

Abstract
Background
The dystonias are a group of hyperkinetic movement disorders whose principal cause is neuron dysfunction at one or more interconnected nodes of the motor system. The study of genes and proteins which cause familial dystonia provides critical information about the cellular pathways involved in this dysfunction which disrupts the motor pathways at systems level. In recent years study of the increasing number of DYT genes has implicated a number of cell functions which appear to be involved in the pathogenesis of dystonia.
Methods
Review of literature published in English language publications available on Pubmed relating to the genetics and cellular pathology of dystonia
Results and Conclusions
Numerous potential pathogenetic mechanisms have been identified. We describe those which fall into three emerging thematic groups: cell cycle and transcriptional regulation in the nucleus, endoplasmic reticulum and nuclear envelope function, and control of synaptic function.
Keywords: DYT genes, Cell cycle, endoplasmic reticulum, Nuclear envelope, synaptic function
Introduction
The dystonias are a heterogeneous group of hyperkinetic movement disorders characterised by sustained involuntary muscle spasms and postures.1 The most common forms are primary, where dystonic movements are the only clinical feature and there is no evidence of neurodegeneration. In the majority of these cases the cellular processes that lead to functional abnormalities of neurons, sufficient to disrupt the finely tuned control of movement, are unknown. However, in recent years the identification of genes that cause rare monogenic familial dystonia has given insight into the neurobiology of dystonia and shed light on molecular mechanisms involved. Further evidence has come from study of more complex forms secondary of dystonia where there is evidence of neurodegeneration or CNS damage and the dystonic movements are part of a more complex neurological phenotype.
The purpose of this review is to summarise the evidence, predominantly from the study of genetic forms of dystonia, and highlight cellular pathways that are important to the genesis of dystonia. In particular, it will focus on areas where there are common themes to the cellular pathogenesis. Most of this information comes from study of monogenic primary dystonias, and the proteins that the various DYT genes encode. The genetic classification of dystonia has subtypes DYT 1-25 and include pure primary dystonias, dystonia-plus syndromes where other features, such as myoclonus or Parkinsonism, are present, and paroxysmal dyskinesias, in which dystonia is often a prominent feature. Table 1 describes the DYT loci, showing key clinical features and information about the protein encoded by the DYT gene. The following sections describe the specific forms of primary and dystonia plus syndromes where the gene and protein has been identified and studied, and which are discussed in this review.
Table 1.
Summary of genes and proteins involved in genetic forms of primary dystonia and dystonia-plus syndromes.
| Locus | Designation | Clinical features | Gene/inheritance | Protein | Putative functions |
|---|---|---|---|---|---|
| Pure Dystonia | |||||
| DYT1 Chr9q34.11 | Early onset primary dystonia | Childhood onset dystonia in limb with generalisation | TOR1A Autosomal Dominant | TorsinA | AAA+ protein, Nuclear envelope, ER secretory and stress response, regulation of synaptic function |
| DYT2 | Early onset dystonia | Adolescent onset segmental or generalised | Autosomal Recessive | Unknown | |
| DYT4 Chr19p13.3 | Whispering dysphonia | Childhood onset laryngeal abductor spasm with cervical dystonia | TUBB4a Autosomal dominant | beta-tubulin 4a | Structural cytoskeleton protein |
| DYT6 Chr 8p11.21 | AD early onset focal dystonia | Early-Onset dystonia with prominent cervical and laryngeal involvement | THAP1 Autosomal Dominant | Thanatos-associated domain-containing apoptosis associated protein 1 | Atypical zinc-finger protein. THAP domain is chromatin binding factor and regulates transcrption |
| DYT7 Chr 8p | Familial focal dystonia | Adult onset focal dystonia | Unknown Autosomal dominant | Unknown | |
| DYT13 Chr1p36.32-p36.13 | Familial cranio-cervical dystonia | Focal or segmental dystonia of cranio-cervical region and upper limbs | Unknown Autosomal dominant | Unknown | |
| DYT17 Chr 20p11.2-2q13.12 | Early onset AR dystonia | Early onset focal dystonia progressing to generalized with dysphonia and dysarthria | Unknown Autosomal recessive | Unknown | |
| DYT21 Chr 2q14.3-q21.3 | Late onset dystonia | Late onset multifocal and generalized dystonia | Unknown Autosomal Dominant | Unknown | |
| DYT23 Chr9q34.11 | Cervical dystonia | Late-onset primary cervical dystonia | CIZ1 Autosomal Dominant | Cip1-interacting zinc finger | Regulation of G1-S cell cycle and DNA replication |
| protein 1 | |||||
| DYT24 Chr11p14.2 | Late onset dystonoa | Cranial and cervical dystonia | ANO3 Autosomal Dominant | Anoctamin3 | Calcium gated chloride channel |
| DYT25 Chr18p | Cervical dystonia with local spread | Predominantly late onset primary cervical dystonia with spread to face | GNAL Autosomal dominant | alpha subunit of G protein | Probable interaction with D1 and Adenosine 2A receptors. |
| Dystonia plus syndromes | |||||
| DYT3 Xq13.1 | X-linked dystonia (Lubag) | Segmental or generalised dystonia with parkinsonism | TAF1 X-linked | TATA box-binding protein associated factor 1 | Regulation of transcription initiation and cell cycle |
| DYT5/14 Chr2q13.2 | Dopa-responsive dystonia | Dystonia with parkinsonism, diurnal variation, very good response to L-dopa | GCH1 Autosomal Dominant | GTP cyclohydrolase 1 | Rate limiting enzyme in synthesis of tetrahydrobiopterin, key co-factor in monoamine synthesis. Results in deficient dopamine synthesis |
| DYT11 Chr7q21.3 | Myoclonic dystonia syndrome | Upper body myoclonic jerks with dystonia. Responsive to alcohol | SGCE Autosomal Dominant | Epsilon sarcoglycan | Cell membrane protein that may act as structural platform for other protein interactions |
| DYT12 Chr19q13.2 | Rapid onset dystonia parkinsonism | Acute onset generalised dystonia with parkinsonism, rostrocaudal gradient of symptoms | ATP1A3 Autosomal Dominant | Alpha 3 subunit of Na/K ATPase | Subunit of Na/K ATPase on neuronal membrane |
| DYT15 Chr 18p11DYT16 Chr2q31.2 | Myoclonus dystonia DRD and parkinsonism | Childhood onset, myoclonus with variable dystonia Dystonia-parkinsonism with bulbar involvement and bradykinesia | Unknown Autosomal Dominant PRKRA Autosomal Recessive | Unknown Protein kinase, interferon-inducible double stranded RNA dependent activator | PRKRA is activator of proteins involved in response to cell stress |
| Paroxysmal Dystonia | |||||
| DYT8 Chr2q35 | Paroxysmal non-kinesigenic | Attacks of dystonia or | MR-1 Autosomal | Myofibrillo-genesis | Affect neuronal stress response |
| dyskinesia (PKND) | choreoathetosis. Precipitants;stress, alcohol, fatigue, coffee | dominant | regulator 1 | pathways | |
| DYT9 and DYT18 Chr1p34.2 | Paroxysmal exercise induced dystonia. Episodic ataxia | Dystonia (usually limb) brought on by exercise. Ataxia/spasticity between attacks | GLUT1 Autosomal dominant | Glucose transporter type 1 | Reduced glucose transport to neurons with unstable metabolic state |
| DYT10 and DYT19 Chr16p11.2 | Proxysmal kinesigenic choreoathetosis (PKC) | Attacks of dystonia or choreoathetosis precipitated by sudden movement. Associated with migraine and epilepsy | PRRT2 Autosomal dominant | Proline-rich trans-membrane protein 2 | Interacts with SNAP25 and may affect synaptic functions/neuronal excitability |
| DYT20 Chr 2q31 | Canadian PKND | PKND phenotype | Unknown Autosomal dominant | Unknown |
DYT1 dystonia: TorsinA
DYT1 dystonia is caused by a heterozygous 3-bp GAG deletion in the TOR1A gene.2 TorsinA is 332 amino acid protein typical of the AAA+ (ATPases associated with a variety of cellular activities) protein family; these proteins typically function as oligomers and use the energy of ATP hydrolysis for many functions including protein trafficking, membrane fusion, protein refolding and degradation.3 The pathogenic codon deletion (ΔE) in torsinA removes a glutamic acid residue from a C-terminal alpha helix believed to be critical for oligomerization and/or tertiary structure.4 TorsinA is ubiquitously expressed in numerous cell types, including neurons and glia within the CNS.5-8 TorsinA mRNA is expressed in sensorimotor regions of brain including cerebral cortex, striatum, substantia nigra pars compacta, thalamus, hippocampus, midbrain, pons, cerebellum, and spinal cord.5-8 It has been suggested that the neuron specific effect of mutant torsinA occurs as there is low expression of other forms of torsin, notably torsinB, and cannot therefore correct for the presence of dysfunctional torsinA.9
DYT4 dystonia: Beta-tubulin 4a
Study of a large family with autosomal dominant whispering dysphonia and generalized dystonia revealed a single cosegregating mutation in the beta-tubulin 4a gene.10 The mutation in the beta-tubulin autoregulatory domain was highly expressed in the nervous system and implicates the cytoskeleton in dystonia pathogenesis.
