expressed by prostate cancer cells, and stimulates osteoblasts in an endocrine manner to secrete factors such as receptor activator of nuclear factor-κB ligand (RANKL), IL-6, C-C chemokine ligand (CCL)-2, and vascular endothelial growth factor (VEGF)-A within the bone microenvironment (16–18). Subsequently, PTHrP-induced cytokines have the ability to trigger cascades of unfavorable events (e.g. signaling pathways leading to potentiation of CD11b+Gr1+ bone marrow cells) within the bone marrow, contributing to tumor progression. Overall, the central hypothesis of this study was prostate cancer-derived PTHrP potentiates CD11b+Gr1+ cells within the bone marrow, contributing to angiogenesis and tumor growth.
MATERIALS AND METHODS
Cells
Two luciferase-labeled PC-3 clones expressing high and low levels of PTHrP were selected from previously established stable-shRNA clones targeting PTHLH (19), designating PTHrPHi and PTHrPLo, respectively. Ace-1 canine prostate carcinoma cells, expressing undetectable basal levels of PTHrP, were stably transfected with a pcDNA3.1 vector expressing full-length mouse/rat PTHrP (17). An empty-vector transfectant was used as a control. Expression of PTHrP was confirmed from the culture supernatant using an immunoradiometric assay kit (Diagnostic laboratories). PC-3 clones were regularly authenticated and matched short tandem repeat DNA profiles of the original PC-3 cell line (last tested on August 28, 2012).
Mice and in vivo tumors
All mouse experiments were approved by the Institutional Animal Care and Use Committees of the University of Michigan and Vanderbilt University. For in vivo tumors, 1×106 prostate tumor cells were suspended in 100μl Hank’s balanced salt solution and 1:1 mixed with growth factor-reduced Matrigel (BD Biosciences), followed by subcutaneous injection into male athymic mice (Harlan Laboratories) as previously described (20,21). Mice were regularly monitored for morbidity or tumor growth, and tumor size was calculated using an equation: Volume = ½×a×b2, where a=long diameter and b=short diameter measured with a caliper (22). Anti-human PTHrP 1–33 monoclonal antibodies (hybridoma 158) were produced and generously gifted by Dr. Richard Kremer (McGill University) (18). Mice were treated with anti-PTHrP monoclonal antibody (200μl) or mouse IgG (Sigma-Aldrich) by every other day IP injection for the first three weeks, followed by daily injection for one week prior to euthanasia.
Flow cytometry
For analyses of CD11b+Gr1+ cells in the tumor tissue, tumors were mechanically dissociated, followed by digestion in complete RPMI-1640 media supplemented with type I collagenase (5mg/ml; Sigma-Aldrich). Viable cells were counted and resuspended in FACS buffer containing combinations of antibodies including FITC-conjugated anti-mouse CD11b, PE-conjugated anti-mouse Gr1, or isotype controls. For analyses or sorting CD11b+Gr1+ cells from the bone marrow, the femoral bone marrow was flushed and dissociated, followed by antibody staining and flow cytometry (23). For analyses of phospho-Y418 SFK, the bone marrow cells were fixed, permeabilized, stained and analyzed according to the BD PhosFlow Cell Signaling protocols. All materials were from BD Biosciences.
Immunohistochemistry
Tumors were surgically removed and bisected, a portion fixed in formalin and a portion snap-frozen. Murine endothelial cell-specific CD31/PECAM immunostaining (clone MEC13.3, BD Biosciences) was performed according to a previously described method (24). Rat anti-mouse CD11b (clone M1/70, BD Biosciences) and anti-mouse Ly-6G (clone RB6-8C5, eBioscience) were fluorescently labeled and used to detect CD11b+Gr1+ cells in the tumor tissue. Three to five randomly selected microscopic images per sample were obtained, and positively-stained cells were counted using ImageJ software.
Quantitative PCR
mRNA samples were prepared from the bone marrow or tumor tissues using TRIzol® reagent (Invitrogen), followed by reverse transcription-quantitative PCR (25). All quantitative PCR probes and reagents were from Applied Biosystems.
Statistical Analyses
All in vivo data sets were tested for normality by Shapiro-Wilk test. Statistical analyses were performed by GraphPad™ Prism software. Student’s t-test or Mann-Whitney U test were used to compare two groups and all statistical tests were two-sided.
