

Abstract
Delusions are maladaptive beliefs about the world. Based upon experimental evidence that prediction error—a mismatch between expectancy and outcome—drives belief formation, this study examined the possibility that delusions form because of disrupted prediction-error processing. We used fMRI to determine prediction-error-related brain responses in 12 healthy subjects and 12 individuals (7 males) with delusional beliefs. Frontal cortex responses in the patient group were suggestive of disrupted prediction-error processing. Furthermore, across subjects, the extent of disruption was significantly related to an individual’s propensity to delusion formation. Our results support a neurobiological theory of delusion formation that implicates aberrant prediction-error signalling, disrupted attentional allocation and associative learning in the formation of delusional beliefs.
Keywords: prediction error, associative learning, fMRI, delusions, psychosis
Introduction
Delusions are fixed and irrational beliefs. Establishing their neurobiological basis is a major challenge, given their complex and insidious nature (Gilleen and David, 2005; Corlett et al., in press). Advances in our understanding of the neural bases of learning and belief formation may help us to meet this challenge. Theories of psychiatric illness that implicate the formation of abnormal associations between ideas have a long history (Locke, 1690/1976; Hartley, 1801; Pavlov, 1928). Indeed, the earliest theories of schizophrenia suggest that the formation of such aberrant associations is a core disease process (Bleuler, 1911/1950; Schneider, 1930). These views have been refined and, latterly, embedded in the neural architecture of learning and association formation (Miller, 1976; Beninger, 1988; Gray et al., 1991; Vinogradov et al., 1992; Hemsley, 1994; Kapur, 2003; Corlett et al., in press).
Prediction error, the mismatch between expectancy and experience, plays a direct role in forming and strengthening associations (Rescorla and Wagner, 1972; Dickinson, 2001) and has an indirect impact upon learning through the allocation of attention to stimuli in the environment. Organisms attend to events that violate their expectancies, which in turn facilitates associative learning (Mackintosh, 1975; Pearce and Hall, 1980; Grossberg, 1982). Psychotic patients describe how, in the early stages of their illness, irrelevant details capture their attention and are imbued with inappropriate significance (Matussek, 1952; McGhie and Chapman, 1961; Chapman, 1966). The attempt to rationalize and account for these bizarre experiences may result in the formation of delusions (Maher, 1974; Gray et al., 1991; Hemsley, 1994; Kapur, 2003; Corlett et al., in press).
Data from laboratory animals indicates that prediction errors are signalled by midbrain dopamine neurons (Schultz, 2000; Waelti et al., 2001). These neurons send dense projections to the basal ganglia and prefrontal cortex, forming the mesocorticolimbic dopamine system (Mann, 1986). Given that this system is strongly implicated in the pathophysiology of schizophrenia (Robbins, 1990; Grace, 1993; Laruelle et al., 2003), and that delusions are theorised to result from abnormal formation of associations, it has been suggested that dysfunction of the mesocorticolimbic dopamine system causes delusion formation via disrupted prediction-error signalling (Gray et al., 1991; Hemsley, 1994; Kapur, 2003; Corlett et al., in press).
We have previously identified a reliable marker for prediction-error processing in right prefrontal cortex (rPFC) using fMRI (Fletcher et al., 2001; Corlett et al., 2004; Turner et al., 2004) and have recently shown that ketamine, a drug which causes a transient psychosis, perturbs this brain response in healthy individuals. Furthermore, the magnitude of the prediction-error response under placebo predicts an individual’s likelihood of experiencing delusional beliefs under ketamine (Corlett et al., 2006). While these data are consistent with a link between disrupted prediction-error signalling and delusional beliefs, direct evidence is needed from individuals suffering a psychotic illness. In the present study, we therefore used the associative causal learning task that we used previously (Corlett et al., 2004, 2006) to evaluate prediction-error signal in such individuals, relating this signal, on an individual basis, to delusion severity.
Experimental procedure
Subjects
Twenty-eight subjects were recruited for the study. Fourteen healthy volunteers were identified from within the local community by advertisement. Fourteen psychotic patient volunteers were identified from the Cambridge-based CAMEO early intervention in psychosis service (http://www.cameo.nhs.uk). All patients had a diagnosis of first-episode psychosis according to the following criteria; clinical presentation with psychotic symptoms for the first time; presentation with psychotic symptoms with suspected untreated episodes in the past, or following previous treatment with antipsychotic medication for less than 6 months (Barnett et al., 2005).
Two healthy volunteers and two CAMEO patients were unable to comply with the scanning procedure and so each group was comprised of 12 subjects (Healthy control; 8 males and 4 females and CAMEO patients; 7 males and 5 females). All subjects gave written informed consent prior to their involvement in the study and received an honorarium for their participation. The study was approved by the Addenbrooke’s NHS Trust Local Research Ethics Committee and was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). The groups were matched for age, handedness and IQ as measured using the National Adult Reading Test (Nelson, 1982). The mean age was 26 years (standard deviation, SD = 3) for both groups, the mean IQ for the control group was 116 (SD = 5); for the patient group it was 111 (SD = 12). Both psychotic and healthy subjects had normal structural MR brain scans, as confirmed by neuroradiological assessment. Control subjects were without a history of psychiatric or physical illness (particularly cardiovascular or neurological disorders), head injury, any history of drug or alcohol dependence, based on subjects’ responses during a locally devised telephone screening interview. Both patient and control subjects were without contra-indications for fMRI scanning.
Patient clinical information
At the time of scanning, patients underwent a psychiatric assessment using the Brief Psychiatric Ratings Scale (BPRS; Ventura et al., 1993). This assessment revealed both positive symptoms (e.g. hallucinations, delusions and thought disorder, BPRS positive symptom score, 14 ± 3) and negative symptoms (e.g. anhedonia and affective flattening, BPRS negative symptom score, 6 ± 3). Eight of the 12 patients were stabilized on atypical antipsychotic medication: see Table 1 (chlorpromazine equivalent dose, 181 ± 70 mg/day (Woods, 2003).