DYT6 dystonia: THAP1
Dystonia-causing mutations in THAP1 (Thanatos-associated protein domain containing apoptosis associated protein 1) have been widely reported.11,12 THAP1 is a transcription factor and a member of THAP protein family that contains an evolutionarily conserved zinc-dependent DNA-binding domain. Mutations in THAP1 include several reported deletions and nonsense mutations, including one that removes the start codon. Many of the mutations occur in the DNA-binding domain and several appear to disrupt the nuclear localization signal.13-15 In addition, at least one mutation appears to cause disease through recessive inheritance.16 Considered together, these observations suggest that THAP1 may cause dystonia through a loss of function mechanism.
Before its identification as a dystonia-causing gene, THAP1 was reported to regulate cell proliferation via regulation of pRB/E2F cell cycle target genes and another report pointed to a potential pro-apoptotic role related to localization in PML (promyelocytic leukaemia) nuclear bodies.17,18
DYT11 myoclonus dystonia: epsilon-sarcoglycan (SGCE)
SGCE is a member of the sarcoglycan protein family that comprise plasma transmembrane proteins. Most SGCE mutations are deletions or nonsense mutations that eliminate gene function.19 The SGCE gene is maternally imprinted, meaning that individuals are dependent on expression from the paternal allele. Nearly all myoclonus-dystonia patients inherit a loss-of-function allele from their father, and therefore lack functional SGCE protein.
Sarcoglycans are components of the dystrophin-glycoprotein complex (DGC), a membrane-spanning complex that makes connections with the extracellular matrix and the intracellular actin cytoskeleton.20,21 While DGC function is typically described in striated muscle where its dysfunction is linked to several muscular dystrophies, DGC proteins are also expressed in the CNS. SGCE is found in many brain regions and is associated with dopaminergic neurons in the substantia nigra and ventral tegmental areas. 22 In neurons, DGC proteins (including SGCE) concentrate at postsynaptic sites, and have been localized to GABAergic inhibitory synapses.23,24 This association with GABAergic synapses may contribute to the deficient inhibition that is observed in many forms of primary dystonia.
DYT16 dystonia-parkinsonism: PRKRA
DYT16 is less prevalent than DYT1 and 6, having been described in only a small number of families.25 The gene encodes the protein PRKRA (protein kinase, interferon-inducible double stranded RNA dependent activator), also known as PACT, which regulates activity of protein-kinase R.26
DYT23 dystonia: Cip1-interacting zinc finger protein 1: CIZ1
A recent study has identified a CIZ1 (c.790A>G, p.S264G) mutation in a large Caucasian pedigree with late-onset primary cervical dystonia.27 Screening in subjects with adult-onset cervical dystonia identified two additional missense mutations in CIZ1 (p.P47S and p.R672M).
CIZ1 encodes Cip1-interacting zinc finger protein 1, a DNA replication factor. CIZ1 was first identified through its interaction with p21Cip1/Waf1 (CDKN1A), a cyclin-dependent kinase inhibitor involved in G1-S cell-cycle regulation and cellular differentiation.27 CIZ1 is expressed in the cerebellum, cerebral cortex, substantia nigra, and putamen of adult human brain.28
DYT24 dystonia: Anoctamin 3 (ANO3)
Exome sequencing in an autosomal dominant family with cranio-cervical dystonia identified a putative mutation in the ANO3 gene, and further mutations were found in other familial and sporadic cases of cervical dystonia.29 ANO3 is expressed highly in the striatum, but also the neocortex, hippocampus and amygdala. Anoctamin 3 is believed to act as a calcium gated chloride channel and functional studies using Ca2+ imaging in case and control fibroblasts demonstrated abnormalities in endoplasmic-reticulum-dependent Ca2+ signalling.
DYT25 dystonia: GNAL
Recently, mutations in GNAL have been identified in a 11 multiplex families (autosomal dominant) predominantly with cervical and segmental dystonia.30,31GNAL encodes the stimulatory alpha subunit, Gαolf , first identified as a G protein that mediates odorant signaling in the olfactory epithelium, coupling D1 and A2a receptors to adenylyl cyclase and histone H3 phosphorylation.
The following sections will focus on mechanisms which have been derived from study of a number of the studied genes described above, but will also consider pathogenesis of selected forms of secondary dystonia. Three key areas can be identified where there appear to be shared molecular pathways:
Cell cycle and transcription
The nuclear envelope/endoplasmic reticulum (ER) interface and ER secretory pathways
Synaptic function
Defects at the G1-S Cell-Cycle Checkpoint
The eukaryotic cell cycle consists of four distinct phases: Gap 1 (G1) phase, Synthesis (S), Gap 2 (G2), and Mitosis (M). In addition, terminally differentiated cells like neurons enter a quiescent state call Gap 0 or G0. The G1 phase is commonly known as the growth phase. During G1 cells prepare for the DNA synthesis that will occur in the S phase. DNA is synthesized and chromosomes are replicated during the S phase. The G1-S cell-cycle checkpoint pathway controls the transition from the end of G1 to S. The G1-S cell-cycle checkpoint ensures that the cell is fully prepared for DNA synthesis. Numerous factors such as DNA damage from irradiation, contact inhibition, TGFβ, and oxidative stress can inhibit transition from G1 to S, and the G1-S checkpoint has also been implicated in the molecular pathology of dystonia.
CIZ1
The cellular role and neural localization of CIZ1 are compatible with current themes in dystonia research. CIZ1, TOR1A, THAP1 and genes associated with neurodegenerative dystonia seen in DYT3 dystonia (TAF1) and ataxia telangiectasia (ATM) are involved in G1-S cell-cycle regulation (Fig 1).
Figure 1.

G1/S cell-cycle checkpoint and dystonia. Dystonia-associated proteins are shaded red. Indirect, multi-step and putative pathways are denoted with hashed lines. In general, arrows indicate excitatory interactions and stops mark inhibitory interactions. However, some relationships are non-linear and the result of combinatorial actions of heteromeric complexes and post-translational modifications. +p, phosphorylation. ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; CDC25A, cell division cycle 25 homolog A; CDK2, cyclin-dependent kinase 2; CDK4, cyclin-dependent kinase 4; CDK6, cyclin-dependent kinase 6; CDKN1A, p21/WAF1 or cyclin-dependent kinase inhibitor 1; CHK1, checkpoint kinase 1; CIZ1, Cip1-interacting zinc finger protein; E2F, transcription factor E2F; G1 Phase, Gap 1 phase of the cell cycle; p53, protein 53; R, restriction point; Rb, retinoblastoma protein; SMAD2, mothers against decapentaplegic homolog 2; SMAD3, mothers against decapentaplegic homolog 3; S Phase, synthesis phase. TAF1, transcription initiation factor TFIID subunit 1; TFDP1, transcription factor Dp1; TGFβ, transforming growth factor β.
In cell-free systems, CIZ1 is able to promote DNA replication after replication complex formation.32 The C-terminal domain anchors CIZ1 to the non-chromatin nuclear matrix, whereas DNA replication activity resides in the N-terminal half of the protein. The C-terminal domain of CIZ1 also recognizes the consensus DNA sequence ARYSR(0-2)YYAC.33 Studies of GFP-tagged CIZ1 have shown that formation of subnuclear particles or foci requires both the N- and C-terminal domains.34
The N-terminal region of CIZ1, including the first zinc finger motif, binds to the N-terminal CDK2-interacting domain of CDKN1A and this interaction is disrupted by overexpression of CDK2.35 When CIZ1 and CDKN1A are individually overexpressed, they localize primarily to the nucleus. However, overexpression of CIZ1 can induce cytoplasmic distribution of CDKN1A, suggesting that CIZ1 regulates the cellular localization of CDKN1A.
In cell-free experiments, CIZ1 increases the number of nuclei that initiate DNA replication and, in intact wild-type and CDKN1A -null cells, CIZ1 stimulates DNA synthesis. Consistent with a role in DNA replication, endogenous CIZ1 was found to co-localize with proliferating cell nuclear antigen (PCNA) during S phase, and targeted depletion of CIZ1 retrains cell proliferation by inhibiting entry into S phase.31 CIZ1-depleted cells accumulate chromatin-bound minichromosome maintenance complex component 3 MCM3 and PCNA but fail to synthesize DNA efficiently.31
CIZ1 is an estrogen-responsive gene.36 CIZ1 co-regulates estrogen receptor alpha (ERα) by enhancing ERα transactivation activity and promoting the recruitment of the ERα complex to target genes. CIZ1 overexpression confers estrogen hypersensitivity and promotes growth rate, anchorage independence, and tumorigenic properties in breast cancer cells. It is tempting to speculate that aberrant interactions between ERα and CIZ1 or other, as yet unidentified dystonia-related proteins, contribute to the relative increased prevalence of cervical dystonia in females.