RESULTS
Reduction of PTHrP in PC-3 prostate tumors decreased CD11b+Gr1+ bone marrow cell recruitment and angiogenesis valid
As a first approach to investigate the role of PTHrP in the potential crosstalk between tumor and the bone marrow, the PTHLH gene (encoding PTHrP) was targeted via lentiviral shRNA vectors in PC-3, human prostate cancer cells (19). Two clones expressing high and low levels of PTHrP (961.8±12.8 vs. 457.8±4.1 pg ml−1 1×106 cells−1 48h−1; measured in the culture supernatant by immunoradiometric assays) were selected and designated PTHrPHi and PTHrPLo, respectively. PTHrP is well known to regulate tumor growth via autocrine, intracrine and paracrine manners (17–19,26,27), hence alterations in the host response (e.g. recruitment of host-derived cells) could simply be secondary to the differences in the tumor size, not in PTHrP expression levels. Therefore, PTHrPLo tumors were grown for a longer period until they reached a similar mean tumor volume as PTHrPHi tumors to circumvent the direct tumor-size effects in the subsequent analyses (Fig. 1A and B). Flow cytometric analyses of the tumor tissues revealed that PTHrPLo tumors had significantly reduced percentages of CD11b+Gr1+ cells in the tumor tissue compared with PTHrPHi tumors (Fig. 1C). Immunohistological analyses showed PTHrP levels correlated with mean vessel density and vessel area of PC-3 tumors (Fig. 1D–F). A well characterized mechanism of MDSC-dependent tumor angiogenesis is through the expression of MMP-9 (7,28). Accordingly, tumor tissues were analyzed for expression of host-derived MMP-9 expression as well as tumor-derived VEGF-A (Fig. 1G and H) using species-specific PCR probes. PTHrPLo tumors had significantly reduced host-derived (i.e. murine) Mmp9 expression, while no significant reduction in the tumor-derived (i.e. human) VEGFA was observed. Collectively, reduction of PTHrP in PC-3 prostate tumors decreased CD11b+Gr1+ cell recruitment and tumor angiogenesis, in association with reduced expression of host MMP-9 but not of tumor VEGF-A.
Fig. 1. Reduction of PTHrP in PC-3 prostate tumors decreased CD11b+Gr1+ MDSC recruitment and angiogenesis.

(A) Tumor growth curve of two PC-3 clones expressing high or low levels of PTHrP (n=9 for PTHrPHi and n=10 for PTHrPLo). PTHrPLo tumors were grown for a longer period (57 days) to reach a similar mean tumor volume as PTHrPHi tumors (38 days).
(B) PTHrPHi and Lo tumors were surgically dissected on the same day and photographed. Mean tumor volume between the two groups was not significantly different (P=0.68, Student’s t-test). Scale bar: 1cm.
(C) Percentages of CD11b+Gr1+ double-positive cells in the tumor tissues were analyzed by flow cytometry.
(D) Tumor tissues were sectioned for H&E and murine CD31/PECAM immunohistochemical staining. Original magnification, ×20. Scale bars: 50μm
(E and F) Microscopic images were analyzed for tumor mean vessel density (MVD) or CD31+ vascular area with normalization to total nuclear area.
(G and H) Host-derived (i.e. murine) Mmp9 and tumor-derived (i.e. human) VEGFA mRNA levels were measured by quantitative RT-PCR using species specific probes (n=9~10 per group).
All P values are from Student’s t-test. NS stands for not-significant. Data in all graphs are mean±SEM.
Ectopic PTHrP increased the recruitment of CD11b+Gr1+ cells in prostate tumor tissue
An additional prostate tumor model was utilized to establish the causal relationship between PTHrP and CD11b+Gr1+ cells. Ace-1 prostate cancer cells produce predominantly osteoblastic lesions in vivo, a phenotype that recapitulates human prostate cancer more realistically than the majority of currently available prostate cancer cell lines (17,29). Ace-1 cells, expressing undetectable basal levels of PTHrP, were stably transfected with PTHrP overexpression (designated PTHrP OE) or empty control (designated pcDNA) vectors. In the same approach as the PC-3 tumor model (i.e. growth in differential periods), two groups of similarly-sized tumors, PTHrP OE and pcDNA control, were produced. To directly examine the effects of systemic PTHrP on CD11b+Gr1+ cell recruitment, one group of mice carrying pcDNA control tumors was treated with recombinant PTHrP (amino acids 1–34, a ligand-binding fragment) for 7 days before harvest (Fig. 2A and Supplemental Fig. 1). Both PTHrP OE and recombinant PTHrP-treated groups had significantly increased CD11b+Gr1+ cells in the tumor tissue compared with pcDNA control tumors (Fig. 2B). While mice burdened with PTHrP OE tumors had significantly increased percentages of CD11b+Gr1+ cells in the bone marrow (Fig. 2C), recombinant PTHrP treatment failed to show such an increase in the bone marrow. This may be explained by either the different modes of PTHrP administration (i.e. intermittent injection vs. continuous expression) or the reduced duration (7 days) of PTHrP treatment compared with tumor burden (21 days). Immunohistochemical analyses of tumor tissue showed that both PTHrP OE and recombinant PTHrP tumors had significantly increased evidence of angiogenesis (Fig. 2D and Supplemental Fig. 1B). In addition, host-derived Mmp9 expression was significantly increased in PTHrP OE tumor tissue (Fig. 2E), suggesting contribution of the CD11b+Gr1+ cell recruitment, at least in part, to angiogenesis. Collectively, data in Fig. 1 and 2 suggest that prostate cancer-derived PTHrP is a crucial regulator of CD11b+Gr1+ cells.
Fig. 2. Ectopic PTHrP increased CD11b+Gr1+ cells in prostate tumors and in the bone marrow.