Table 1. Patient clinical information.
| Subject | Medication | Age (years) | Age at first symptoms |
BPRS unusual thought content |
BPRS hallucinations |
BPRS negative symptoms |
BPRS conceptual disorganization |
DSM IV diagnosis |
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 34 | 33 | 3 | 5 | 3 | 1 | Schizophrenia |
| 2 | 100 | 28 | 23 | 3 | 2 | 7 | 1 | Schizophrenia |
| 3 | 0 | 20 | 16 | 6 | 1 | 13 | 3 | Schizophrenia |
| 4 | 200 | 25 | 22 | 5 | 3 | 6 | 1 | Schizophrenia |
| 5 | 200 | 26 | 24 | 4 | 1 | 3 | 1 | Schizophrenia |
| 6 | 300 | 23 | 22 | 2 | 5 | 3 | 1 | Schizophrenia |
| 7 | 200 | 22 | 21 | 6 | 1 | 6 | 1 | Schizophrenia |
| 8 | 200 | 26 | 25 | 6 | 5 | 10 | 1 | Bipolar disorder |
| 9 | 0 | 24 | 23 | 6 | 1 | 4 | 1 | Schizophrenia |
| 10 | 0 | 27 | 17 | 3 | 3 | 7 | 1 | Psychosis not otherwise specified |
| 11 | 50 | 21 | 20 | 4 | 4 | 3 | 1 | Schizophrenia |
| 12 | 200 | 33 | 33 | 1 | 3 | 3 | 1 | Schizophrenia |
Note:This table summarizes patients’ clinical information.Medication level is given in chlorpromazine equivalent doses in mg (Woods, 2003). BPRS scores are taken from the Brief Psychiatric Rating Scale (Ventura et al., 1993) and were acquired on the day of scanning. DSM IV diagnoses were obtained 12 months after scanning and correspond to criteria outlined in the Diagnostic and Statistical Manual of Mental Disorders IV – Text Revision (2000).
Associative learning task
We used the task reported by Corlett et al. (2004), a retrospective revaluation paradigm in which engendered expectations are violated to produce a prediction error. The task design is summarized in Table 2. Subjects were asked to imagine themselves working as an allergist confronted with a new patient ‘Mr X’, who suffers allergic reactions following some meals but not others. Their task was to work out which foods caused allergic reactions by observing the consequences of eating various foods. The task consisted of a series of trials each of which had the general structure summarized in Fig. 1. Trials comprised presentation of a food picture (representing a meal eaten by Mr X), a predictive response by the subject and, following this, an outcome. Subjects’ behavioural responses indicated both the outcome that they predicted for a particular meal (which button that they pushed) and their confidence in that prediction (how long they held down the button).
Table 2. Summary of experimental design.
| Stage 1 (12) |
Stage 2 (6) |
Stage 3 (6) |
|
|---|---|---|---|
| A1B1+ | A1+ | B1+ | Backward blocking (violation) |
| A2B2+ | A2+ | B2− | Backward blocking (confirmation) |
| C1D1+ | C1− | D1+ | Unovershadowing (confirmation) |
| C2D2+ | C2− | D2− | Unovershadowing (violation) |
| I+ | I+ | I+ | Control for violation of backward blocking |
| J− | J− | J− | Control for violation of unovershadowing |
Note: Each letter represents a different food picture (counterbalanced across subjects), + indicates the presence of an allergic reaction, − indicates the absence of an allergic reaction. Numbers in parentheses indicate the number of repetitions of each trial type for that stage.
Fig. 1. Trial structure.

On each trial, subjects were presented with a meal that their patient had eaten, and then they made a predictive response. Finally they were informed of the effect of that meal on their patient.
Experimental structure
The key manipulations relevant to the question under study are summarized in Fig. 2. Each subject was trained concurrently on a number of different contingencies between foods and allergic reactions. Learning occurred over three stages: Stage 1, Training; Stage 2, Retrospective revaluation and, finally, Stage 3, Violation. This design is clarified with examples in Fig. 2 and Table 2. In summary expectancies were set using an initial training phase. Retrospective revaluation (unovershadowing and backward blocking) occurred, engendering revalued expectancies about particular foods and, at the critical stage, we explored the impact on brain activity of violation of those revalued expectancies, relative to events that confirmed subjects’ expectancies (Table 2), in which a consistent relationship between a stimulus and an outcome (or non-outcome) was maintained throughout the course of the experiment.
Fig. 2. Experimental stages.
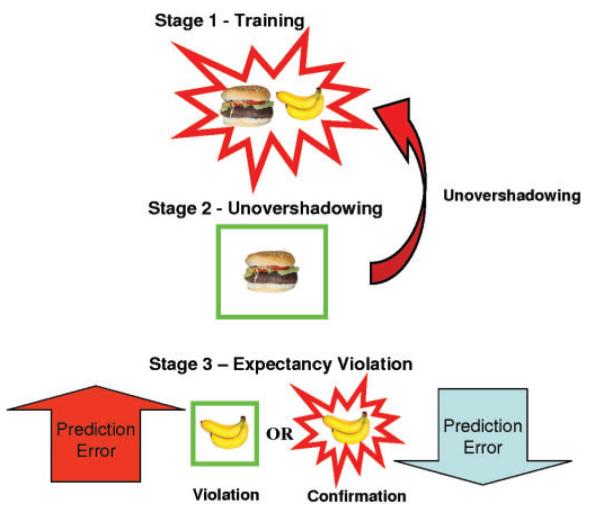
Stage 1—Training: This preliminary stage set up the expectancies. The key trial types in this were pairs of foods (in this case, hamburger paired with banana) in which subjects learned to expect an allergic response. Stage 2—Unovershadowing: In the unovershadowing condition, one cue from the pair (here, Hamburger) that had previously been paired with an allergy was presented without an allergy outcome. The aim was to engender an augmented expectancy that the other cue from the pair (Banana) would cause an allergy. This process of increasing the expectancy associated with the absent food is called unovershadowing. Stage 3—Violation of learned expectancies: This was the critical stage of the experiment. It involved items that were absent at stage 2 but subject to unovershadowing (represented here by the banana). Half of these items were presented in association with an allergic reaction, half with no allergic reaction. Critically, the trials on which no outcome was presented following unovershadowing should violate the expectation engendered by retrospective revaluation during stage 2. Items associated with an allergic reaction following unovershadowing should fulfill the prediction engendered by the revaluation process. Thus the occurrence of no allergic reaction following banana, should be surprising (i.e. should engender a prediction error).
fMRI data acquisition
A Bruker MedSpec 30/100 (Ettlingen, Germany) operating at 3 Tesla was used to collect imaging data. Gradient-echo echo planar T2*-weighted images depicting BOLD contrast were acquired from 21 non-contiguous near axial planes: TR = 1.1 s, TE = 27.5 ms, flip angle = 66°, in-plane resolution = 3.1 × 3.1 mm, matrix size 64 × 64, field of view 20 × 20 cm, bandwidth 100 kHz. A total of 705 volumes per subject were acquired in stage 1 and 893 volumes per subject across stages 2 and 3 (21 slices each of 4 mm thickness, interslice gap 1 mm). The first 6 volumes were discarded to allow for T1 equilibration effects leaving 887 volumes.
