

Abstract
The taccalonolides are highly acetylated steroids that stabilize cellular microtubules and overcome multiple mechanisms of taxane resistance. Recently, two potent taccalonolides, AF and AJ, were identified that bind tubulin directly and enhance microtubule polymerization. Extensive studies were conducted to characterize these new taccalonolides. AF and AJ caused aberrant mitotic spindles and bundling of interphase microtubules that differed from the effects of either paclitaxel or laulimalide. AJ also distinctly affected microtubule polymerization in that it enhanced the rate and extent of polymerization in the absence of any noticeable effect on microtubule nucleation. Additionally, the resulting microtubules were found to be profoundly cold stable. These data, along with studies showing synergistic antiproliferative effects between AJ and either paclitaxel or laulimalide, suggest a distinct binding site. Direct binding studies demonstrated that AJ could not be displaced from microtubules by paclitaxel, laulimalide or denaturing conditions, suggesting irreversible binding of AJ to microtubules. Mass spectrometry confirmed a covalent interaction of AJ with a peptide of β-tubulin containing the cyclostreptin binding sites. Importantly, AJ imparts strong inter-protofilament stability in a manner different from other microtubule stabilizers that covalently bind tubulin, consistent with the distinct effects of the taccalonolides as compared to other stabilizers. AF was found to be a potent and effective antitumor agent that caused tumor regression in the MDA-MB-231 breast cancer xenograft model. The antitumor efficacy of some taccalonolides, which stabilize microtubules in a manner different from other microtubule stabilizers, provides the impetus to explore the therapeutic potential of this site.
Keywords: taccalonolide, microtubule, paclitaxel, microtubule stabilizer, tubulin
Introduction
Microtubule stabilizers, including paclitaxel (PTX), docetaxel (DTX) and ixabepilone, are highly effective in the treatment of many solid tumors, however, innate and acquired drug resistance limits their efficacy [1-3]. The search for new microtubule stabilizers that can overcome these limitations has been intense. The taccalonolides are a novel class of microtubule stabilizers isolated from tropical plants of the genus Tacca. Many of the cellular effects of the taccalonolides are similar to all other microtubule stabilizers in that they increase the density of cellular microtubules and interrupt mitotic progression leading to apoptosis [4]. The most abundant naturally occurring taccalonolide, A (Fig. 1A), is substantially less potent than other microtubule stabilizers in vitro with an IC50 value for inhibition of proliferation of 5 μM [5], however it exhibits antitumor effects in animal models [6, 7] and is able to overcome multiple mechanisms of drug resistance including mutations in the taxane binding site [4], the expression of MRP7 [7], βIII-tubulin [7], and P-glycoprotein (Pgp) both in vitro and in vivo [4, 7]. Other cellular effects of taccalonolide A that differentiate it from PTX include its high cellular persistence and the ability to initiate microtubule bundling at the IC50 for inhibition of proliferation, while PTX requires concentrations 20-times higher than its IC50 [8]. Recently, potent new taccalonolides, including taccalonolides AF and AJ, (AF, AJ) that have IC50 values for inhibition of proliferation of 23 and 4 nM respectively in HeLa cells, were identified [5]. The potency of these taccalonolides is comparable to PTX and laulimalide (LAU), which each have IC50 values of 1-3nM in HeLa cells. AF and AJ cause microtubule bundling in cells and robustly stimulate the polymerization of purified tubulin, indicating for the first time a direct interaction of taccalonolides with tubulin [5].
Figure 1.