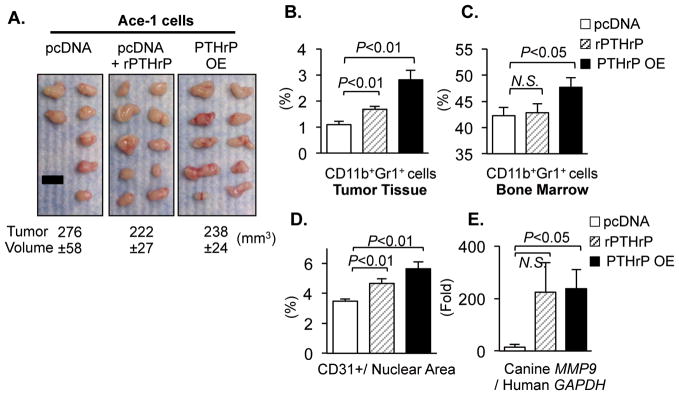
Ace-1 prostate cancer cells, expressing undetectable basal levels of PTHrP, were engineered to overexpress PTHrP (PTHrP OE) with a vector-alone transfectant control (pcDNA). Cells were implanted subcutaneously in male athymic mice.
(A) PTHrP OE (n=10; right panel) were grown for a shorter period (17 days) to produce similarly sized tumors as pcDNA control tumors (n=7; grown for 32 days; left panel). In addition, one group of mice carrying pcDNA tumors (n=10; middle panel) was administered recombinant PTHrP (1–34) once daily for 7 days prior to euthanasia (designated rPTHrP). Mean tumor volume±SEM (mm3) for each group is indicated. No statistical significance in tumor volume (P>0.05 by Student’s t-test) in any pair of groups. Scale bar: 1cm.
(B and C) Percentages of CD11b+Gr1+ cells in the tumor tissues or in the bone marrow were analyzed by flow cytometry.
(D) Tumor tissues were analyzed for quantification of CD31+ vessel area with normalization to total nuclear area (n=7~10/group).
(E) Tumor-derived (i.e. canine) MMP9 mRNA levels were measured (n=7~10/group).
All P values are from Student’s t-test. NS stands for not-significant. Data in all graphs are mean±SEM.
CD11b+Gr1+ cells promoted tumor growth in vivo
The pro-tumorigenic functions of CD11b+Gr1+ cells are relatively well characterized using multiple tumor models (5,7,30,31). To more rigorously examine the effects of CD11b+Gr1+ cells on tumor growth in the prostate tumor model, two fractions of bone marrow cells, i.e. CD11b/Gr1-double positive or negative cells, were isolated and co-implanted with parental Ace-1 tumor cells in vivo (Fig. 3A). Increasing numbers of CD11b+Gr1+ cells mixed with tumor cells correspondingly increased the tumor size within 15 days (Fig. 3B and C). More importantly, Ace-1 tumor co-implanted with 0.5×106 CD11b+Gr1+ cells grew significantly larger than tumors co-implanted with the same number of CD11b−Gr1− cells, suggesting that altered tumor size in Fig. 1 and 2 were secondary to the altered recruitment of CD11b+Gr1+ cells in the tumor tissue.
Fig. 3. CD11b+Gr1+ cells promoted tumor growth in vivo.

(A) Schematic representation of the experiment. Male athymic mice were implanted with PTHrP overexpressing Ace-1 tumors for 21 days prior to harvesting femoral bone marrow cells. Flow cytometric sorting resulted in two fractions of CD11b/Gr1-double positive or double negative cells, followed by their co-injection with parental Ace-1 cells subcutaneously in male athymic mice (n=8 per group).
(B) Individual tumor size was measured 15 days after xenograft and plotted on a graph. Dots: individual tumor volumes (mm3). Horizontal lines: medians. P values are from Student’s t-test.
(C) Tumor growth curve was plotted to compare growth kinetics. Data are mean±SEM. * P<0.05 and ** P<0.01, Student’s t-test
Tumor-derived PTHrP confers increased angiogenic potential to CD11b+Gr1+ cells
To examine whether tumor-derived PTHrP regulates CD11b+Gr1+ cells within the bone marrow of tumor hosts, CD11b+Gr1+ bone marrow cells were isolated from two groups of mice bearing either PTHrP-overexpressing or pcDNA control tumors for 3 weeks, resulting in two fractions of CD11b+Gr1+ cells (i.e. PTHrP-activated vs. control). Parental Ace-1 tumor cells were mixed with the isolated CD11b+Gr1+ cells and xenografted into male athymic mice (Fig. 4A). Tumors co-implanted with PTHrP-activated CD11b+Gr1+ cells were significantly larger than the tumors with control CD11b+Gr1+ cells (Fig. 4B), potentially due to increased MMP9 and angiogenesis as determined by immunohistochemistry (Fig. 4C, D and Supplemental Fig. 2).
Fig. 4. PTHrP promoted tumorigenic function of CD11b+Gr1+ bone marrow cells.