fMRI data analysis
fMRI data were analysed using statistical parametric mapping in the SPM2 programme (Wellcome Department of Cognitive Neurology, London, UK; http://www.fil.ion.ucl.ac.uk/spm). Images were realigned, spatially normalized to a standard template and spatially smoothed with a Gaussian kernel (8 mm). The time series in each session were high-pass filtered (to a maximum of 1/120 Hz) and serial autocorrelations were estimated using an AR(1) model. The average haemodynamic response to each event type was designated as beginning at the presentation of the food stimulus and lasting throughout the duration of the outcome presentation (total of 4 s). This modelling strategy was chosen in order to take into account neural responses to the predictive cues and the production of a behavioural response. By using subtractive analyses, we attempted to avoid any deleterious contributions of cue processing or button-push responses to our final estimates of neural responses to outcomes during learning, without over fitting the data by separately modelling multicollinear events (Cairo et al., 2004). Each trial was modelled using a canonical, synthetic haemodynamic response function (Friston et al., 1998). This function was used as a covariate in a general linear model and a parameter estimate was generated for each voxel for each event type. The parameter estimate, derived from the mean least squares fit of the model to the data, reflects the strength of covariance between the data and the canonical response function for a given condition. Individuals’ contrast images, derived from the pair-wise comparisons between key events, were then entered into a second level group analysis for each of the stages. At this stage, for each subject, we derived parameter estimates, averaged across each of the five regions within a mask from our previous study using the same experimental design (Corlett et al., 2004, 2006; see Fig. 4). The five regions were right lateral prefrontal cortex (rPFC), left and right nucleus accumbens and left and right substantia nigra. These regions of interest were identified in SPM2 using the PickAtlas tool (Maldjian et al., 2003) with anatomical definitions (nucleus accumbens and substantia nigra) and a spherical region of interest, radius 10 mm, centred on co-ordinates defined from previous data sets (Corlett et al., 2004, 2006). Averaged parameter estimates for each region for each subject were then entered into two sample t-tests within the Statistical Package for Social Sciences 13.0 for Windows (SPSS). A Bonferonni correction was applied to adjust for the five multiple regions that entered analyses.
Fig. 4. Group differences in brain response to the acquisition of causal associations.

The maximum intensity projection depicts between-group differences in brain responses to basic associative learning. The rendered images show the position of specific regional differences and next to those renderings, the plots display average parameter estimates from those regions for the controls (C) and patients (P).
Planned comparisons
Patient versus Control differences in
Brain activations accompanying basic associative learning (stage 1)
For each of the ROIs, we examined responses to the acquisition of causal associative relationships across groups to a low-level fixation baseline, to establish whether there were any significant differences in brain responses to initial associative learning.
Activation to unovershadowing versus well-learned control trials (stage 2)
Based on previous data (Corlett et al., 2004) and computational models (Aitken and Dickinson, 2005), unovershadowing is the more powerful retrospective revaluation effect. It is for this reason that we confine our stage 2 analyses to the comparison of unovershadowing trials (Fig. 2) with well-learned control items (J- in Table 2).
Activation at stage 3 occurring as a consequence of violation of learned expectancies
The retrospective revaluation process in stage 2 engenders a modified expectancy of the causal strength of the unovershadowed items that had been presented in stage 1 and were absent in stage 2. Re-presenting these stimuli singly in stage 3 enabled us to determine brain responses to violations of this modified expectancy. For this analysis, we compared events in which unovershadowing-based expectancy was violated (D- in Table 2, see Fig. 2) with well-learned control items (J- Table 2). Since these well-learned trials are consistently presented without an allergic outcome throughout training, subjects learn to expect the absence of an allergic outcome in their patient following these meals. The comparison of unovershadowing violation trials with well-learned control items should therefore provide a reliable neurophysiological measure of subjects’ prediction-error response (Corlett et al., 2006), which is spatially consistent with prediction-error responses engendered in the context of a range of human causal learning phenomena (Fletcher et al., 2001; Corlett et al., 2004; Turner et al., 2004).
Investigating the relationship between delusion scores and brain response to associative learning in psychotic patients
Demonstrating that prediction-error-driven associative learning is disrupted in psychotic patients would provide some support for associative theories of symptom formation. However, more compelling evidence in favour of such theories would be the demonstration of a relationship between the extent to which prediction-error processing is disrupted in psychotic patients and the severity of their symptomatology. The relationship between positive and negative symptoms at the time scanning, and brain responses to simple associative learning, unovershadowing and prediction-error processing were assessed using backward multiple regression (Altman, 1991). This process begins by fitting the full model of all potentially explanatory symptom variables and proceeds by removing unimportant variables, one at a time, until all of those remaining in the model contribute significantly. The symptom variables entered into the model were BPRS unusual thought content; grandiosity; suspiciousness; hallucinations; conceptual disorganization and negative symptom scores (Ventura et al., 1993).
Effects of medication
Of the 12 psychotic patients included in the final analysis, 8 were medicated on stable doses of atypical antipsychotics, 4 were unmedicated. The effect of antipsychotic medication upon brain activation in the context of this task was determined by computing the correlation, across subjects, between brain activation in the regions of interest at stages 1, 2 and 3 and chlorpromazine equivalent dose of medication (Woods, 2003).
A dynamic assessment of brain changes in response to changing prediction error
Prediction errors change with training and, in a secondary analysis of the data, we explored further the nature of aberrant prediction-error processing in individuals with psychosis by exploring the relationship between trial-by-trial behavioural predictions and brain responses to outcome presentation. Taking the data from the learning stage, we calculated, for each individual and each trial type, trial-by-trial estimates of prediction error based upon individually specified behavioural responses. Brain responses were modelled as canonical haemodynamic responses occurring at the presentation of feedback in each trial parametrically modulated by the behavioural response and the confidence asserted by the individual with respect to this response. Put simply, this analysis was based upon the hypothesis that prediction error should diminish as, across trials, individuals made correct responses with increasing confidence and that brain regions responding to prediction error should show decreases with this increasing confidence. For patients and controls separately, we analysed the nature of relationship between activity [in regions within the previously specified mask (see earlier)] and this trial-by-trial regressor using one-sample t-tests. We then compared the relationships across groups using a two-sample t-test. Our hypothesis was that, complementary to the findings from the subtraction analysis above, there would be a significant relationship between prediction error and rPFC response in the controls and that this would be attenuated in patients. In a further analysis, confined to the patient group, we explored whether the relationship was modulated by the BPRS measure of unusual thought content. In this case, we sought to determine whether a perturbed prediction-error response in rPFC was correlated with delusional symptoms.