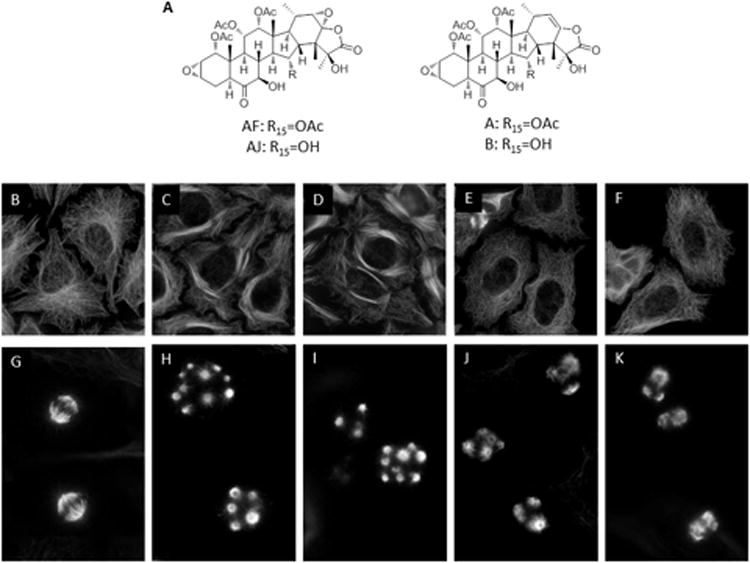
Effects of diverse microtubule stabilizers on microtubule structures.(A) Chemical structure of AF and AJ, which differ from one another at the C15 position. The structures of taccalonolides A and B are also included for reference. HeLa cells were treated with vehicle, (EtOH), (B, G), 100 nM AF (C, H), 30 nM AJ (D, I), 15 nM LAU (E, J) or 12.5 nM PTX (F, K) and microtubule structures in interphase (B-F) and mitotic (G-K) cells were visualized by indirect immunofluorescence.
Two major, non-overlapping microtubule stabilizer binding sites on microtubules have been identified; the taxane site in the interior lumen of the microtubule and the peloruside A/laulimalide site on the exterior of the microtubule [9, 10]. A combination of methods mapped PTX binding to a pocket on β-tubulin in the microtubule lumen with contacts at R282, H227 and V23 [10-13]. Detailed binding and synergism studies demonstrated that overlapping, non-identical drug binding sites exist within the taxane binding pocket, which allows chemically diverse agents including PTX, DTX, the epothilones, discodermolide and the dictyostatins to elicit similar effects on microtubule stability [14-18]. These drugs enhance microtubule stability by strengthening lateral protofilament interactions, often by inducing the otherwise unstructured M-loop on β-tubulin into an ordered helical structure [19-21]. Increased microtubule stability can also result from stabilization of lateral protofilament interactions on α-tubulin as seen for discodermolide, which does not affect the M-loop [22]. The multiple orientations and interactions of diverse compounds within the taxane pocket have recently been expanded to include the covalent binding of zampanolide/dactylolide to β-tubulin residues N226 and H227, which also causes M-loop stabilization [21, 23]. In addition to the taxane site, the second major stabilizer binding site of peloruside A and laulimalide was mapped to the exterior of the microtubule in a site containing residues F294, R306, N337 and Y340 on β-tubulin [9]. Although the taxane and laulimalide sites are non-overlapping, binding to either site initiates nearly identical effects on microtubule polymerization and stability [9, 24].
More recent evidence also demonstrates that some taxane site binding agents can also bind weakly to a low affinity site near the microtubule pore they may use to gain access to the interior of the microtubule [25]. The microbial metabolite cyclostreptin, which covalently binds to T218 of β-tubulin deep within this pore, was instrumental in the characterization of this site, which is referred to as the “gatekeeper” site [26]. Cyclostreptin, which causes microtubule stabilization in both biochemical and cellular assays, can also bind covalently to N226 within the taxane pocket if it traverses the pore to reach the microtubule lumen [27-29].
In this manuscript, the biochemical, cellular and in vivo antitumor activities of AJ and AF were evaluated. We determined that they bind covalently to microtubules in a similar manner to cyclostreptin. Striking inter-protofilament stability that did not involve M-loop stabilization was initiated by the covalent binding of AJ to microtubules. The ability to covalently bind to and stabilize microtubules sheds light on many of the distinct properties of the taccalonolides, including their cellular persistence, ability to overcome multiple drug resistance mechanisms and their potent in vivo effects.