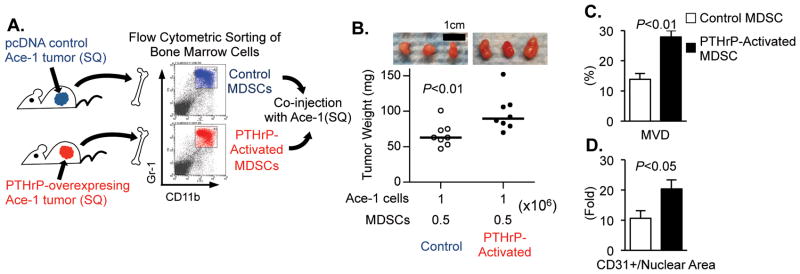
(A) Schematic representation of the experiment. Two groups of male athymic mice (n=3 per group) were implanted with PTHrP-overexpressing or control tumors for 21 days prior to flow cytometric sorting of CD11b+Gr1+ bone marrow cells. Subsequently, parental Ace-1 prostate tumor cells were co-injected with the primed CD11b+Gr1+ cells into male athymic mice (n=8 per group).
(B) Individual tumor weight was measured and plotted, and photographs of three representative tumors are shown. Dots: individual tumor weight (mg). Horizontal lines: median. (n=8 per group)
(C and D) Tumor tissues were analyzed for quantification of CD31+ mean vessel density or vessel area with normalization to total nuclear area. Data are mean±SEM. All P values are from Student’s t-test.
PTHrP increased expression of phospho-[Y418] Src family kinases in CD11b+Gr1+ cells
The molecular mechanism for the observed PTHrP-dependent CD11b+Gr1+ cell potentiation was subsequently investigated. Recently, Liang et al. demonstrated that dasatinib, a Src family kinase (SFK) inhibitor, suppressed prostate tumor growth as well as the numbers of CD11b+ myeloid cells in tumor tissues (32). Accordingly, the effects of PTHrP administration on SFK in CD11b+Gr1+ cells were investigated. A single administration of PTHrP (1–34) to male athymic mice significantly increased the activating phosphorylation of Tyr-418 residue of SFK in CD11b+Gr1+ cells (Fig. 5A). As SFK activation requires intra-molecular conformational changes and interaction with activated receptor kinases via the SH-2 domain, phosphorylation of [Y418] in the SH-2 domain indicates the status of full activation. On the other hand, since CD11b+Gr1+ cells do not express receptors for PTHrP (as determined by quantitative RT-PCR for Pthr1, Supplemental Fig. 3), phosphorylation of [Y418] SFK was reasoned to be indirect through cytokines from osteoblasts, the predominant cells expressing the PTH/PTHrP receptor (PTHR1) in the bone marrow. Potential candidate cytokines from PTHrP-stimulated osteoblasts included IL-6, VEGF-A, C-C chemokine ligand (CCL)-2 and receptor activator of NF-κB ligand (RANKL) (16,33–35). Therefore, CD11b+Gr1+ cells were isolated from femoral bone marrow and treated with these osteoblastic cytokines. Although all four cytokines (IL-6, VEGF-A, CCL-2 and RANKL) have been shown to up-regulate SFKs (36–38), only IL-6 and VEGF-A increased the expression of phospho-[Y418] SFK in MDSCs (Fig. 5B).
Fig. 5. PTHrP phosphorylated [Y418] Src family kinases in CD11b+Gr1+ cells.
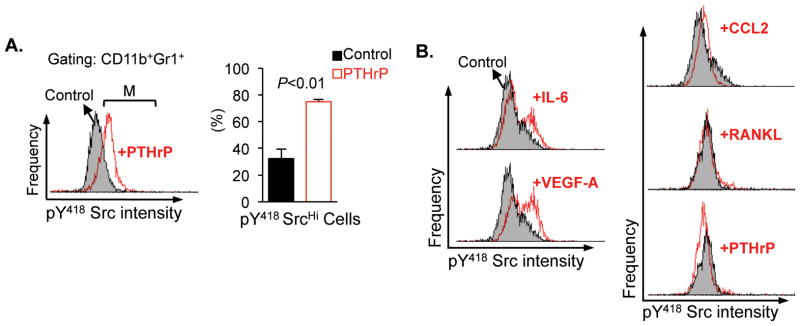
(A) Male athymic mice (n=3 per group) were stimulated with a single administration of PTHrP (1–34) or saline control, 8 hours prior to sacrifice and flow cytometric analyses of phospho-[Y418] Src family kinase expression levels in CD11b+Gr1+ bone marrow cells. Representative histograms from the control group (shaded) and the PTHrP-stimulated group (open) were overlapped to show the intensity of phospho-[Y418] Src expression. CD11b+Gr1+ cells expressing high levels of phospho-[Y418] Src family kinases (indicated by a bracket ‘M’) were quantified and plotted. Data are mean±SEM. P<0.01, Student’s t-test.
(B) CD11b/Gr1 double positive cells were sorted from the femoral bone marrow of male athymic mice, followed by treatment with saline, IL-6, VEGF-A, CCL-2, RANKL or PTHrP (all 100ng/ml for 0.5×106 cells) for 1 hour at 37°C (n=3/group). Representative histograms (open) were overlapped onto un-stimulated controls (shaded) to show the intensity of staining.