Results
Behavioural results
Figure 3 illustrates subjects’ behavioural responses across the three training phases. It is clear from the plots that both patients and controls rapidly learn to predict the outcome of each meal, with increasing confidence. Analyses of variance with group (patient and control) as a between-subjects factor and training trial as a repeated measure, were conducted upon these data using SPSS 13.0 for Windows. The stage 1 analysis revealed a significant effect of training trial [F(5,118) = 57.9, P = 0.0001] but no group by training interaction [F(5,118) = 0.642, P = 0.681]. The findings were similar at stage 2; a significant effect of training [F(4,85) = 28.3, P<0.0001] but no significant group by training interaction [F(4,85) = 0.31, P = 0.873]. These findings suggest that both psychotic patients and controls did indeed acquire the associative relationships between foods and outcomes.
Fig. 3. Behavioural performance.

Subjects’ predictive responses are charted (y-axis shows scores based on prediction and confidence as described in the text: a positive value reflects a tendency to predict an outcome, a negative value to predict no outcome; the number refers to the ‘confidence’ expressed in terms of the length of button-push). The evolution of prediction and confidence is depicted across trials (x-axis). An upward deflection represents a tendency to predict with increasing confidence, that a food will cause an allergic reaction. A downward deflection represents a prediction of no allergy. Control data are presented on the left, patient data on the right. Stage 1—Training: The average behavioural response to meals associated with an allergy (red line) and no allergy (green line) are presented. Stage 2—Unovershadowing: Plots show subjects’ predictive responses to single items not paired with an allergy, that were previously presented as part of a pair during stage 1. Stage 3—Predictive response to first presentation during stage 3 of unovershadowed items: This measure is taken as an estimate of the occurrence of unovershadowing during the previous stage. Data on controls and patients are shown.
Unovershadowing was similar in both groups, confirmed by subjects’ initial predictive responses at stage 3. Subjects’ initial choice (allergy or no allergy) and their confidence in that choice, about revalued items at trial 1 of stage 3 relative to well-learned control items, provides a measure of what they learned about the absent food. These data are depicted in Fig. 3. Analysis of variance revealed that unovershadowing had indeed taken place, since both patients and control subjects predict an allergic response following unovershadowed items relative to well-learned items [main effect of task, F(1,22) = 84.37, P<0.001]. There was no significant group by learning interaction [F(1,22) = 0.357, P = 0.556].
Overall, the behavioural observations show that both patients and controls were capable of acquiring basic associations and of revaluating these associations as a result of additional information. This matched performance is important to interpreting any brain differences since, clearly, it would be difficult to interpret differences in prediction-error responses if subjects were making different predictions (Price and Friston, 1999).
Neuroimaging results
Patient versus control analyses
Stage 1 (training)
Table 3 summarizes the group differences in brain response to stage 1 training. This stage represents the basic acquisition of associative relationships between causal food cues and allergy/no allergy outcomes. As Fig. 4 shows, psychotic patients failed to activate the left caudate relative to controls.
Table 3. Group differences in brain response to acquisition (stage 1), revaluation (stage 2) and violation (stage 3) of causal relationships.
| Stage | Region | t-value | P-value |
|---|---|---|---|
| 1 | L Striatum | 3.15 | 0.01 |
| 2 | L substantia nigra | 3.02 | 0.02 |
| 2 | R substantia nigra | 3.20 | 0.01 |
| 2 | R prefrontal cortex | 2.94 | 0.02 |
| 3 | R prefrontal cortex | 2.74 | 0.03 |
Note: The table depicts the results of region of interest analyses. Only significant (P<0.05 Bonferroni corrected) differences between patients and controls are reported
Stage 2 (retrospective revaluation)
Between-group differences in the neurophysiological response to unovershadowing are detailed in Table 3 and Fig. 5. In psychotic patients, bilateral substantia nigra and rPFC were not significantly more active for unovershadowed items than well-learned control items.
Fig. 5. Group differences in brain response to retrospective revaluation of causal associations.

The maximum intensity projection depicts between-group differences in brain responses to unovershadowing. The rendered images show the position of specific regional differences and next to those renderings, the plots display average parameter estimates from those regions for the controls (C) and patients (P), for unovershadowed (Unover) and control items.
Stage 3 (violation of expectancies)
The characteristic response to prediction error in rPFC (Fletcher et al., 2001; Corlett et al., 2004; Turner et al., 2004; Corlett et al., 2006) was perturbed in patients. Figure 6 and Table 3 depict this effect. The significant group-by-expectancy-violation interaction seems to be being driven by two effects; first, an attenuation of the rPFC activation to the unexpected event and secondly, an augmentation of the rPFC response to predictable events.
Fig. 6. Group differences in brain response to prediction error.

The maximum intensity projection depicts between-group differences in brain response to prediction error. The rendered image shows the specific position of regional differences. In the adjacent panel, the plot displays average parameter estimates from those regions for the controls (C) and patients (P) for violation and control items.
Relating brain prediction-error responses to delusions in the patient group
Brain response to expectancy violation is disrupted in first-episode psychosis patients, consistent with the associative theory outlined earlier. This finding was further supported by an observation that, following backward linear regression analysis as described earlier, the current level of unusual thought content on BPRS was the only surviving symptom correlating with rPFC response to expectancy violation at stage 3. Specifically, the greater the level of current unusual thought content, the less likely that rPFC activation distinguished violation and fulfillment of expectancy (Fig. 7).
Fig. 7. Relating brain response to prediction error with delusion formation.

The rendered image highlights a region of right lateral prefrontal cortex, the activity of which correlates across patients with their delusion severity at the time of scanning. The plot represents this significant relationship. The y-axis represents BPRS unusual thought content score at the time of scanning. The x-axis represents brain activation difference between expected and unexpected events.
Effects of medication
There was a significant (P<0.05, r = −0.6) correlation between activation in left ventral striatum activation during the acquisition of causal associations at stage 1 and chlorpromazine equivalent dose of antipsychotic medication. There were no other significant correlations between brain activity and medication level at stages 2 and 3. Thus, the group difference noted at stage 1 should be treated with caution as a potential effect of medication. It will not be discussed further, particularly given that the key experimental manipulations were at stages 2 and 3 (Table 2).
Post hoc analyses
Regions more active in patients than controls in response to expectancy violation
Since the behavioural performance of individuals with psychosis and healthy controls did not differ significantly (see earlier), it is possible that individuals with psychosis engaged compensatory mechanisms. To examine this possibility, we contrasted the brain responses of patients and controls to expectancy violation at stage 3, weighting the contrast to identify regions that were more active in patients than controls. At a whole-brain uncorrected threshold of P<0.05 we found a region of right middle temporal cortex that was more active in patients than controls. However, activation in this region did not correlate with behavioural performance on the task, as measured by subjects’ behavioural confidence and prediction about the first presentation of the item that had been subject to unovershadowing (Pearson’s r = 0.173, P = 0.59).