Materials and Methods
Cell culture
HeLa cells [American Type Culture Collection (ATCC) ] were maintained in Basal Medium Eagle and MDA-MB-231 cells (ATCC) were maintained in Improved Modified Eagle Medium, both supplemented with 10% fetal bovine serum. Cells were passaged for fewer than 6 months after resuscitation from liquid nitrogen.
Immunofluorescence
HeLa cells were treated for 18 h with vehicle (EtOH), PTX, LAU, AF or AJ at the minimal concentrations that caused maximum G2/M arrest. The cells were fixed and microtubules visualized by indirect immunofluorescence techniques with a β-tubulin antibody as previously described [4].
Tubulin Assays
Tubulin polymerization was evaluated turbidimetrically at 37°C in a Spectramax plate reader with purified porcine brain tubulin (Cytoskeleton Inc.). Tubulin (2 mg/ml) in GPEM buffer (80 mM Pipes pH 6.8, 1 mM MgCl2, 1 mM EGTA) containing 10% glycerol and 1mM GTP was incubated with vehicle (0.5% v/v) or drug in a final volume of 200 μl. Cold-induced tubulin depolymerization was evaluated by cooling the plates at 4° or -20°C in a commercial refrigerator or freezer respectively. No freezing of the reactions was observed. Effects on steady state tubulin polymerization levels were analyzed by detection of tubulin by SDS-PAGE of the soluble or pellet fractions after centrifugationat 25°C.
Electron Microscopy
Samples were collected from the tubulin assays described above. Aliquots of the samples were taken 4, 11, 30 or 60 min after warming to 37° C as well as after chilling and recovery and mixed with equal volumes of a 4% gluteraldehyde solution. Microtubules were then mounted on 200 mesh copper grids, washed with a 10% cytochrome C solution, negatively stained with 8% uranyl acetate and visualized on a JEOL100CX transmission electron microscope.
Cellular Growth Inhibition and Synergism
Antiproliferative effects were evaluated using the sulforhodamine B (SRB) assay as described previously [4]. The synergism of drug combinations was evaluated with CompuSyn software to generate isobolograms and combination indices as described by Chou and Talalay [30].
Drug Binding, Displacement and Stability
Purified porcine brain tubulin at a concentration of 20 μMin GPEM buffer containing 10% glycerol and 1 mM GTP was incubated with 10 μM drug(s) for 1 h at 37°C in a final volume of 100 μl. After incubation, the reactions were centrifuged at 20,000 × g for 30 min at 37°C to separate the bound and unbound drug. The pellet was resuspended in 6 M urea, 50 mM Tris (pH 8) and 0.03% beta-mercaptoethanol and heated to 80°C for 30 min. Drugs were extracted from the supernatant and pellet fractions with ethyl acetate and detected using an Agilent 6224 Accurate–Mass TOF LC/MS. The quantity of drug(s) in each sample was determined from a standard curve generated using identical experimental conditions. Each sample was evaluated in triplicate using MassHunter Workstation software. Drug stability over time in pH 7 PBS was determined by LC/MS.
Structural Mass Spectrometry
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) methods as described in detail previously [9] were used to explore the conformational effects of AJ on microtubules. Briefly, 60 μM bovine brain tubulin (Cytoskeleton) was assembled into stabilized microtubules using 1 mM GMPCPP as a non-hydrolyzable GTP analog and treated with either 125 μM AJ or DTX. Samples were then processed with a conventional HDX protocol and the resulting peptides were analyzed by LC/MS on a Qstar Pulsar i using a chilled LC apparatus as previously described [31]. Deuterium incorporation for each was quantified with Hydra [32], and significant drug-induced alterations of deuteration were determined from quadruplicate data sets for each state. The levels of altered deuteration were color coded per peptide on segments of microtubule structures (PDB 2XRP) [33]. Structures were rendered in all figures using Pymol (http://pymol.sourceforge.net) and all residue numbering is based on bovine sequences in UniProt (α-tubulin: P81948, β-tubulin: Q6B856). The remaining digests from the HDX analysis were depleted of deuterium at neutral pH, and re-analyzed by nanoLC-MS/MS on an OrbitrapVelos (Thermo Scientific). Peptides with significantly reduced intensity upon drug treatment triggered a reanalysis of the digest, as this suggested covalent binding of the drug, and new peaks were sequenced with both collisionally-induced dissociation and electron-transfer dissociation, to identify the host peptide and study the nature of the covalent interaction.