Phospho-[Y418] SFK by osteoblastic VEGF-A and IL-6 increased MMP-9 expression in CD11b+Gr1+ cells
To further investigate the functional significance of phospho-[Y418] SFKs in MDSCs, several published markers of CD11b+Gr1+ cell activation were examined in combination with PTHrP-dependent osteoblastic cytokines and a SFK selective inhibitor, PP2 (5,13,28). Only VEGF-A and IL-6 increased Mmp9 gene expression, while Cxcr2, Cxcr4 or Itgb1 expression remained unaffected in CD11b+Gr1+ cells, and this increase was reversed by PP2 treatment (Fig. 6A–D). Furthermore, to confirm the requirement of osteoblasts in PTHrP-dependent potentiation of CD11b+Gr1+ cells, primary osteoblasts were established from murine calvaria and treated with PTHrP (1–34) or saline for 24 hours and conditioned media harvested (39). CD11b+Gr1+ cells were isolated from femoral bone marrow and stimulated with osteoblast-derived control- or PTHrP-conditioned media in combination with neutralizing antibodies against VEGF-A and/or IL-6. Consistent with the previous data, PTHrP-conditioned media from osteoblast cultures increased Mmp9 gene expression (Fig. 6E) and functional MMP9 (Fig. 6F) in the MDSCs, and these effects were blocked by anti-VEGF-A and/or -IL-6 neutralizing antibodies. Furthermore, the effect of PTHrP-conditioned media on MMP9 expression was suppressed by PP2 (Fig. 6G).
Fig. 6. Phosphorylation of Src family kinases by osteoblastic VEGF-A and IL-6 increased MMP-9 expression in CD11b+Gr1+ cells.

(A–D) CD11b+Gr1+ cells were sorted from the femoral bone marrow of male athymic mice via flow cytometry, followed by treatment with saline (control), VEGF-A, IL-6, PTHrP or CCL-2 (100ng/ml for 0.5×106 cells) in combination with PP2 (an SFK inhibitor; 100nM) or DMSO control for 1 hour at 37°C (n=3/group). mRNA levels of Mmp9, Cxcr4, Cxcr2, and Itgb1 were determined via quantitative RT-PCR. Data are mean±SEM. Asterisks indicate P<0.01 compared to the DMSO control group. No other combination had statistical significance (P>0.05, Student’s t-test).
(E–G) Conditioned media were collected from primary calvarial osteoblasts stimulated with saline (control) or PTHrP (1–34). Femoral bone marrow CD11b+Gr1+ cells were treated with the control or PTHrP-conditioned media in combination with neutralizing antibodies against VEGF-A and/or IL-6, followed by (E) quantitative PCR for Mmp9 gene expression (n=3/group) or (F) zymography. Data are mean±SEM. Asterisks represent P<0.05 and NS stands for not-significant by Student’s t-test. (G) CD11b+Gr1+ cells were treated with control or PTHrP-conditioned media in combination with PP2 or DMSO control, followed by zymography.
Anti-PTHrP monoclonal antibody treatment decreased MDSC recruitment in PC-3 tumors
Lastly, to more rigorously determine the causal relationship between PTHrP and MDSC recruitment, mice bearing PTHrPHi PC-3 tumors were treated with non-specific control IgG or anti-human PTHrP monoclonal antibodies. Anti-PTHrP antibodies significantly suppressed tumor growth, but not to the level of PTHrPLo tumors (Fig. 7A and B). As anti-PTHrP monoclonal antibodies potentially suppress tumor growth via inhibition of autocrine PTHrP effects on tumor cells (Supplemental Fig. 4), tumor tissues were analyzed for MDSC recruitment by immunofluorescence co-localization of CD11b+Gr1+ cells (Fig. 7C and D). Numbers of CD11b+Gr1+ cells were decreased in anti-PTHrP antibody-treated or PTHrPLo tumor tissues, suggesting that reduced PTHrP is causal to decreased MDSCs found in tumor tissues. Serum calcium levels were correlated with PTHrP levels, indicating the functional activity of PTHrP (Fig. 7E). Quantitative RT-PCR analysis in tumor tissues revealed that shRNA-mediated PTHrP-knockdown was stable in PC-3 tumor cells during in vivo tumor growth, and the correlation between PTHrP and Mmp9 gene expression (Fig. 7F and G).
Fig. 7. Anti-human PTHrP monoclonal antibody decreased MDSC recruitment in PC-3 tumors.

(A) Tumor growth curve of PC-3 PTHrPHi tumors treated with control IgG or anti-PTHrP monoclonal antibodies (mAb), and PC-3 PTHrP Lo tumors (n=10/group). Both P values are from linear regression comparison with PC-3 PTHrPHi IgG tumor group. Data are mean±SEM.
(B) Individual tumor weight was measured upon necropsy and plotted. Dots: individual measurements (mg). Horizontal lines: mean±SEM. (n=10/group).