THAP1
THAP1 plays an important role in transcriptional regulation in the context of cell proliferation and pRb/E2F cell cycle pathways.18,37,38 Both RNAi silencing and overexpression of THAP1 inhibit G1-S progression. Overexpression of THAP1 in primary human endothelial cells inhibited proliferation, and gene expression profiling showed that this effect was due to repression of pRB/E2F cell-cycle target genes. The anti-proliferative effects of THAP1 on endothelial cells were not dependent on apoptosis. THAP1 appears to inhibit cell-cycle progression at the G1-S transition, localizes to promyelocytic leukemia nuclear bodies with the pro-apoptotic leucine-zipper protein Par-4 and potentiates TNFα-induced apoptosis.17
Clouaire and colleagues (2005) identified a consensus DNA-binding sequence (AGTACGGGCAA) recognized by the THAP domain of THAP1.39 Nucleotide positions upstream of the core motif appear to modulate the strength and affinity of the GGCA/THAP interaction. Other THAP zinc fingers, including human THAP2 and THAP3, share structural homology, but do not recognize the same DNA target sequence. Protein-protein interactions, including multimerization, mediated by the coiled-coil domain of THAP1 may increase binding of the THAP zinc finger. Human THAP1, human THAP9 and Drosophila THAP bind DNA through a bipartite interaction using both the major and minor grooves.40
Overexpression of THAP1 in endothelial cells has been used as an indirect means of identifying THAP1 targets.18 A total of 16 genes were up-regulated >1.5 fold, and 80 down-regulated. Of the latter group, the genes were concentrated in classes related to cell-cycle/cell proliferation and the majority was also regulated by the pRB/E2F pathway. The cellular effects of THAP1 knock-down with RNAi were similar to the effects of overexpression. In addition, THAP1 knock-down was associated with decreased expression of 8 pRB/E2F cell-cycle target genes: RRM1, Mad2, survivin, HMMR, RRM2, CDC2, cyclin B1, and DLG7. RRM1 was shown to be a direct transcriptional target of THAP1. Potential THAP1 binding sites were also identified in the promoters of other genes: RRM2, BIRC2, survivin, and cyclin B1.
Studies have also focused on a potential functional interaction with torsinA. THAP1 was reported to bind to and activate the promoter of the Tor1a gene that encodes torsinA, although these studies did not demonstrate changes in torsinA mRNA following manipulation of THAP1.41,42 Moreover, there do not appear to be sequence alterations in the genes encoding THAP1 or torsinA that explain disease penetrance in DYT1 dystonia.43,44
Two-hybrid studies have identified several protein interactions for THAP1.17,45 Analysis of the set of interacting proteins suggests that THAP1 may contribute to transcription, splicing, and, possibly, RNA transport. Interestingly, several interacting proteins indirectly implicate THAP1 in cerebellar development. For example, NKAP is a transcriptional repressor acting on Notch target genes and is required for T cell development. Notch 1 is required for neuronal and glial differentiation in the cerebellum.46 FXR2, a homologue of the fragile × mental retardation protein (FMRP), is involved in mRNA transport. Deletion of FMRP in mice is associated with cerebellar ultrastructural abnormalities and motor learning deficits.47 Finally, DVL2 appears to be involved in the Wnt signaling pathway which is critical for cerebellar morphogenesis.48 There is an increasing body of evidence to implicate the cerebellum in the genesis of dystonic movements,49 and these data suggest that THAP1 could lead to subtle functional abnormalities of cerebellar functions.
While often considered a neurodegenerative disorder, some patients with Lubag (DYT3) manifest isolated focal (including cervical) or segmental dystonia years before the development of Parkinsonism.50 DYT3 is associated with deficiency of a neuronal isoform of TAF1 (N-TAF1) which is expressed preferentially in medium spiny neurons of the striosome compartment.51,52 TAF1 (TATA box-binding protein [TBP] associated factor 1) forms part of the TFIID transcriptional complex that is composed of TBP and up to 13 additional TAFs. TFIID binds to TATA boxes and participates in transcriptional initiation. Deficient of defective TAF1 could exert deleterious effects on cell-cycle control via multiple mechanisms. For instance, TAF1 induces G1-S progression by phosphorylating p53 at threonine-55.53 In TAF1-defective cell lines, ATR localizes to subnuclear foci and contributes to phosphorylation of downstream targets such as p53 and CHK1, which induces cell cycle arrest.54 Finally, TAF1 has been shown to undergo alternative splicing in response to developmental or DNA damage signals.55
TorsinA
TorsinA binds the KASH domain of nesprin-3, which spans the NE outer membrane and TA is concentrated at the nuclear envelope in its mutant form. TorsinA appears to play a role in interactions between the nucleus and cytoskeleton.26 These interactions may be important for cell polarity and/or transcription.56,57 Alternatively, torsinA may participate in transcriptional regulation and/or G1-S cell-cycle regulation via the TGFβ pathway.58, 59 Drosophila larvae that overexpress mutant ΔE torsinA exhibit overt ultrastructural defects at the neuromuscular junction similar to defects reported for mutants with defective TGFβ signaling.60,61 Overexpression of SMAD2, a downstream effector of the TGFβ pathway corrects morphological and behavioral defects associated expression of mutant torsinA.58
Variant ataxia-telangiectasia due to recessive mutations in ATM may present with dystonia.62 This is similar to THAP1 dystonia with frequent craniocervical and upper limb involvement, and can occur in the absence of cerebellar atrophy on MRI or ataxia on clinical examination. ATM (ataxia telangiectasia mutated) is a serine/threonine kinase activated by DNA double-strand breaks and can phosphorylate p53, effector kinase CHK2, and inhibitor of p53, MDM2.63 CHK2 also phosphorylates p53. Activation of p53 leads to increased expression of the Cdk inhibitor p21 and contributes to maintenance of cell-cycle arrest.
GNAL
Gα(olf) is found widely in brain with particularly robust expression in the striatum and cerebellar Purkinje cells.31 In the striatum, Gα(olf) may contribute to cell-cycle control via its role in phosphorylation of histone H3.64 At the transcriptional level, mutations in GNAL have been associated with up-regulation of genes involved in cell-cycle control and development.31
Neuronal death is an important phenomenon in the development of the nervous system and the above suggests one or more dystonia-associated proteins (CIZ1, THAP1, torsinA, N-TAF1, Gα(olf), and ATM) may contribute to this process.7,12,65 For example, cell-cycle control and the G1-S checkpoint are known to be critical to olivocerebellar development.66 In the cerebellar cortex, for instance, cell death may maintain numerical matching between granule and Purkinje cells (175:1). The downstream consequences of mutant or deficient dystonia-associated proteins could be aberrant cell-cycle reentry in terminally differentiated neurons, leading to numerical mismatch between specific neuronal populations, or ultrastructural defects of specific neuronal types such Purkinje cells or medium spiny neurons.67-70
Endoplasmic Reticulum/Nuclear Envelope Associated Dystonia Proteins
Another area where there appears to be convergence in pathogenic mechanisms in dystonia is the endoplasmic reticular/nuclear envelope (ER/NE) endomembrane system. Several genes that cause dystonia when mutated encode proteins associated with the (ER/NE) system. Included in this group are torsinA, epsilon sarcoglycan and THAP1. The ER/NE membrane system is located in the cell interior and, critically, all proteins must pass through this organelle to become membrane embedded. Membrane embedded proteins such as ion channels and neurotransmitter receptors and transporters are precisely the types of molecules that would be expected to cause neural dysfunction seen in primary dystonia.
TorsinA
TorsinA resides in the ER lumen and several observations support a role for TorsinA in membrane trafficking. It can inhibit trafficking of polytopic membranebound proteins, including the dopamine transporter, an effect suppressed by the ΔE mutation.71 Similarly, protein processing through the secretory pathway is defective in fibroblasts from patients with DYT1 dystonia, an abnormality that is rescued by down-regulating the mutant protein.72,73 TorsinA function in the ER also appears to impact the ability of a neuron to withstand cellular stress. Overexpression of torsinA protects cells from ER stress.74 Conversely torsinA knockdown or the ΔE mutation sensitize cells to ER stress.73 These effects may relate to a role for torsinA in endoplasmic reticular-associated degradation (ERAD), a process whereby misfolded proteins are removed from the ER lumen and degraded by the proteasome.75,76 Interestingly, the DYT16 protein PRKRA (PACT) may also play a role in regulating ER stress, via induction of Protein kinase R.77
TorsinA also functions at the nuclear envelope.78,79 A “substrate trap” version of torsinA that prevents it from uncoupling from protein partners causes it to accumulate abnormally in the NE.80-82 Similarly, disease mutant ΔE-torsinA concentrates abnormally at the nuclear membrane indicating that the DYT1 mutation may lead to an abnormal interaction with an NE partner,80,83 possibly lamina-associated polypeptide 1 (LAP1).84 The neuronal nuclear membranes from torsinA null of ΔE-homozygous knock-in mice exhibit abnormal nuclear envelope membranes, providing in vivo evidence for a NE role for torsinA.80 Considered together, these observations suggest that the DYT1 mutation impairs normal torsinA function.
The role of torsinA within the NE is not well understood, but may involve regulating connections between proteins that tether the nucleus to the cytoskeletal networker transcriptional regulation at the inner NE membrane.78,85 These functions may intersect with that of epsilon sarcoglycan, a protein that participates in nucleo-cytoskeletal connections, and the transcription factor THAP1 (Figure 2).
Figure 2.