Relating behavioural performance with delusion severity
We examined the possibility of a relationship between subject’s learning performance and their delusion severity, regressing patients’ delusion scores (as measured by their BPRS unusual thought content score) on their behavioural performance (as measured by their prediction and confidence about the unovershadowed item at the first trial of stage 3). There was no significant correlation between subjects behavioural learning and their delusion severity (Pearson’s r = 0.125, P = 0.698).
Analysis of brain changes in response to trial-by-trial changes in prediction-error: controls
As expected, with decreasing prediction error (estimated by increasing confidence in correct responses), activity in rPFC was significantly reduced (x, y, z = 42, 16, 20, z = 3.6; x, y, z = 54, 26, 18, z = 3.2). This provides strong confirmatory evidence for the relationship between activity in rPFC and prediction error.
Patients
There was no evidence of a significant relationship between rPFC and the dynamic measure of prediction error in the patient group as a whole.
Controls versus patients
There was some, albeit subtle, evidence of a significant group by prediction-error interaction, with the relationship between rPFC and prediction error being significantly stronger in controls than patients (x, y, z = 42, 16, 20, z = 2.3 and 44, 14, 16; z = 2.9). These data are summarized in Fig. 8a.
Fig. 8. Dynamic changes in rPFC with trial-by-trial changes in prediction error.

(a) The region of right lateral PFC in which there was a significant relationship between prediction-error changes (as estimated by changing behavioural response) and activation in controls (shown in red). Superimposed on this is the region (shown in yellow) in which there were group differences in this relationship. Specifically, there was an attenuation of this right PFC-prediction-error relationship in the patient group. (b) A surface rendering showing the region of right lateral PFC in which there was a significant inverse correlation between PFC prediction-error response (using the trial-by-trial estimate as for Fig. 8a) and symptoms scores (unusual thought content on BPRS). The plot in Fig. 8c shows this relationship graphically. Specifically, we have plotted across individuals, the BPRS scores against the extent to which lateral PFC activity correlated with out estimate of trial-by-trial prediction error. As can be seen, individuals in whom this relationship was most strongly positive, showed lower scores on this symptom (Pearson’s r = 0.81).
Relationship between trial-by-trial prediction-error responses and delusional severity
The BPRS score on unusual thought content, an estimate of delusional severity, was significantly related to the rPFC-prediction-error pattern in the patient group. Those individuals showing lack of the predicted relationship were the ones showing delusional scores of greatest severity on the day of scanning (x, y, z = 34, 34, 26, Pearson’s r = 0.81, P<0.001). See Fig. 8b and c.
Discussion
We determined brain responses to the acquisition, revaluation and violation of associations in psychotic patients. In addition to a general attenuation in prediction-error-related signal in rPFC, the patient group showed a significant relationship between this attenuation and their current experience of odd beliefs (as measured by their unusual thought content score on the BPRS). Given the consistent relationship between rPFC response and the violation of learned expectancies in causal learning (Fletcher et al., 2001; Corlett et al., 2004; Turner et al., 2004; Corlett et al., 2006); this perturbation, in psychotic patients, of rPFC response and its correlation with delusion score provides experimental support for learning-based accounts of delusion formation (Beninger, 1988; Gray et al., 1991; Vinogradov et al., 1992; Hemsley, 1994; Kapur, 2003; Corlett et al., in press). Our findings are consistent with the prior suggestion of fronto-basal-ganglia disruption in psychosis (Robbins, 1990), a suggestion which has already received support from neuropsychological (Elliott et al., 1995; Joyce et al., 1996; Pantelis et al., 1997; Hutton et al., 1998) and functional brain imaging studies (Bertolino et al., 1999; Biver et al., 1995; Buchsbaum et al., 1999; Manoach et al., 2000; Meyer-Lindenberg et al., 2002). We extend these studies by demonstrating a relationship between a specific psychological process (prediction-error processing), its neural instantiation (rPFC) and a psychotic symptom (delusions).
It is important to note that we have taken a symptom-rather than syndrome-approach to psychosis. Consequently, our findings are applicable to delusions in general but are not diagnostically specific. Our experimental approach to understanding delusions rests upon a series of studies in healthy controls implicating a frontostriatal system in prediction-error-dependent causal associative learning (Fletcher et al., 2001; Corlett et al., 2004; Turner et al., 2004; Corlett et al., 2006).
The present data are consistent with the possibility that in psychotic patients, prediction errors are signalled inappropriately and those errors maladaptively update the prefrontal representation of the world with irrelevant information. In this respect, it is noteworthy that brain responses to violations of learning (at stage 3) correlated with delusions. Furthermore, our use of individual- and trial-specific regressors to estimate dynamic brain changes corroborated the link between prediction error signal and delusions: those patients in whom there was a relatively preserved link between rPFC activation and prediction error showed lowest delusional scores (Fig. 8). This is further evidence that an aberration in this frontally mediated inferential processing relates to delusion formation. We should note that it is perfectly possible that the rPFC is not the ‘site’ of prediction error per se but may be concerned rather with inferences that are made as a consequence of prediction-error signal. This observation may offer an explanation of why it is possible to have, as we observed here, apparently preserved learning of associations in the face of such frontal perturbation. Specifically, subjects could learn simple associative relationships but show a change in the extent or nature of inference about the food–allergy relationships as a whole. This would account for the apparently normal behavioural learning profiles under these conditions. Further work would be required to establish whether inferential processing in patients is truly altered in the patients.
Extending this argument, it is relevant to note that humans often employ short-cuts or heuristics when making causal judgements (Kahneman et al., 1982). One basis for causal inference is the ease with which a plausible scenario can be constructed mentally. The prefrontal associative learning mechanism outlined by Daw and colleagues might underlie such construction or simulation (Daw et al., 2005). In response to a prediction-error signal, the prefrontal cortex may interrogate the possible associative chains of causal cues that may have led to the unexpected outcome and drive, where necessary, the formation of novel cue-outcome associations. Hyperactive engagement of this interrogation process may lead psychotic patients to form and strengthen inappropriate causal associations during delusion formation (Maher, 1974; Kilhlstrom and Hoyt, 1988; Gray et al., 1991; Hemsley, 1994; Corlett et al., in press). Of course, the theory under test here is relevant to the early formation of delusions and it does not attempt to account for the complexity and fixity of such beliefs.