(C) Tumor tissues were sectioned and stained for CD11b (Alexa-Fluor 488), Gr1 (Alexa-Fluor 546), and DAPI. Original magnification, ×40. Scale bars: 50μm
(D) Immunofluorescent images were merged and analyzed for CD11b+Gr1+ cell per microscopic field. Three positively stained non-necrotic tumor areas were randomly selected for quantification (5 tumors/group).
(E) Sera were collected upon necropsy, followed by calcium assay.
(F and G) PTHLH or Mmp9 mRNA levels in the pulverized tumor tissue were measured by quantitative RT-PCR (n=10/group). Data in all bar graphs are mean±SEM. All P values, unless indicated otherwise, are from Student’s t-test.
DISCUSSION
This study provides new evidence that distant tumors stimulate the bone marrow to increase critical component cells in the tumor microenvironment. In brief, prostate cancer-derived PTHrP circulates to potentiate CD11b+Gr1+ cells within the bone marrow via up-regulation of IL-6 and VEGF-A in osteoblasts, contributing to tumor growth and angiogenesis (Fig. 8). As a proposed mechanism of CD11b+Gr1+ cell potentiation, these data demonstrate that PTHrP increased activating phosphorylation of SFKs that subsequently increased Mmp9 gene expression in CD11b+Gr1+ cells, supporting that CD11b+Gr1+ cell-dependent tumor growth is, at least in part, mediated by MMP-9 expression and angiogenesis.
Fig. 8. Proposed model of CD11b+Gr1+ cell activation within the bone marrow of prostate tumor hosts via PTHrP.

Prostate tumor-derived PTHrP circulates to stimulate VEGF-A and IL-6 expression by osteoblasts, leading to SFK phosphorylation of CD11b+Gr1+ MDSCs. Activation of SFK confers angiogenic potential of MDSCs via increased MMP-9 expression, contributing to prostate cancer growth and angiogenesis.
Increasing evidence now clearly supports the critical functions of CD11b+Gr1+ MDSCs in the tumor microenvironment (40). However, the majority of previous studies have been focused either on the roles of MDSCs within the tumor microenvironment (i.e. immune suppression and angiogenesis) or the mechanism of MDSC recruitment to the tumor (41,42). Given that bone is an essential partner for tumor progression by supplying numerous bone marrow-derived stromal cells, primary tumor cells are speculated to have active mechanisms to interact with the bone/bone marrow (43,44). The data in this study demonstrate that PTHrP serves as a messenger between the primary tumor and the bone marrow, conferring MDSCs with increased angiogenic potential. PTHrP is a potent bone modulating cytokine expressed by multiple types of tumor cells such as prostate, breast, lung and colorectal cancers (45–48). In addition, PTHrP is a key regulator of the ‘vicious cycle’ hypothesis of metastatic tumor-bone interactions (49). However, given that MDSCs are currently considered universally essential components of the tumor microenvironment, not all tumor types express PTHrP, suggesting that PTHrP is not the only factor mediating the interactions between tumor and bone.
The molecular mechanisms of MDSC activation, expansion and/or mobilization, and ultimately therapeutic approaches targeting the key signaling mechanisms, warrant extensive further investigation. Interestingly, the preliminary studies shown in Supplemental Fig. 5 suggest that PTHrP induces a series of alterations in the bone marrow to mobilize and/or expand MDSCs. Still, questions remain regarding whether and how PTHrP stimulates differentiation of MDSCs from bone marrow precursors. Nevertheless, this work provides a biological rationale for the clinical application of SFK inhibitors in targeting two compartments (i.e. tumor and the microenvironment) simultaneously, of which the mechanism requires further studies. The data in this study demonstrate that activation of SFKs is one of the key signal transduction mechanisms of MDSCs’ angiogenic potential, in addition to two other kinases, STAT3 and PI3K, that have previously been shown to be implicated in MDSC functions (13,31). Indeed, SFKs mediate crucial regulatory functions in both tumor cells and stromal cells (e.g. endothelial cells and osteoclasts), suggesting that SFKs are promising therapeutic targets for the suppression of tumor as well as stromal compartments (24,50).
In conclusion, this study provides evidence that prostate cancers positively regulate the bone marrow microenvironment via PTHrP, IL-6, VEGF-A and SFKs, thereby increasing the angiogenic potential of CD11b+Gr1+ MDSCs, leading to increased tumor growth.
Supplementary Material
Acknowledgments
Financial Supports: This work was financially supported by the Department of Defense Prostate Cancer Research Program (W81XWH-10-1-0546 and WX81XWH-12-1-0348 to S.I.P.) and the National Cancer Institute Program Project (P01CA093900 to L.K.M.). Flow cytometric analyses were supported in part by the National Cancer Institute Cancer Center Support (P30CA068485) to Vanderbilt-Ingram Cancer Center.
The authors thank Drs. Evan Keller, Russell Taichman and Kenneth Pienta for valuable discussion; and Dr. Richard Kremer for providing anti-PTHrP monoclonal antibody.