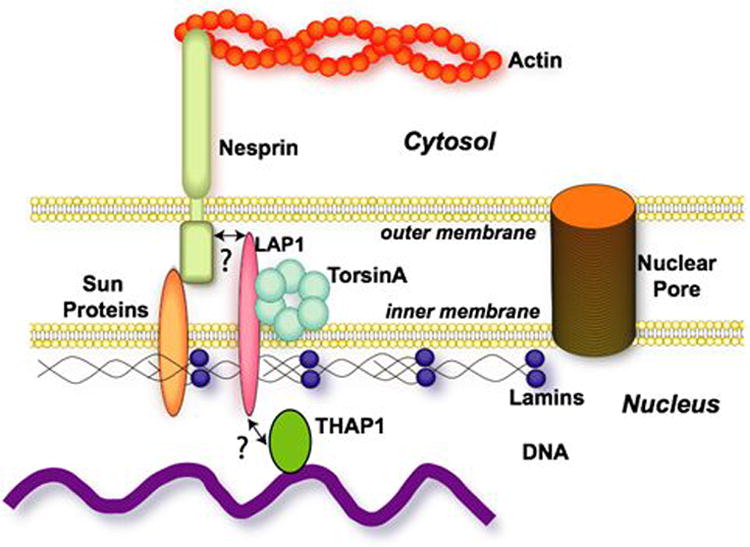
Potential interactions of torsinA and THAP1 at the nuclear envelope.
Epsilon Sarcoglycan (SCGE)
The connection of SCGE function to the actin cytoskeleton, which itself has links to the nuclear membrane, points to a possible connection between torsinA and SCGE. TorsinA has been implicated in regulating connections between the nucleus and the cytoskeleton, in particular through its reported interactions with the nesprins, a family of outer NE proteins that themselves can interact with the actin cytoskeleton. TorsinA has been reported to interact with nesprin-3, which mislocalizes in cells lacking torsinA or that harbor DYT1 mutant torsinA.86 Several reports demonstrate that nesprin-3 can connect to the actin cytoskeleton, consistent with a potential functional connection between torsinA and SCGE. This connection between torsinA and SCGE may also be in part direct, as torsinA has been reported to interact with and promote the degradation of SGCE mutants in the ER.87 Abnormalities of the actin cytoskeleton could account for observations of neural process abnormalities in cell lines overexpressing torsinA.88, 89
THAP1
Whilst THAP1 does not appear to have a direct effect on torsinA expression, there are other ways that nuclear-localized THAP1 could potentially interact with torsinA-related pathways. The nuclear membrane is increasingly recognized as a site of transcriptional regulation through several mechanisms.90,91 It is possible that THAP1 targets are dysregulated in DYT1 dystonia, in which mutant torsinA mislocalizes to the nuclear membrane. It is also possible that THAP1 and torsinA pathways connect via pRB, as this is a THAP1 binding partner that also indirectly connects to torsinA via laminA and LAP1. Direct testing of these possibilities awaits the identification of THAP1 target genes in neurons, which can then be assessed in DYT1 and forms of primary dystonia.
Synaptic function
Primary dystonia and the dystonia plus syndromes are characterized by absence of neurodegeneration implying a functional neuronal defect that leads to the abnormal movement command. This may be a neurochemical defect, possibly for multiple neurotransmitter pathways. The hypothesis that primary dystonia is due to disturbance of neurotransmitters or synaptic transmission is an attractive one and there is evidence to support this, particularly involving the dopaminergic pathway. The clearest example is from study of dopa-responsive dystonia where mutations encoding proteins critical for dopamine (DA) biosynthesis, including GTP-cyclohydrolase 1 and tyrosine hydroxylase cause dystonia.92,93 Most cases of DRD are due to GTPCH1 mutations; GTPCH1 is the rate-limiting step in production of tetrahydrobiopterin, which is a key co-factor in the synthesis of monoamines, particularly dopamine. Reduced levels of functional GTPCH1 lead to reduced DA levels and dystonia. DRD, therefore, responds very well L-dopa therapy which corrects the presynaptic deficit of DA.94
D2 receptor antagonists can cause tardive dystonia 95 and dystonia is also seen in Parkinson's disease, particularly young-onset,96 and Lesch-Nyhan syndrome where there is preferential loss of nigral dopaminergic neurons.97 Furthermore, other DA-related abnormalities have been found in various forms of focal primary dystonia, including reduced basal ganglia D2 receptor binding in imaging studies and possible association of dystonia with polymorphisms in the D5 receptor gene.98,99
TorsinA
The finding of torsinA mRNA in neurons of the substantia nigra led to neurochemical analysis of dopamine and its metabolites in post-mortem brain tissue.5,100 However, these studies were inconclusive.101,102 Similarly, studies in various transgenic mouse models provided inconsistent results regarding metabolite levels.103-106 However, more recent work in a model using TH promoter to drive expression of wild-type or mutant torsinA has suggested a defect in DA reuptake implicating the DA transporter (DAT).107,108 This has been supported by findings or reduced DA reuptake in hMT-CMV mice 109 and a recent study in DYT knock-in mice showing that TH positive substantia nigra neurons were slightly reduced in numbers and increased in size, 109 and evidence of a direct interaction between torsinA and DAT71 as well as vesicle monoamine transporter 2 (VMAT2).111 Recent work using more sensitive voltammetry to measure extracellular DA, however, found evidence to suggest that it was release not reuptake that was impaired, arguing against DAT dysfunction.107,112
Further evidence for involvement of presynaptic vesicles comes from cellular studies. TorsinA has been detected associated with vesicles in axons and presynaptic terminals, and biochemical fractionation analysis showed enrichment of torsinA in the fraction containing synaptosomal membranes.113 TorsinA has also been found to co-localise with snapin (SNARE-associated protein) on dense-core granules at the tips of differentiated PC12 cells.114 Functional analysis in SH-SY5Y cells expressing wild-type or mutant torsinA has shown that it regulates the degradation of snapin and stonin-2 (synaptotagmin specific endocytic adaptor) by the proteosome, with mutant torsinA leading to reduced levels and compromised synaptic vesicle recycling.115
Abnormalities in synaptic vesicle recycling has been supported by recent work in cultured hippocampal neurons from a knock-in mouse model of DYT1 dystonia which suggested that torsinA regulates recycling at a level at or upstream of the rise in calcium concentration in nerve terminals, and the regulation is influenced by neuronal activity.116 Furthermore, using patch-clamp electrophysiology, it was found that neurons with mutant-torsinA had more frequent miniature glutamate release, which may underlie the excitability of the CNS in DYT1 dystonia.117 The authors have not looked at other neurons, potentially more relevant to basal ganglia dysfunction.
Post-synaptic defects in hMT-CMV derived striatal slices showed that activation of D2 receptors (D2R) led to abnormal activation and inappropriate firing of cholinergic interneurons118 and GABAergic medium spiny neurons.119 In addition, medium spiny neurons from hMT-CMV mice had decreased expression of surface D2R with impaired G protein coupling, despite normal levels of D2R mRNA.120 It was suggested that there was a post-translational defect in receptor processing. This may occur in the ER for TA, where it acts as a molecular chaperone, leading to abnormal folding or oligomerization of the D2R, and a direct interaction between TA and the D2R has previously been demonstrated.71 In support of this hypothesis is data from [11C]-raclopride PET studies which showed reduced levels of D2R binding in patients with DYT1 and DYT6 dystonia, suggesting a pathogenetic link between these two forms and a D2R defect.121,122 For DYT1, reductions in radioligand binding were found in the caudate, putamen and ventrolateral thalamus.123 Further work needs to be performed to clarify this area, not least whether the potential D2R defect in DYT6 dystonia is also at a post-translational level or relates to disrupted transcription.
The abnormal synaptic plasticity in transgenic models of primary dystonia has been reviewed recently and highlighted a disruption of synaptic scaling, with facilitation of synaptic potentiation, together with loss of synaptic inhibitory processes.124 In the hMT-CMV model described above, impaired D2R postsynaptic function was suggested by inability of D2R agonist to re-establish normal corticostriatal synaptic plasticity. Interestingly, blockade of A2A receptors fully restored the impairment of synaptic plasticity. A2A receptors and D2 receptors oppose each other in the induction of bidirectional synaptic plasticity, with D2R promoting long term depression, and A2A receptors favouring induction of long term potentiation. The effect of the A2AR antagonist suggests the deficit in D2R function in the model can be reverted by eliminating the negative tone exerted by A2ARs on this. The possible role of A2ARs in genetic dystonia is also implicated from preliminary work on GNAL.
GNAL
G proteins link seven-transmembrane-domain receptors to downstream effectors and function as heterotrimers composed of α,β and γ subunits. Evidence suggests that Gα(olf) acts in medium spiny neurons to couple dopamine type 1 (D1R) receptors of the direct pathway and adenosine A2A receptors (A2AR) of the indirect pathway to the activation of adenylate cyclase type 5.125 A2AR and Gαolf are also expressed in striatal cholinergic interneurons. The mutations identified in GNAL appear to lead to loss of function and implicate abnormalities in D1R and/or A2AR transmission in the pathogenesis of dystonia.