An alternative possibility, one which we attempt to address empirically, is that patients perform the task with an alternative approach, underpinned by different neural mechanisms, and, using this mechanism, they achieve matched performance with control subjects. We did identify a region of right middle temporal cortex that was more active in patients than in controls, providing a candidate alternative neural mechanism. However, this region was identified at a less stringent statistical threshold and its activity in patients was not related to successful task performance. Based on these limitations on interpretation, we maintain that fMRI responses to associative learning and expectancy violation provide a more sensitive assessment of performance than subjects’ behavioural predictions, a position at which we have arrived based on own empirical work using this task (Corlett et al., 2004, 2006) and the neuroimaging findings of others (see Fletcher, 2004).
Violation of learned expectancy is also inherent in quite different tasks that have been used to explore psychosis and schizophrenia. Studies exploring models of cognitive control emphasize the interaction between phasic responses in subcortical dopamine neurons and more sustained firing in the prefrontal cortex (Cohen et al., 1996; Braver and Cohen, 1999; Miller and Cohen, 2001; Rougier et al., 2005). The prediction-error signal from the midbrain is responsible for ‘gating’ the access of information to the prefrontal cortex. Noise in this gating system would cause disruptions in manipulation and maintenance of information necessary for goal-directed behaviour and would disrupt attention and motivation as is observed in schizophrenia (Braver and Cohen, 1999). Indeed the processing of cortical noise as relevant signal has been implicated as a pathophysiological mechanism in schizophrenia (Winterer and Weinberger, 2004; Winterer, 2006).
There is a wealth of evidence implicating mesocorticolimbic dopamine neurotransmission both in prediction-error processing (Schultz, 2000; Waelti et al., 2001) and in psychosis (Carlsson and Lindqvist, 1963; Anden et al., 1970; Nyback and Sedvall, 1970; Creese et al., 1976; Seeman et al., 1976). However, glutamate function may also be involved in both the pathophysiology of psychosis (Javitt and Zukin, 1991; Krystal et al., 1994; Jentsch and Roth, 1999; Goff and Coyle, 2001; Laruelle et al., 2003) as well as in the signalling of prediction errors (Lavin et al., 2005). Furthermore, these two neurotransmitter systems are intimately involved with each other, such that a disruption to one can have profound effects on the other, precipitating psychotic symptoms (Grace, 1991, 1993; Moore et al., 1999). The pattern and regional specificity of disruption to prediction-error processing in psychotic patients (Fig. 6) is strikingly redolent of our previously observed (Corlett et al., 2006) disruption of rPFC response under low dose ketamine (an NMDA receptor antagonist that is increasingly being employed as an experimental model of psychosis, see Krystal et al., 2002, for review). The strong similarity between the current findings and our previous study of ketamine provide support for the NMDA receptor hypo-function model of psychosis (Javitt and Zukin, 1991; Olney and Farber, 1995; Carlsson et al., 1999; Olney et al., 1999). However, the inferences are not straightforward. A challenge is to understand why, if glutamate is critical to prediction-error signal, NMDA hypofunction is associated with abnormally high prediction-error-related firing. One possibility is that NMDA receptor hypofunction increases extracellular glutamate levels in PFC via GABAergic disinhibition of cortical afferents including subcortical and midbrain dopamine neurons (Moghaddam et al., 1997). This effect would induce stimulation of cortical AMPA receptors, a possibility that finds support in the observation that an NMDA antagonist increases the number of randomly distributed single spikes in prefrontal neurons of awake rats (Jackson et al., 2004). Jackson and colleagues posit that this increased random spiking is induced by AMPA receptor stimulation.
Bringing these observations together, if it is indeed the case that NMDA hypofunction (under conditions of psychosis or ketamine), increases cortical noise via AMPA receptor stimulation thus impairing the filtering of irrelevant information and promoting the transmission of misinformation (Lisman, 1997), these effects would be consistent with our current and previous low dose ketamine findings (Corlett et al., 2006): an augmentation of the rPFC response to task irrelevant control items (a result of the increased prefrontal noise due to random spiking).
While we have focused upon the relationship between prediction error and the earliest emergence of false beliefs, it should be noted that other broader models have also explained other symptoms of psychosis, notably hallucinations, in terms of comparable underlying mechanisms (e.g. Kapur, 2003). In this respect, it is noteworthy that while, in the current study, perturbed prediction-error signal was related statistically to delusions, the same relationship was not found for hallucinations. However, given that our experimental design and analysis focused primarily on false beliefs, we are very cautious about interpreting the latter negative finding.
In summary, we used a brain marker for prediction-error-dependent causal learning to explore aberrant responses in psychosis. Psychotic patients demonstrate a disruption in prediction error, the magnitude of which correlates, across patients, with the severity of their delusions. Our findings provide support for associative models of delusion formation and provide a possible mechanism for the disruptions that underlie the emergence of psychotic beliefs. This mechanism incorporates disrupted neurotransmission in the mesocorticolimbic dopamine system, particularly the mesocortical pathway from VTA to PFC, known to corealease both dopamine and glutamate at its terminals in response to salient environmental events (Lavin et al., 2005). The present data suggest that this signalling process may be impaired in individuals with psychosis, such that the prefrontal cortex responds to physiological noise as if it were salient biological signal and drives the attribution of salience and attention to irrelevant and inconsequential environmental events.
The observation that disrupted prefrontal processing of expectancy violations correlates with delusion severity in our patient sample [in a manner redolent of that observed in healthy individuals administered ketamine (Corlett et al., 2006)], implicates aberrant prediction-error processing in delusion formation and suggests that glutamatergic neurotransmission contributes to endogenous psychosis.