Footnotes
Conflict of Interest: All authors declare no financial conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Gene Dev. 2011;25:2559–72. doi: 10.1101/gad.169029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park SI, Soki FN, McCauley LK. Roles of bone marrow cells in skeletal metastases: no longer bystanders. Cancer Microenviron. 2011;4:237–46. doi: 10.1007/s12307-011-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SI, Liao J, Berry JE, Li X, Koh AJ, Michalski ME, et al. Cyclophosphamide creates a receptive microenvironment for prostate cancer skeletal metastasis. Cancer Res. 2012;72:2522–32. doi: 10.1158/0008-5472.CAN-11-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Edwards CM, Mundy GR. Gr-1+CD11b+ myeloid-derived suppressor cells: formidable partners in tumor metastasis. J Bone Miner Res. 2010;25:1701–6. doi: 10.1002/jbmr.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn J-I, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–75. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 8.Zhao F, Obermann S, Wasielewski von R, Haile L, Manns MP, Korangy F, et al. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology. 2009;128:141–9. doi: 10.1111/j.1365-2567.2009.03105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahn G-O, Brown JM. Influence of bone marrow-derived hematopoietic cells on the tumor response to radiotherapy: experimental models and clinical perspectives. Cell Cycle. 2009;8:970–6. doi: 10.4161/cc.8.7.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahn G-O, Tseng D, Liao C-H, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 12.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19:715–27. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dayyani F, Gallick GE, Logothetis CJ, Corn PG. Novel therapies for metastatic castrate-resistant prostate cancer. J Natl Cancer I. 2011;103:1665–75. doi: 10.1093/jnci/djr362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11:411–25. doi: 10.1038/nrc3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Loberg R, Liao J, Ying C, Snyder LA, Pienta KJ, et al. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009;69:1685–92. doi: 10.1158/0008-5472.CAN-08-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao J, Li X, Koh AJ, Berry JE, Thudi N, Rosol TJ, et al. Tumor expressed PTHrP facilitates prostate cancer-induced osteoblastic lesions. Int J Cancer. 2008;123:2267–78. doi: 10.1002/ijc.23602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Karaplis AC, Huang DC, Siegel PM, Camirand A, Yang XF, et al. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Invest. 2011;121:4655–69. doi: 10.1172/JCI46134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park SI, McCauley LK. Nuclear localization of parathyroid hormone-related peptide confers resistance to anoikis in prostate cancer cells. Endocr Relat Cancer. 2012;19:243–54. doi: 10.1530/ERC-11-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park SI, Kim SJ, McCauley LK, Gallick GE. Pre-clinical mouse models of human prostate cancer and their utility in drug discovery. Curr Protoc Pharmacol. 2010;Chapter 14(Unit14):15. doi: 10.1002/0471141755.ph1415s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung Y, Shiozawa Y, Wang J, McGregor N, Dai J, Park SI, et al. Prevalence of prostate cancer metastases after intravenous inoculation provides clues into the molecular basis of dormancy in the bone marrow microenvironment. Neoplasia. 2012;14:429–39. doi: 10.1596/neo.111740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomayko MMM, Reynolds CPC. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–54. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 23.Pirih FQ, Michalski MN, Cho SW, Koh AJ, Berry JE, Ghaname E, et al. Parathyroid hormone mediates hematopoietic cell expansion through interleukin-6. PLoS One. 2010;5:e13657. doi: 10.1371/journal.pone.0013657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–33. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 25.Novince CM, Koh AJ, Michalski MN, Marchesan JT, Wang J, Jung Y, et al. Proteoglycan 4, a novel immunomodulatory factor, regulates parathyroid hormone actions on hematopoietic cells. Am J Pathol. 2011;179:2431–42. doi: 10.1016/j.ajpath.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dougherty KM, Blomme EA, Koh AJ, Henderson JE, Pienta KJ, Rosol TJ, et al. Parathyroid hormone-related protein as a growth regulator of prostate carcinoma. Cancer Res. 1999;59:6015–22. [PubMed] [Google Scholar]
- 27.Tovar Sepulveda VA, Falzon M. Parathyroid hormone-related protein enhances PC-3 prostate cancer cell growth via both autocrine/paracrine and intracrine pathways. Regul Pept. 2002;105:109–20. doi: 10.1016/s0167-0115(02)00007-1. [DOI] [PubMed] [Google Scholar]
- 28.Ahn G-O, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeRoy BE, Thudi NK, Nadella MVP, Toribio RE, Tannehill-Gregg SH, van Bokhoven A, et al. New bone formation and osteolysis by a metastatic, highly invasive canine prostate carcinoma xenograft. Prostate. 2006;66:1213–22. doi: 10.1002/pros.20408. [DOI] [PubMed] [Google Scholar]