DYT12: alpha-3 subunit of the Na+/K+ ATPase
Neuronal dysfunction leading to abnormal neurophysiology has been implicated for the DYT12 protein, alpha-3 subunit of the Na+/K+ ATPase, which belongs to the group of P-type ATPases, which utilise energy liberated during the hydrolysis of ATP for active transport of cations across cell membranes. Biochemical enzyme assays have revealed that mutations in α3 cause a reduction in both Na+ affinity and extrusion of intracellular Na+ leading to disrupted electrochemical ionic gradients across the neuronal cell membrane.126 It is possible that this may lead to downstream abnormalities of synaptic function. A phenotypic model of RDP was recently generated by chemically inhibiting the α3-isoform of the ATPase function in selected brain regions using the targeted infusion of ouabain, which selectively reduces α3-ATPase function in a dose-dependent manner in genetically wild-type mice.127 Ouabain infusions in the basal ganglia and cerebellum induced a parkinsonism-like or dystonic-like phenotype, respectively but only concomitant infusions in both structures yielded a stress-inducible phenotype resembling features of RDP. In the mouse model dystonic postures were reduced following transient inhibition of cerebellar input by GABA injection again underlining the emerging importance of cerebellar dysfunction in Dystonia and RDP.
Further evidence to support a dopaminergic aetiology for dystonia comes from study of the dopamine transporter deficiency syndrome.
Dopamine transporter deficiency syndrome
Dopamine transporter deficiency syndrome is an autosomal recessive condition caused by loss of function mutations in the DAT gene 128,129. DAT is a transmembrane protein exclusively expressed in dopaminergic neurons where it mediates the re-uptake of dopamine into pre-synaptic terminals after synaptic transmission. This rapid recycling of neurotransmitter is crucial to synaptic function as it replenishes dopamine stores in the presynaptic terminal and prevents desensitization of the postsynaptic terminal. Children presented in infancy with either hyperkinesia, parkinsonism, or a mixed hyperkinetic and hypokinetic movement disorder. Some individuals had previously been misdiagnosed with cerebral palsy. During childhood they developed severe dystonia- parkinsonism associated with an eye movement disorder and pyramidal tract features. Investigations revealed raised ratios of homovanillic acid to 5-hydroxyindoleacetic acid in cerebrospinal fluid. DAT SCAN imaging in one patient showed complete loss of dopamine transporter activity in the basal nuclei. Although a trial of L-dopa had no effect on either the patients' symptoms, or on CSF parameters.
Common themes in dystonia molecular pathways
The increasing knowledge of proteins whose mutant forms cause dystonia has implicated a large number of neurobiological pathways that lead to dystonic movements. A number of themes have emerged which have been identified in this review from abnormal transcription and cell cycle due to the nuclear effects of dystonia genes, to ER dysfunction and synaptic abnormalities. It is right to seek common pathways that may represent targets for therapeutic strategies for this group of incurable movement disorders. However, it may also lead to oversimplification in the search for unifying mechanisms. Most cases of dystonia are primary and not associated with neuronal death. Thus the pathogenic mechanisms may be subtle and only cause relatively mild defects in the relevant pathways, leading to abnormal processing of the motor command within the CNS.70
There is increasing awareness for the role of abnormal inhibition and plasticity affecting sensorimotor pathways in dystonia.1 This may be a template laid down in early life supporting the view that primary dystonia is a neurodevelopmental circuit disorder. In support of this are the developmental patterns of expression of the best studied primary dystonia gene products: torsinA and THAP1. TorsinA expression in the mouse was highest during prenatal and early postnatal development, particularly in the cortex, striatum, thalamus and cerebellum.130 In human brain torsinA protein is detectable at 4-8 weeks postnatally in the cerebellum (Purkinje cells), substantia nigra hippocampus and basal ganglia.131 Similarly, THAP1 is expressed in the rat in early development, particularly in the cerebellum (Purkinje cells), cortical pyramidal neurons, relay neurons in thalamus, medium and cholinergic striatal neurons, dopaminergic substantia nigra neurons and hippocampal neurons.132 This developmentally regulated expression of two dystonia associated proteins suggest a role in terminal regulation and establishment of key circuits involved in motor control. The themes identified in this review would have a significant effect on neurodevelopment in terms of altered transcription of key genes at nuclear level, protein processing and trafficking through the ER, or altered ER stress response in important periods of neural cell differentiation, through to abnormal synaptic function affecting neurotransmission or synaptic plasticity. Any or all of the above mechanisms could lead to abnormal patterns or responsiveness of sensorimotor circuits leading to a susceptibility to developing an abnormal “dystonic state”.
It may be that with the increasing number of DYT loci, and better understanding of recently identified genes encoding CIZ1, GNAL and ANO3, the key cellular pathogenetic mechanisms involved in the genesis of dystonic movement will become clearer.
Figure 3.
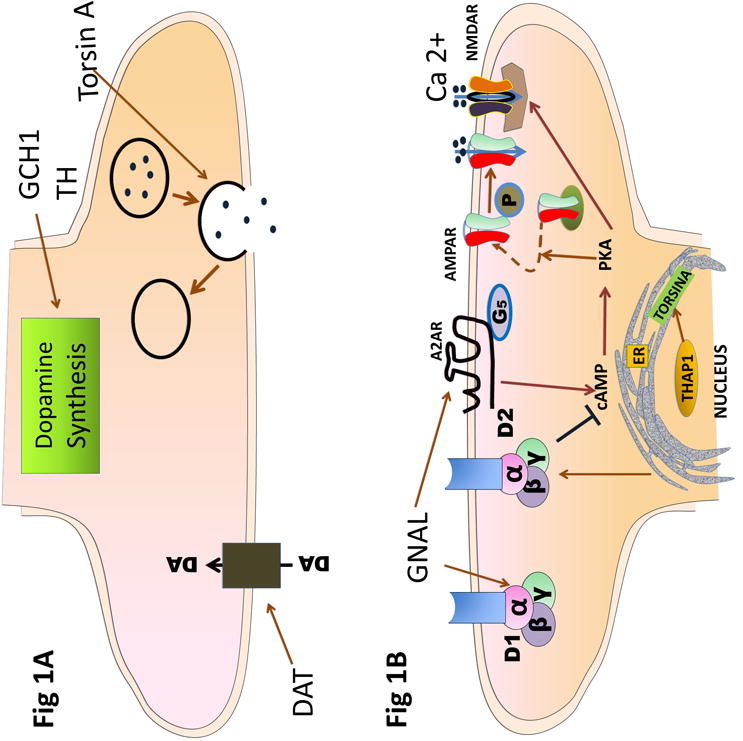
Schematic diagram of synapse indicating potential impact of proteins whose mutant forms cause dystonia. 1A: presynaptic terminal showing interaction of torsinA, DAT and mutations in GCH1 and TH on synaptic vesicle recycling, dopamine uptake and synthesis respectively. 1B: postsynaptic terminal. TorsinA and THaP1 may affect dopamine receptor expression of function. D2R and A2AR function may be influenced by these proteins or directly by GNAL influencing PKA phosphorylation and promoting delivery of GluA1-containing AMPARs to extrasynaptic pools increasing their availability for subsequent synaptic recruitment upon LTP induction and NMDAR activation. Calcium influx through synaptic NMDARs is endogenously facilitated by A2AR, but it can also be inhibited by activation of D2R through PKA-dependent mechanisms.
α, β, γ: subunits of G protein, DA: Dopamine, D2: Dopamine type2 receptor, D1: Dopamine type 1 receptor, GCH1: GTPcyclohydrolase 1, GNAL: stimulatory a subunit of G protein, TH: tyrosine hydroxylase, ER: endoplasmic reticulum, A2AR adenosine 2 receptor, AMPAR: AMPA receptor, NMDAR: NMDA receptor.
Acknowledgments
Dr. LeDoux has been supported by the Dystonia Medical Research Foundation, Bachmann-Strauss Dystonia & Parkinson Foundation, Tyler's Hope for a Dystonia Cure, Prana, CHDI, Merz and NIH grants R01NS048458, R01NS060887, R01NS057722, R01NS058850, 5U01NS052592, 5U01AT000613 and U54NS065701.
Professor Dauer is supported by the Dystonia Medical Research Foundation, Tyler's Hope for a Dystonia Cure and the NINDS (P50 NS038370)
Professor Warner receives support from Brain Research Trust, Cure Huntington's Disease Initiative and has received grants from Wellcome Trust, The Dystonia Society, NHS Innovations Inc, NIH, Bachmann-Strauss Dystonia and Parkinson Foundation.
Footnotes
Relevant conflicts of interest/financial disclosures: Nothing to report.
Author Roles: MLD: 1A, 1C, 3A, 3B WTD: 1A, 1C, 3A, 3B TTW: 1A, 1B, 1C, 3A, 3B
Financial disclosure. Dr. LeDoux serves on the speakers' bureaus for Lundbeck and Teva Neuroscience; serves on the Xenazine Advisory Board for Lundbeck and Azilect Advisory Board for Teva, receives research support from the NIH, Dystonia Medical Research Foundation, Tyler's Hope for a Dystonia Cure, Prana, CHDI and Merz; and receives royalty payments for Animal Models of Movement Disorders (Elsevier).