In Vivo Antitumor Trial
Female athymic nude mice were maintained in an AAALAC approved facility and provided food and water ad libitum. 3 × 106 MDA-MB-231 cells supplemented with Matrigel were bilaterally injected s.c. into each flank. Mice were randomized into treatment groups (n = 5 mice, 10 tumors) and drug treatments initiated when a median tumor volume of 60 mg was reached. The optimized formulation of each drug was compared to untreated tumored mice. AF and AJ were administered i.p. in 5% EtOH in PBS and PTX in 5% Cremophor in PBS with a total volume of 0.2ml per injection. Tumor mass was calculated using the formula: mass (mg) = [length (mm) × width (mm)2]/2. One way ANOVA with a Dunnett's post test was applied to determine statistical significance between control and taccalonolide treatment groups.
Results
Effects of diverse microtubule stabilizers on interphase and mitotic microtubules
The effects of the new potent taccalonolides, AF and AJ (Fig. 1A), on interphase and mitotic microtubules were evaluated and compared with the effects initiated by PTX and LAU. For direct comparison, the minimum concentration of each drug that caused maximal G2/M arrest was used. AF and AJ, but not PTX or LAU, caused bundling of interphase microtubules at their respective G2/M arrest concentrations (Fig. 1B - F). Additionally, the mitotic spindle asters formed by AF or AJ were more numerous and compact as compared to those induced by PTX or LAU (Fig. 1G – K). These effects are similar to those previously observed with less potent taccalonolides [8].
Effects of diverse microtubule stabilizers on tubulin polymerization
To further evaluate differences between the taccalonolides and other microtubule stabilizers, the effects of the most potent taccalonolide, AJ, on the kinetics and extent of tubulin polymerization were compared with PTX and LAU over a range of concentrations (Fig. 2). In the vehicle controls, a 9 min lag period was observed prior to initiation of tubulin polymerization, after which the rate and extent of polymerization were monitored and normalized to 1. At the lowest concentrations that affected tubulin polymerization, PTX (0.1 μM) or LAU (0.25 μM) caused only modest increases in the total microtubule polymer over vehicle-treated controls yet they decreased the lag period from 9 to 4 min (Fig.2A, B, D). With higher concentrations of PTX or LAU, a dose-dependent decrease in lag period was measured and polymerization occurred immediately after the addition of 5 μM of either drug with no measurable lag period. In contrast, a lag period of at least 5 min was observed with every concentration of AJ evaluated, including those where the rate and extent of total tubulin polymerization were greater than that observed with any other treatment (Fig. 2C, D). The persistence of a lag period for polymerization was observed over the entire range of AJ concentrations and is a property of all taccalonolides with IC50 values for inhibition of cellular proliferation in the low nM range, including AF [5]. This finding suggests that the taccalonolides are not as efficient at initiating microtubule polymerization as compared to PTX or LAU.
Figure 2.