- 30.Abe F, Dafferner AJ, Donkor M, Westphal SN, Scholar EM, Solheim JC, et al. Myeloid-derived suppressor cells in mammary tumor progression in FVB Neu transgenic mice. Cancer Immunol Immunother. 2010;59:47–62. doi: 10.1007/s00262-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367–77. doi: 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang W, Kujawski M, Wu J, Lu J, Herrmann A, Loera S, et al. Antitumor activity of targeting SRC kinases in endothelial and myeloid cell compartments of the tumor microenvironment. Clin Cancer Res. 2010;16:924–35. doi: 10.1158/1078-0432.CCR-09-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang YF, Harrison JR, Lorenzo JA, Kream BE. Parathyroid hormone induces interleukin-6 heterogeneous nuclear and messenger RNA expression in murine calvarial organ cultures. Bone. 1998;23:327–32. doi: 10.1016/s8756-3282(98)00115-x. [DOI] [PubMed] [Google Scholar]
- 34.Lowik CW, van der Pluijm G, Bloys H, Hoekman K, Bijvoet OL, Aarden LA, et al. Parathyroid hormone (PTH) and PTH-like protein (PLP) stimulate interleukin-6 production by osteogenic cells: a possible role of interleukin-6 in osteoclastogenesis. Biochem Biophys Res Commun. 1989;162:1546–52. doi: 10.1016/0006-291x(89)90851-6. [DOI] [PubMed] [Google Scholar]
- 35.Esbrit P, Alvarez-Arroyo MV, De Miguel F, Martin O, Martinez ME, Caramelo C. C-terminal parathyroid hormone-related protein increases vascular endothelial growth factor in human osteoblastic cells. J Am Soc Nephrol. 2000;11:1085–92. doi: 10.1681/ASN.V1161085. [DOI] [PubMed] [Google Scholar]
- 36.Sabbota AL, Kim H-RC, Zhe X, Fridman R, Bonfil RD, Cher ML. Shedding of RANKL by tumor-associated MT1-MMP activates Src-dependent prostate cancer cell migration. Cancer Res. 2010;70:5558–66. doi: 10.1158/0008-5472.CAN-09-4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hallek M, Neumann C, Schaffer M, Danhauser-Riedl S, Bubnoff von N, de Vos G, et al. Signal transduction of interleukin-6 involves tyrosine phosphorylation of multiple cytosolic proteins and activation of Src-family kinases Fyn, Hck, and Lyn in multiple myeloma cell lines. Exp Hematol. (1997) 1997;25:1367–77. [PubMed] [Google Scholar]
- 38.Inngjerdingen M, Torgersen KM, Maghazachi AA. Lck is required for stromal cell-derived factor 1 alpha (CXCL12)-induced lymphoid cell chemotaxis. Blood. 2002;99:4318–25. doi: 10.1182/blood.v99.12.4318. [DOI] [PubMed] [Google Scholar]
- 39.Jin R, Sterling JA, Edwards JR, DeGraff DJ, Lee C, Park SI, et al. Activation of NF-kappa B Signaling Promotes Growth of Prostate Cancer Cells in Bone. Wang QJ, editor PLoS One. 2013;8:e60983. doi: 10.1371/journal.pone.0060983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youn J-I, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Nature Immunology. Nature Publishing Group; 2013. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer; pp. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coffelt SB, Lewis CE, Naldini L, Brown JM, Ferrara N, De Palma M. Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am J Pathol. 2010;176:1564–76. doi: 10.2353/ajpath.2010.090786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shojaei F, Zhong C, Wu X, Yu L, Ferrara N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008;18:372–8. doi: 10.1016/j.tcb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Koh AJ, Wang Z, Soki FN, Park SI, Pienta KJ, et al. Inhibitory effects of megakaryocytic cells in prostate cancer skeletal metastasis. J Bone Miner Res. 2011;26:125–34. doi: 10.1002/jbmr.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X, Liao J, Park SI, Koh AJ, Sadler WD, Pienta KJ, et al. Drugs which inhibit osteoclast function suppress tumor growth through calcium reduction in bone. Bone. 2011;48:1354–61. doi: 10.1016/j.bone.2011.03.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kremer R, Li J, Camirand A, Karaplis AC. Parathyroid Hormone Related Protein (PTHrP) in Tumor Progression. Adv Exp Med Biol. (2011) 2011;720:145–60. doi: 10.1007/978-1-4614-0254-1_12. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H, Yu C, Dai J, Keller JM, Hua A, Sottnik JL, et al. Oncogene. Nature Publishing Group; 2013. Parathyroid hormone-related protein inhibits DKK1 expression through c-Jun-mediated inhibition of b-catenin activationof the DKK1 promoter in prostate cancer; pp. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCauley LK, Martin TJ. Twenty-five years of PTHrP progress: From cancer hormone to multifunctional cytokine. J Bone Miner Res. 2012;27:1231–9. doi: 10.1002/jbmr.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho SW, Pirih FQ, Koh AJ, Michalski M, Eber MR, Ritchie K, et al. The Soluble Interleukin-6 Receptor Is a Mediator of Hematopoietic and Skeletal Actions of Parathyroid Hormone. J Biol Chem. 2013;288:6814–25. doi: 10.1074/jbc.M112.393363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, et al. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–9. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335:249–59. doi: 10.1007/s00441-008-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.