References
- 1.Phukan J, Albanese A, Gasser T, Warner T. Primary dystonia and dystonia-plus syndromes: clinical characteristics, diagnosis, and pathogenesis. Lancet Neurol. 2011;10:1074–1085. doi: 10.1016/S1474-4422(11)70232-0. [DOI] [PubMed] [Google Scholar]
- 2.Ozelius L, Kramer P, Page CE, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nature Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 3.Hanson PI, Whiteheart SW. AAA+proteins: have engine will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 4.Kock N, Naismith TV, Boston HE, et al. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum Mol Genet. 2006;15:1355–1364. doi: 10.1093/hmg/ddl055. [DOI] [PubMed] [Google Scholar]
- 5.Augood SJ, Penney JB, Friberg IK, et al. Expression of the early onset torsion dystonia gene (DYT1) in human brain. Ann Neurol. 1998;43:669–673. doi: 10.1002/ana.410430518. [DOI] [PubMed] [Google Scholar]
- 6.Rostasy K, Augood SJ, Hewett JW, et al. TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol Dis. 2003;12:11–24. doi: 10.1016/s0969-9961(02)00010-4. [DOI] [PubMed] [Google Scholar]
- 7.Xiao J, Gong S, Zhao Y, LeDoux MS. Developmental expression of rat torsinA transcript and protein. Brain Res Dev Brain Res. 2004;152:47–60. doi: 10.1016/j.devbrainres.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Xiao J, Ueda M, Wang Y, Hines M, Nowak TS, Jr, LeDoux MS. Glial elements contribute to stress-induced torsinA expression in the central and peripheral nervous systems. Neuroscience. 2008;155:439–453. doi: 10.1016/j.neuroscience.2008.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jungwirth M, Dear ML, Brown P, Holbrook K, Goodchild R. Relative expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum Mol Genet. 2010;19:888–900. doi: 10.1093/hmg/ddp557. [DOI] [PubMed] [Google Scholar]
- 10.Hersheson J, Menacci NE, Davis M, et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann Neurol. 2013 doi: 10.1002/ana.23832. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blanchard A, Ea V, Roubertie A, et al. DYT6 dystonia: review of the literature and creation of the UMD Locus-Specific Database (LSDB) for mutations in the THAP1 gene. Hum Mutat. 2011;32(11):1213–1224. doi: 10.1002/humu.21564. [DOI] [PubMed] [Google Scholar]
- 12.LeDoux MS, Xiao J, Rudzińska M, et al. Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat Disord. 2012;18:414–425. doi: 10.1016/j.parkreldis.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- 14.Djarmati A, Schneider SA, Lohmann K, et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study. Lancet Neurol. 2009;8:447–452. doi: 10.1016/S1474-4422(09)70083-3. [DOI] [PubMed] [Google Scholar]
- 15.Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol. 2009;8:441–446. doi: 10.1016/S1474-4422(09)70081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider SA, Ramirez A, Shafiee K, et al. Homozygous THAP1 mutations as cause of early-onset generalized dystonia. Mov Disord. 2011;26:858–861. doi: 10.1002/mds.23561. [DOI] [PubMed] [Google Scholar]
- 17.Roussigne M, Cayrol C, Clouaire T, Amalric F, Girard JP. THAP1 is a nuclear proapoptotic factor that links prostate-apoptosis-response-4 (Par-4) to PML nuclear bodies. Oncogene. 2003;22:2432–2442. doi: 10.1038/sj.onc.1206271. [DOI] [PubMed] [Google Scholar]
- 18.Cayrol C, Lacroix C, Mathe C, et al. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood. 2007;109:584–594. doi: 10.1182/blood-2006-03-012013. [DOI] [PubMed] [Google Scholar]
- 19.Zimprich A, Grabowski M, Asmus F, et al. T Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29:66–69. doi: 10.1038/ng709. [DOI] [PubMed] [Google Scholar]
- 20.Sunada Y, Campbell KP. Dystrophin-glycoprotein complex: molecular organization and critical roles in skeletal muscle. Curr Opin Neurol. 1995;8:379–384. [PubMed] [Google Scholar]
- 21.Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- 22.Chan P, Gonzalez-Maeso J, Ruf F, et al. Epsilon-sarcoglycan immunoreactivity and mRNA expression in mouse brain. J Comp Neurol. 2005;482:50–75. doi: 10.1002/cne.20377. [DOI] [PubMed] [Google Scholar]
- 23.Brunig I, Suter A, Knuesel I, Luscher B, Fritschy JM. GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J Neurosci. 2002;22:4805–4813. doi: 10.1523/JNEUROSCI.22-12-04805.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci. 2002;22:4274–4285. doi: 10.1523/JNEUROSCI.22-11-04274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camargos S, Scholz S, Simon-Sanchez J, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress response protein PRKRA. Lancet Neurology. 2008;7:207–215. doi: 10.1016/S1474-4422(08)70022-X. [DOI] [PubMed] [Google Scholar]
- 26.Bragg DC, Armata IA, Nery FC, et al. Molecular pathways in dystonia. Neurobiol Dis. 2011;42:136–147. doi: 10.1016/j.nbd.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao J, Uitti RJ, Zhao Y, et al. Mutations in CIZ1 cause adult-onset primary cervical dystonia. Ann Neurol. 2012;71:458–469. doi: 10.1002/ana.23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitsui K, Matsumoto A, Ohtsuka S, et al. Cloning and characterization of a novel p21(Cip1/Waf1)-interacting zinc finger protein, ciz1. Biochem Biophys Res Commun. 1999;264:457–464. doi: 10.1006/bbrc.1999.1516. [DOI] [PubMed] [Google Scholar]
- 29.Charlesworth G, Plagnol V, Holmstrom KM, et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet. 2012;91:1041–1050. doi: 10.1016/j.ajhg.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuchs T, Saunders-Pullman R, Masuho I, et al. Mutations in GNAL cause primary torsion dystonia. Nature Genet. 2013;45:88–92. doi: 10.1038/ng.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vemula SR, Puschmann A, Xiao J, et al. Role of Ga(Olf) in familial and sporadic adult onset primary dystonia. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt102. epub ahead of press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coverley D, Marr J, Ainscough J. Ciz1 promotes mammalian DNA replication. J Cell Sci. 2005;118:101–112. doi: 10.1242/jcs.01599. [DOI] [PubMed] [Google Scholar]
- 33.Warder DE, Keherly MJ. Ciz1, Cip1 interacting zinc finger protein 1 binds the consensus DNA sequence ARYSR(0 2)YYAC. J Biomed Sci. 2003;10:406–417. doi: 10.1007/BF02256432. [DOI] [PubMed] [Google Scholar]
- 34.Ainscough JF, Rahman FA, Sercombe H, et al. C-terminal domains deliver the DNA replication factor Ciz1 to the nuclear matrix. J Cell Sci. 2007;120:115–124. doi: 10.1242/jcs.03327. [DOI] [PubMed] [Google Scholar]
- 35.Copeland NA, Sercombe HE, Ainscough JF, Coverley D. Ciz1 cooperates with cyclin-A-CDK2 to activate mammalian DNA replication in vitro. J Cell Sci. 2010;123:1108–1115. doi: 10.1242/jcs.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.den Hollander P, Rayala SK, Coverley D, Kumar R. Ciz1, a Novel DNA-binding coactivator of the estrogen receptor alpha, confers hypersensitivity to estrogen action. Cancer Res. 2006;66:11021–11029. doi: 10.1158/0008-5472.CAN-06-2336. [DOI] [PubMed] [Google Scholar]
- 37.Bessiere D, Lacroix C, Campagne S, et al. Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J Biol Chem. 2008;283:4352–4363. doi: 10.1074/jbc.M707537200. [DOI] [PubMed] [Google Scholar]
- 38.Campagne S, Saurel O, Gervais V, Milon A. Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res. 2010;38:3466–3476. doi: 10.1093/nar/gkq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clouaire T, Roussigne M, Ecochard V, Mathe C, Amalric F, Girard JP. The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc Natl Acad Sci U S A. 2005;102:6907–6912. doi: 10.1073/pnas.0406882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat Struct Mol Biol. 2010;17:117–123. doi: 10.1038/nsmb.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavarini S, Cayrol C, Fuchs T, Lyons N, Ehrlich ME, Girard JP, Ozelius LJ. Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann Neurol. 2010;68(4):549–553. doi: 10.1002/ana.22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaiser FJ, Osmanoric A, Rakovic, et al. The dystonia gene DYT1 is repressed by the transcription factor THAP1 (DYT6) Ann Neurol. 2010;68(4):554–559. doi: 10.1002/ana.22157. [DOI] [PubMed] [Google Scholar]
- 43.Palada V, Stiern S, Glockle, et al. K Lack of sequence variations in THAP1 gene and THAP1-binding sites in TOR1A promoter of DYT1 patients. Mov Disord. 2012;27(7):917. doi: 10.1002/mds.24974. [DOI] [PubMed] [Google Scholar]
- 44.Kamm C, Uflacker N, Asmus F, et al. No evidence for THAP1/DYT6 variants as disease modifiers in DYT1 dystonia. Mov Disord. 2011;26(11):2136–2137. doi: 10.1002/mds.23777. [DOI] [PubMed] [Google Scholar]
- 45.Rual JF, Venkatesan K, Hao T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–8. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 46.Lütolf S, Radtke F, Aguet M, Suter U, Taylor V. Notch1 is required for neuronal and glial differentiation in the cerebellum. Development. 2002;129:373–85. doi: 10.1242/dev.129.2.373. [DOI] [PubMed] [Google Scholar]