Acknowledgements
This work was supported by the Wellcome Trust and conducted within the University of Cambridge Behavioural and Clinical Neuroscience Institute, supported by a joint award from the Medical Research Council and the Wellcome Trust. GKM is supported by the Department of Health. Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Abbreviations
- rPFC
right prefrontal cortex
References
- Diagnostic and statistical manual of mental disorders - IV. American Psychiatric Association; 2000. [Google Scholar]
- Aitken MR, Dickinson A. Simulations of a modified SOP model applied to retrospective revaluation of human causal learning. Learn Behav. 2005;33:147–59. doi: 10.3758/bf03196059. [DOI] [PubMed] [Google Scholar]
- Altman DG. Practical statistics for medical research. Chapman & Hall/CRC; London: 1991. [Google Scholar]
- Anden NE, Butcher SG, Corrodi H, Fuxe K, Ungerstedt U. Receptor activity and turnover of dopamine and noradrenaline after neuroleptics. Eur J Pharmacol. 1970;11:303–14. doi: 10.1016/0014-2999(70)90006-3. [DOI] [PubMed] [Google Scholar]
- Barnett JH, Sahakian BJ, Werners U, Hill KE, Brazil R, Gallagher O, et al. Visuospatial learning and executive function are independently impaired in first-episode psychosis. Psychol Med. 2005;35:1031–41. doi: 10.1017/s0033291704004301. [DOI] [PubMed] [Google Scholar]
- Beninger RJ. The slow therapeutic action of antipsychotic drugs: a possible mechanism involving the role of dopamine in incentive learning. Karger; 1988. [Google Scholar]
- Bertolino A, Knable MB, Saunders RC, Callicott JH, Kolachana B, Mattay VS, et al. The relationship between dorsolateral prefrontal N-acetylaspartate measures and striatal dopamine activity in schizophrenia. Biol Psychiatry. 1999;45:660–7. doi: 10.1016/s0006-3223(98)00380-1. [DOI] [PubMed] [Google Scholar]
- Biver F, Goldman S, Luxen A, Delvenne V, De Maertelaer V, De La Fuente J, et al. Altered frontostriatal relationship in unmedicated schizophrenic patients. Psychiatry Res. 1995;61:161–71. doi: 10.1016/0925-4927(95)02672-k. [DOI] [PubMed] [Google Scholar]
- Bleuler E. Dementia Praecox or the group of Schizophrenias. International University Press; New York: 1911/1950. [Google Scholar]
- Braver TS, Cohen JD. Dopamine, cognitive control, and schizophrenia: the gating model. Prog Brain Res. 1999;121:327–49. doi: 10.1016/s0079-6123(08)63082-4. [DOI] [PubMed] [Google Scholar]
- Buchsbaum MS, Hazlett EA, Haznedar MM, Spiegel-Cohen J, Wei TC. Visualizing fronto-striatal circuitry and neuroleptic effects in schizophrenia. Acta Psychiatr Scand Suppl. 1999;395:129–37. doi: 10.1111/j.1600-0447.1999.tb05992.x. [DOI] [PubMed] [Google Scholar]
- Cairo TA, Liddle PF, Woodward TS, Ngan ET. The influence of working memory load on phase specific patterns of cortical activity. Brain Res Cogn Brain Res. 2004;21:377–87. doi: 10.1016/j.cogbrainres.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Hansson LO, Waters N, Carlsson ML. A glutamatergic deficiency model of schizophrenia. Br J Psychiatry Suppl. 1999:2–6. [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and Normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963;20:140–4. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- Chapman J. The early symptoms of schizophrenia. Br J Psychiatry. 1966;112:225–51. doi: 10.1192/bjp.112.484.225. [DOI] [PubMed] [Google Scholar]
- Cohen JD, Braver TS, O’Reilly RC. A computational approach to prefrontal cortex, cognitive control and schizophrenia: recent developments and current challenges. Philos Trans R Soc Lond B Biol Sci. 1996;351:1515–27. doi: 10.1098/rstb.1996.0138. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Aitken MR, Dickinson A, Shanks DR, Honey GD, Honey RA, et al. Prediction error during retrospective revaluation of causal associations in humans: fMRI evidence in favor of an associative model of learning. Neuron. 2004;44:877–88. doi: 10.1016/j.neuron.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Honey GD, Aitken MR, Dickinson A, Shanks DR, Absalom AR, et al. Frontal responses during learning predict vulnerability to the psychotogenic effects of ketamine: linking cognition, brain activity, and psychosis. Arch Gen Psychiatry. 2006;63:611–21. doi: 10.1001/archpsyc.63.6.611. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Honey GD, Fletcher PC. From prediction error to psychosis: ketamine as a pharmacological model of delusions. J Psychopharmacol. 2007;21:238–52. doi: 10.1177/0269881107077716. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–3. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Dickinson A. The 28th Bartlett Memorial Lecture. Causal learning: an associative analysis. Q J Exp Psychol B. 2001;54:3–25. doi: 10.1080/02724990042000010. [DOI] [PubMed] [Google Scholar]
- Elliott R, McKenna PJ, Robbins TW, Sahakian BJ. Neuropsychological evidence for frontostriatal dysfunction in schizophrenia. Psychol Med. 1995;25:619–30. doi: 10.1017/s0033291700033523. [DOI] [PubMed] [Google Scholar]
- Fletcher PC, Anderson JM, Shanks DR, Honey R, Carpenter TA, Donovan T, et al. Responses of human frontal cortex to surprising events are predicted by formal associative learning theory. Nat Neurosci. 2001;4:1043–8. doi: 10.1038/nn733. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Fletcher P, Josephs O, Holmes A, Rugg MD, Turner R. Event-related fMRI: characterizing differential responses. Neuroimage. 1998;7:30–40. doi: 10.1006/nimg.1997.0306. [DOI] [PubMed] [Google Scholar]
- Gilleen J, David AS. The cognitive neuropsychiatry of delusions: from psychopathology to neuropsychology and back again. Psychol Med. 2005;35:5–12. doi: 10.1017/s0033291704003976. [DOI] [PubMed] [Google Scholar]
- Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158:1367–77. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Grace AA. Cortical regulation of subcortical dopamine systems and its possible relevance to schizophrenia. J Neural Transm Gen Sect. 1993;91:111–34. doi: 10.1007/BF01245228. [DOI] [PubMed] [Google Scholar]
- Gray JA, Feldon J, Rawlins JNP, Hemsley D, Smith AD. The neuropsychology of schizophrenia. Behav Brain Sci. 1991;14:1–84. [Google Scholar]
- Grossberg S. Processing of expected and unexpected events during conditioning and attention: a psychophysiological theory. Psychol Rev. 1982;89:529–72. [PubMed] [Google Scholar]
- Hartley D. Observations on man, his frame, his duty, and his expectations. W. Eyres; London: 1801. [Google Scholar]
- Hemsley DR. Perceptual and cognitive abnormalities as the bases for schizophrenic symptoms. In: David AS, Cutting JC, editors. The neuropsychology of schizophrenia. Lawrence Erlbaum Associates; Hove, UK: 1994. pp. 97–118. [Google Scholar]
- Hutton SB, Puri BK, Duncan LJ, Robbins TW, Barnes TR, Joyce EM. Executive function in first-episode schizophrenia. Psychol Med. 1998;28:463–73. doi: 10.1017/s0033291797006041. [DOI] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci USA. 2004;101:8467–72. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–25. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Joyce EM, Collinson SL, Crichton P. Verbal fluency in schizophrenia: relationship with executive function, semantic memory and clinical alogia. Psychol Med. 1996;26:39–49. doi: 10.1017/s0033291700033705. [DOI] [PubMed] [Google Scholar]