- 47.Koekkoek SK, Yamaguchi K, Milojkovic BA, et al. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron. 2005;47:339–52. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 48.Andersson T, Södersten E, Duckworth JK, et al. CXXC5 is a novel BMP4-regulated modulator of Wnt signaling in neural stem cells. J Biol Chem. 2009;284:3672–81. doi: 10.1074/jbc.M808119200. [DOI] [PubMed] [Google Scholar]
- 49.Sadnicka A, Hoffland BS, Bhatia KP, van de Warrenburg BP, Edwards MJ. The cerebellum in dystonia – help of hindrance. Clin Neurophysiol. 2012;123:65–70. doi: 10.1016/j.clinph.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 50.Evidente VG, Advincula J, Esteban R, et al. Phenomenology of “Lubag” or X-linked dystonia-parkinsonism. Mov Disord. 2002;17:1271–1277. doi: 10.1002/mds.10271. [DOI] [PubMed] [Google Scholar]
- 51.Makino S, Kaji R, Ando S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80:393–406. doi: 10.1086/512129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sako W, Moridaki R, Kaji R, et al. Identification and localization of a neuron-specific isoform of TAF1 in rat brain; implications for neuropathology of DYT3 dystonia. Neuroscience. 2011;189:100–7. doi: 10.1016/j.neuroscience.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li HH, Li AG, Sheppard HM, Liu X. Phosphorylation on Thr-55 by TAF1 mediates degradation of p53: a role for TAF1 in cell G1 progression. Mol Cell. 2004;13:867–878. doi: 10.1016/s1097-2765(04)00123-6. [DOI] [PubMed] [Google Scholar]
- 54.Buchmann AM, Skaar JR, DeCaprio JA. Activation of a DNA damage checkpoint response in a TAF1-defective cell line. Mol Cell Biol. 2004;24:5332–5339. doi: 10.1128/MCB.24.12.5332-5339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katzenberger RJ, Marengo MS, Wassarman DA. ATM and ATR pathways signal alternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. Mol Cell Biol. 2006;26:9256–9267. doi: 10.1128/MCB.01125-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basham SE, Rose LS. The Caenorhabditis elegans polarity gene ooc-5 encodes a Torsin-related protein of the AAA ATPase superfamily. Development. 2001;128:4645–4656. doi: 10.1242/dev.128.22.4645. [DOI] [PubMed] [Google Scholar]
- 57.Baptista MJ, O'Farrell C, Hardy J, Cookson MR. Microarray analysis reveals induction of heat shock proteins mRNAs by the torsion dystonia protein, TorsinA. Neurosci Lett. 2003 May 29;343(1):5–8. doi: 10.1016/s0304-3940(03)00302-1. [DOI] [PubMed] [Google Scholar]
- 58.Koh YH, Rehfeld K, Ganetzky B. A Drosophila model of early onset torsion dystonia suggests impairment in TGF-beta signaling. Hum Mol Genet. 2004;13:2019–2030. doi: 10.1093/hmg/ddh208. [DOI] [PubMed] [Google Scholar]
- 59.Erol A. Genotoxic stress-mediated cell cycle activities for the decision of cellular fate. Cell Cycle. 2011;10:3239–3248. doi: 10.4161/cc.10.19.17460. [DOI] [PubMed] [Google Scholar]
- 60.Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- 61.McCabe BD, Marqués G, Haghighi AP, et al. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–54. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- 62.Saunders-Pullman R, Raymond D, Stoessl AJ, et al. Variant ataxia-telangiectasia presenting as primary-appearing dystonia in Canadian Mennonites. Neurology. 2012;78:649–657. doi: 10.1212/WNL.0b013e3182494d51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pommier Y, Weinstein JN, Aladjem MI, Kohn KW. Chk2 molecular interaction map and rationale for Chk2 inhibitors. Clin Cancer Res. 2006;12:2657–2661. doi: 10.1158/1078-0432.CCR-06-0743. [DOI] [PubMed] [Google Scholar]
- 64.Bertran-Gonzalez J, Håkansson K, Borgkvist A, et al. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology. 2009;34:1710–20. doi: 10.1038/npp.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herrup K, Sunter K. Numerical matching during cerebellar development: quantitative analysis of granule cell death in staggerer mouse chimeras. J Neurosci. 1987;7:829–36. doi: 10.1523/JNEUROSCI.07-03-00829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subkhankulova T, Zhang X, Leung C, Marino S. Bmi1 directly represses p21Waf1/Cip1 in Shh-induced proliferation of cerebellar granule cell progenitors. Mol Cell Neurosci. 2010;45:151–162. doi: 10.1016/j.mcn.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 67.Wang L, Wang R, Herrup K. E2F1 works as a cell cycle suppressor in mature neurons. J Neurosci. 2007;27:12555–12564. doi: 10.1523/JNEUROSCI.3681-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J, Chen J, Vinters HV, Gatti RA, Herrup K. Stable brain ATM message and residual kinase-active ATM protein in ataxia-telangiectasia. J Neurosci. 2011;31:7568–7577. doi: 10.1523/JNEUROSCI.0778-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prudente CN, Pardo CA, Xiao J, Hanfelt J, Hess EJ, Ledoux MS, Jinnah HA. Neuropathology of cervical dystonia. Exp Neurol. 2013;241:95–104. doi: 10.1016/j.expneurol.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Song CH, Bernhard D, Bolarinwa C, Hess EJ, Smith Y, Jinnah HA. Subtle microstructural changes of the striatum in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis. 2013;54:362–71. doi: 10.1016/j.nbd.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci U S A. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hewett JW, Tannous B, Niland BP, Nery FC, Zeng J, Li Y, Breakefield XO. Mutant torsinA interferes with protein processing through the secretory pathway in DYT1 dystonia cells. Proc Natl Acad Sci U S A. 2007;104:7271–7276. doi: 10.1073/pnas.0701185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hewett JW, Nery FC, Niland B, et al. siRNA knock-down of mutant torsinA restores processing through secretory pathway in DYT1 dystonia cells. Hum Mol Genet. 2008;17:1436–1445. doi: 10.1093/hmg/ddn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen P, Burdette AJ, Porter JC, et al. The early-onset torsion dystonia-associated protein, torsinA, is a homeostatic regulator of endoplasmic reticulum stress response. Hum Mol Genet. 2010;19:3502–3515. doi: 10.1093/hmg/ddq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nery FC, Armata IA, Farley JE, et al. TorsinA participates in endoplasmic reticulum-associated degradation. Nat Commun. 2011;2:393. doi: 10.1038/ncomms1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol. 2011;23:176–183. doi: 10.1016/j.ceb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee ES, Tang N, Thompson S, Miller S, Katze MG. The double-stranded RNA-activated protein kinase (PKR) plays a significant role in a sustained ER stress-induced apoptosis. FEBS J. 2007;581:4325–4332. doi: 10.1016/j.febslet.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 78.Gerace L. TorsinA and torsion dystonia: Unraveling the architecture of the nuclear envelope. Proc Natl Acad Sci U S A. 2004;101:8839–8840. doi: 10.1073/pnas.0402441101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci U S A. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goodchild RE, Kim CE, Dauer WT. Loss of the Dystonia-Associated Protein TorsinA Selectively Disrupts the Neuronal Nuclear Envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 81.Naismith TV, Heuser JE, Breakefield XO, Hanson PI. TorsinA in the nuclear envelope. Proc Natl Acad Sci U S A. 2004;101:7612–7617. doi: 10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24:2593–2601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giles LM, Chen J, Li L, Chin LS. Dystonia-associated mutations cause premature degradation of torsinA protein and cell-type-specific mislocalization to the nuclear envelope. Hum Mol Genet. 2008;17:2712–2722. doi: 10.1093/hmg/ddn173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol. 2005;168:855–862. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Worman HJ, Gundersen GG. Here come the SUNs: a nucleocytoskeletal missing link. Trends Cell Biol. 2006;16:67–9. doi: 10.1016/j.tcb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 86.Nery FC, Zeng J, Niland, et al. TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J Cell Sci. 2008;121:3476–3486. doi: 10.1242/jcs.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esapa CT, Waite A, Locke M, et al. SGCE missense mutations that cause myoclonus-dystonia syndrome impair epsilon-sarcoglycan trafficking to the plasma membrane: modulation by ubiquitination and torsinA. Hum Mol Genet. 2007;16:327–342. doi: 10.1093/hmg/ddl472. [DOI] [PubMed] [Google Scholar]
- 88.Ferrari-Toninelli G, Paccioretti S, Francisconi S, Uberti D, Memo M. TorsinA negatively controls neurite outgrowth of SH-SY5Y human neuronal cell line. Brain Res. 2004;1012:75–81. doi: 10.1016/j.brainres.2004.02.080. [DOI] [PubMed] [Google Scholar]
- 89.Hewett JW, Zeng J, Niland BP, Bragg DC, Breakefield XO. Dystonia-causing mutant torsinA inhibits cell adhesion and neurite extension through interference with cytoskeletal dynamics. Neurobiol Dis. 2006;22:98–111. doi: 10.1016/j.nbd.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 90.Srsen V, Korfali N, Schirmer EC. Nuclear envelope influences on cell-cycle progression. Biochem Soc Trans. 2011;39:1742–1746. doi: 10.1042/BST20110656. [DOI] [PubMed] [Google Scholar]
- 91.Egecioglu D, Brickner JH. Gene positioning and expression. Curr Opin Cell Biol. 2011;23:338–345. doi: 10.1016/j.ceb.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236–242. doi: 10.1038/ng1194-236. [DOI] [PubMed] [Google Scholar]
- 93.Knappskog PM, Flatmark T, Mallet J, Ludecke B, Bartholome K. Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet. 1995;4:1209–1212. doi: 10.1093/hmg/4.7.1209. [DOI] [PubMed] [Google Scholar]
- 94.Trender-Gerhard I, Sweeney MG, Schwingenschuh P, et al. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry. 2009;80:839–845. doi: 10.1136/jnnp.2008.155861. [DOI] [PubMed] [Google Scholar]
- 95.Casey DE. Pathophysiology of antipsychotic drug-induced movement disorders. J Clin Psychiatry. 2004;65:25–28. [PubMed] [Google Scholar]
- 96.Tolosa E, Compta Y. Dystonia in Parkinson's disease. J Neurol. 2006;(Suppl 7):VII7–13. doi: 10.1007/s00415-006-7003-6. [DOI] [PubMed] [Google Scholar]
- 97.Egami K, Yitta S, Lewers JC, et al. Basal ganglia dopamine loss due to defect in purine cycling. Neurobiol Dis. 2007;26:391–407. doi: 10.1016/j.nbd.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Perlmutter JS, Stambuk MK, Markham J, Black KJ, McGee-Minnich L, Jankovic J, Moerlein SM. Decreased [18F]spiperone binding in putamen in idiopathic focal dystonia. J Neurosci. 1997;17:843–850. doi: 10.1523/JNEUROSCI.17-02-00843.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Misbahuddin A, Placzek MR, Chaudhuri KR, Wood NW, Bhatia KP, Warner TT. A polymorphism in the dopamine receptor DRD5 is associated with blepharospasm. Neurology. 2002;58:124–126. doi: 10.1212/wnl.58.1.124. [DOI] [PubMed] [Google Scholar]
- 100.Augood SJ, Martin DM, Ozelius LJ, Breakefield XO, Penney JB, Jr, Standaert DG. Distribution of the mRNAs encoding torsinA and torsinB in the normal adult human brain. Ann Neurol. 1999;46:761–769. [PubMed] [Google Scholar]
- 101.Augood SJ, Hollingsworth Z, Albers DS, et al. Dopamine transmission in DYT1 dystonia: a biochemical and autoradiographical study. Neurology. 2002;59:445–458. doi: 10.1212/wnl.59.3.445. [DOI] [PubMed] [Google Scholar]
- 102.Furukawa Y, Hornykiewicz O, Fahn S, Kish SJ. Striatal dopamine in early-onset primary torsion dystonia with the DYT1 mutation. Neurology. 2000;54:1193–1195. doi: 10.1212/wnl.54.5.1193. [DOI] [PubMed] [Google Scholar]
- 103.Shashidharan P, Sandu D, Potla U, et al. Transgenic mouse model of early-onset DYT1 dystonia. Hum Mol Genet. 2005;14:125–133. doi: 10.1093/hmg/ddi012. [DOI] [PubMed] [Google Scholar]
- 104.Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y. Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol. 2005;196:452–463. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 105.Grundmann K, Hubener J, Habig K, et al. Gene expression changes in a transgenic mouse model overexpressing human wild-type and mutant torsinA. Proteomics Clin Appl. 2008;2:720–736. doi: 10.1002/prca.200780053. [DOI] [PubMed] [Google Scholar]
- 106.Zhao Y, Decuypere M, Ledoux MS. Abnormal motor function and dopamine neurotransmission in DYT1 DeltaGAG transgenic mice. Exp Neurol. 2008;210:719–730. doi: 10.1016/j.expneurol.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Page ME, Bao L, Andre P, et al. Cell-autonomous alteration of dopaminergic transmission by wild type and mutant (DeltaE) TorsinA in transgenic mice. Neurobiol Dis. 2010 doi: 10.1016/j.nbd.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balcioglu A, Kim MO, Sharma N, Cha JH, Breakefield XO, Standaert DG. Dopamine release is impaired in a mouse model of DYT1 dystonia. J Neurochem. 2007;102:783–788. doi: 10.1111/j.1471-4159.2007.04590.x. [DOI] [PubMed] [Google Scholar]
- 109.Hewett J, Johanson P, Sharma N, Standaert DG, Balcioglu A. Function of dopamine transporter is compromised in DYT1 transgenic animal model in vivo. J Neurochem. 2010;113:228–235. doi: 10.1111/j.1471-4159.2010.06590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Song CH, Fan X, Exeter CJ, Hess EJ, Jinnah HA. Functional analysis of Dopaminergic system in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis. 2012;48:66–78. doi: 10.1016/j.nbd.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Misbahuddin A, Placzek MR, Taanman JW, Gschmeissner S, Schiavo G, Cooper JM, Warner TT. Mutant torsinA, which causes early-onset primary torsion dystonia, is redistributed to membranous structures enriched in vesicular monoamine transporter in cultured human SH-SY5Y cells. Mov Disord. 2005;20:432–440. doi: 10.1002/mds.20351. [DOI] [PubMed] [Google Scholar]
- 112.Bao L, Patel JC, Walker RH, Shashidharan P, Rice ME. Dysregulation of striatal dopamine release in a mouse model of dystonia. J Neurochem. 2010 doi: 10.1111/j.1471-4159.2010.06890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Augood SJ, Hollingsworth Z, Albers DS, et al. Dopamine transmission in DYT1 dystonia: a biochemical and autoradiographical study. Neurology. 2002;59:445–458. doi: 10.1212/wnl.59.3.445. [DOI] [PubMed] [Google Scholar]
- 114.Granata A, Watson R, Collinson L, Schiavo G, Warner TT. The dystonia-associated protein torsinA modulates synaptic vesicle recycling. J Biol Chem. 2008;283:7568–7579. doi: 10.1074/jbc.M704097200. [DOI] [PubMed] [Google Scholar]
- 115.Granata A, Koo SJ, Haucke V, et al. CSN complex controls the stability of selected synaptic proteins via a torsinA-dependent process. EMBO Journal. 2011;30:181–93. doi: 10.1038/emboj.2010.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kakazu Y, Koh JY, Ho D, Gonzalez-Alegre P, Harata C. Synaptic vesicle recycling is enhanced by torsinA that harbours the DYT1 dystonia mutation. Synapse. 2012;66:453–464. doi: 10.1002/syn.21534. [DOI] [PubMed] [Google Scholar]
- 117.Kakazu Y, Koh JY, Iwabuchi S, Gonzalez-Alegre P, Harata C. Miniature release events of glutamate from hippocampal neurons are influenced by the dystonia-associated protein TorsinA. Synapse. 2012;66:807–822. doi: 10.1002/syn.21571. [DOI] [PubMed] [Google Scholar]
- 118.Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 119.Sciamanna G, Bonsi P, Tassone A, et al. Impaired striatal D2 receptor function leads to enhanced GABA transmission in a mouse model of DYT1 dystonia. Neurobiol Dis. 2009;34:133–145. doi: 10.1016/j.nbd.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Napolitano F, Pasqualetti M, Usiello A, et al. Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiol Dis. 2010;38:434–445. doi: 10.1016/j.nbd.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Asanuma K, Carbon-Correll M, Eidelberg D. Neuroimaging in human dystonia. J Med Invest. 2005;52:272–279. doi: 10.2152/jmi.52.272. [DOI] [PubMed] [Google Scholar]
- 122.Carbon M, Niethammer M, Peng S, Raymond D, Dhawan V, Chaly T, Ma Y, Bressman S, Eidelberg D. Abnormal striatal and thalamic dopamine neurotransmission: Genotype-related features of dystonia. Neurology. 2009;72:2097–2103. doi: 10.1212/WNL.0b013e3181aa538f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carbon M, Argyelan M, Eidelberg D. Functional imaging in hereditary dystonia. Eur J Neurol. 2010;(1):58–64. doi: 10.1111/j.1468-1331.2010.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Quatarone A, Pisani A. Abnormal synaptic plasticity in dystonia: disruption of synaptic homeostasis. Neurobiol Disease. 2011;42:162–170. doi: 10.1016/j.nbd.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 125.Herve D, Le Moine C, Corvol JC, et al. Gaolf levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J Neurosci. 2001;21:4390–4399. doi: 10.1523/JNEUROSCI.21-12-04390.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ashmore LJ, Hrizo SL, Paul SM, Van Voorhies WA, Beitel GJ, Palladino MJ. Novel mutations affecting the Na, K ATPase alpha model complex neurological diseases and implicate the sodium pump in increased longevity. Hum. Genet. 2009;126:431–447. doi: 10.1007/s00439-009-0673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci. 2011;14:357–365. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kurian MA, Zhen J, Cheng SY, et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest. 2009;119:1595–1603. doi: 10.1172/JCI39060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kurian MA, Li Y, Zhen J, et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 2011;10:54–62. doi: 10.1016/S1474-4422(10)70269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vasudevan A, Breakefield XO, Bhide PG. Developmental patterns of torsinA and torsinB expression. Brain Res. 2006:1073–1074. 139–45. doi: 10.1016/j.brainres.2005.12.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Siegert S, Bahn C, Kramer ML, et al. TorsinA expression is detectable in human infants as young as 4 weeks old. Brain Res Dev Brain Res. 2005;757:19–26. doi: 10.1016/j.devbrainres.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 132.Zhao Y, Xiao J, Gong S, Clara JA, Ledoux MD. Neural expression of transcription factor THAP1 during development in rat. Neurosci. 2013;231:282–95. doi: 10.1016/j.neuroscience.2012.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]