- Kahneman D, Slovic P, Tversky A. Judgment under uncertainty: heuristics and biases. Cambridge University Press; New York: 1982. [DOI] [PubMed] [Google Scholar]
- Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- Kilhlstrom JF, Hoyt IP. Hypnosis and the psychology of delusions. Wiley; New York: 1988. [Google Scholar]
- Krystal JH, Anand A, Moghaddam B. Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiat. 2002;59:663–4. doi: 10.1001/archpsyc.59.7.663. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiat. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann N Y Acad Sci. 2003;1003:138–58. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- Lavin A, Nogueira L, Lapish CC, Wightman RM, Phillips PE, Seamans JK. Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci. 2005;25:5013–23. doi: 10.1523/JNEUROSCI.0557-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Locke J. An essay concerning human understanding. Dent; London: 1690/1976. [Google Scholar]
- Mackintosh NJ. A theory of attention: Variations in associability of stimuli with reinforcement. Psychol Rev. 1975;82:276–98. [Google Scholar]
- Maher BA. Delusional thinking and perceptual disorder. J Individ Psychol. 1974;30:98–113. [PubMed] [Google Scholar]
- Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–9. doi: 10.1016/s1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- Mann DMA. Dopamine neurons of the vertebrate brain: some aspects of anatomy and pathology. Manchester University Press; Manchester: 1986. [Google Scholar]
- Manoach DS, Gollub RL, Benson ES, Searl MM, Goff DC, Halpern E, et al. Schizophrenic subjects show aberrant fMRI activation of dorsolateral prefrontal cortex and basal ganglia during working memory performance. Biol Psychiatry. 2000;48:99–109. doi: 10.1016/s0006-3223(00)00227-4. [DOI] [PubMed] [Google Scholar]
- Matussek P. Studies in delusional perception. In: Cutting J, Shepherd M, editors. The clinical roots of the schizophrenia concept. Translations of seminal European contributions on schizophrenia. Cambridge University Press; Cambridge: 1952. pp. 89–103. [Google Scholar]
- McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103–16. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Miletich RS, Kohn PD, Esposito G, Carson RE, Quarantelli M, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267–71. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Miller R. Schizophrenic psychology, associative learning and the role of forebrain dopamine. Med Hypotheses. 1976;2:203–11. doi: 10.1016/0306-9877(76)90040-2. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore H, West AR, Grace AA. The regulation of forebrain dopamine transmission: relevance to the pathophysiology and psychopathology of schizophrenia. Biol Psychiat. 1999;46:40–55. doi: 10.1016/s0006-3223(99)00078-5. [DOI] [PubMed] [Google Scholar]
- Nelson H. National Adult Reading Test Manual. NFER-Nelson; Windsor, UK: 1982. [Google Scholar]
- Nyback H, Sedvall G. Further studies on the accumulation and disappearance of catecholamines formed from tyrosine-14C in mouse brain. Effect of some phenothiazine analogues. Eur J Pharmacol. 1970;10:193–205. doi: 10.1016/0014-2999(70)90273-6. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–33. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Pantelis C, Barnes TR, Nelson HE, Tanner S, Weatherley L, Owen AM, et al. Frontal-striatal cognitive deficits in patients with chronic schizophrenia. Brain. 1997;120(Pt 10):1823–43. doi: 10.1093/brain/120.10.1823. [DOI] [PubMed] [Google Scholar]
- Pavlov IP. Lectures on conditioned reflexes. Vol 1. Lawrence & Wishart; London: 1928. [Google Scholar]
- Pearce JM, Hall G. A model for Pavlovian learning: variations in the effectiveness of conditioned but not of unconditioned stimuli. Psychol Rev. 1980;87:532–52. [PubMed] [Google Scholar]
- Price CJ, Friston KJ. Scanning patients with tasks they can perform. Hum Brain Mapp. 1999;8:102–8. doi: 10.1002/(SICI)1097-0193(1999)8:2/3<102::AID-HBM6>3.0.CO;2-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA, Wagner AR. A theory of Pavlovian conditioning: variations in the effectiveness of reinforcement and non-reinforcement. In: Black AH, Prokasy WF, editors. Classical conditioning II: current research and theory. Appleton-Century-Crofts; New York: 1972. [Google Scholar]
- Robbins TW. The case of frontostriatal dysfunction in schizophrenia. Schizophr Bull. 1990;16:391–402. doi: 10.1093/schbul/16.3.391. [DOI] [PubMed] [Google Scholar]
- Rougier NP, Noelle DC, Braver TS, Cohen JD, O’Reilly RC. Prefrontal cortex and flexible cognitive control: rules without symbols. Proc Natl Acad Sci USA. 2005;102:7338–43. doi: 10.1073/pnas.0502455102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. Die Psychologie der Schizophrenen. Germany Thieme; Leipzig: 1930. [Google Scholar]
- Schultz W, Dickinson A. Neural coding of prediction errors. Ann Rev Neurosci. 2000:473–500. doi: 10.1146/annurev.neuro.23.1.473. [DOI] [PubMed] [Google Scholar]
- Seeman P, Chau-Wong M, Tedesco J, Wong K. Dopamine receptors in human and calf brains, using [3H]apomorphine and an antipsychotic drug. Proc Natl Acad Sci USA. 1976;73:4354–8. doi: 10.1073/pnas.73.12.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DC, Aitken MR, Shanks DR, Sahakian BJ, Robbins TW, Schwarzbauer C, et al. The role of the lateral frontal cortex in causal associative learning: exploring preventative and super-learning. Cereb Cortex. 2004;14:872–80. doi: 10.1093/cercor/bhh046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura J, Green MF, Shaner A, Lieberman RP. Training and quality assurance with the Brief Psychiatric Ratings Scale: “The Drift Buster”. Int J Methods Psychiat Res. 1993;3:221–4. [Google Scholar]
- Vinogradov S, King RJ, Huberman BA. An associationist model of the paranoid process: application of phase transitions in spreading activation networks. Psychiatry. 1992;55:79–94. doi: 10.1080/00332747.1992.11024582. [DOI] [PubMed] [Google Scholar]
- Waelti P, Dickinson A, Schultz W. Dopamine responses comply with basic assumptions of formal learning theory. Nature. 2001;412:43–8. doi: 10.1038/35083500. [DOI] [PubMed] [Google Scholar]
- Winterer G. Cortical microcircuits in schizophrenia–the dopamine hypothesis revisited. Pharmacopsychiatry. 2006;39(Suppl 1):S68–71. doi: 10.1055/s-2006-931498. [DOI] [PubMed] [Google Scholar]
- Winterer G, Weinberger DR. Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci. 2004;27:683–90. doi: 10.1016/j.tins.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Woods SW. Chlorpromazine equivalent doses for the newer atypical antipsychotics. J Clin Psychiatry. 2003;64:663–7. doi: 10.4088/jcp.v64n0607. [DOI] [PubMed] [Google Scholar]