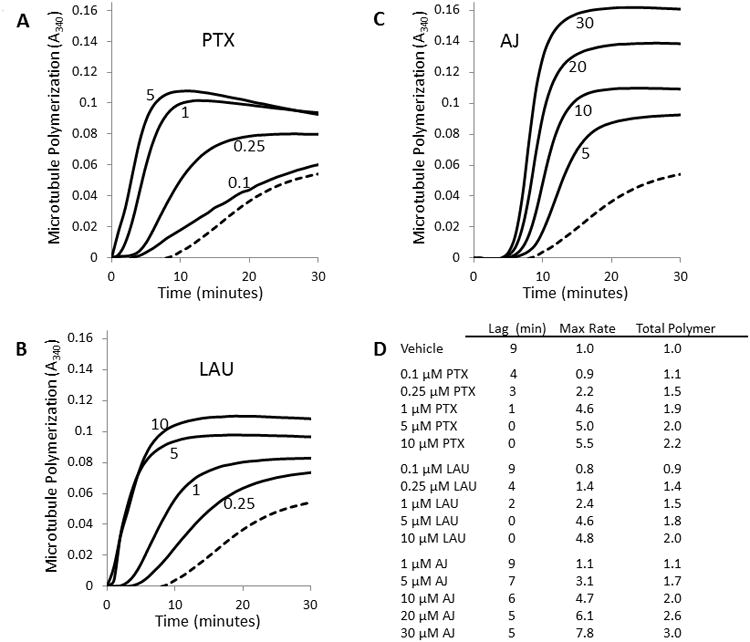
Tubulin polymerization. The kinetics of the polymerization of 2 mg/ml purified tubulin with microtubule stabilizers was monitored turbidimetrically. (A) PTX at 0.1, 0.25, 1 and 5 μM. (B) LAU at 0.25, 1, 5 and 10 μM. (C) AJ at 5, 10, 20 and 30 μM. The lowest, dashed line in each graph indicates microtubule polymerization with vehicle alone. (D) Quantitation of tubulin polymerization parameters. Lag indicates the time required to achieve exponential microtubule polymerization. The maximum relative rate and total polymerization have been normalized to vehicle-treated controls.
The effects of each drug on the maximal rate of polymerization and the total tubulin polymerformed as determined from turbidity measurements were quantified. Dose-dependent increases in both the rate and extent of tubulin polymerization were observed up to 1 μM PTX or 5 μM LAU where the total polymer and maximum rate did not increase significantly with higher concentrations (Fig. 2A, B). For both PTX and LAU, the maximum rate of tubulin polymerization was approximately 5 times greater than vehicle-treated controls with a plateau of 2-fold greater total polymer (Fig. 2D). Even though they bind to distinct sites on tubulin, the almost identical effects of PTX and LAU on microtubule polymerization are consistent with previous findings [24].
AJ also caused a dose-dependent increase in the rate and extent of tubulin polymerization, with 10 μM AJ resulting in a 4.7-fold increase in the rate of polymerization over vehicle and a doubling in total polymerformed (Fig. 2C, D). Although these values are almost identical to the effects of 10 μM PTX or LAU, higher concentrations of 20 or 30 μM AJ, caused further increases (30-66%), in both the rate of polymerization and total polymer formed as measured turbidimetrically.
Since the tubulin polymerization in Fig. 2 was inferred by turbidity measurements, it was important to directly visualize the structures formed to determine whether they were microtubules. The tubulin structures in the presence of 10 μM PTX, LAU or AJ, which showed similar effects on the rate and total extent of tubulin polymerization (Fig. 2), were evaluated 4, 11, 30 and 60 min after polymerization was initiated at 37°C and compared to vehicle-treated controls. Electron micrographs show that PTX and LAU caused a substantial increase in microtubule polymer within 4 min at 37° C, while the low number of microtubules formed in the presence of AJ at this time point was not greater than those observed for vehicle controls (Fig. 3). This is consistent with the lag period observed during AJ-initiated microtubule polymerization (Fig. 2C). The amount of microtubule polymer increased over time in all conditions in a manner that was consistent with turbidity measurements (Fig. 3). No major differences between the microtubule structures induced by the three drugs were observed, even at higher magnifications (Supplementary Fig. S1). Microtubule structures were also observed at higher concentrations of AJ, which were required to observe maximum polymerization. Interestingly, the microtubules formed in the presence 30 μM AJ did appear somewhat morerigid (Supplementary Fig. S2), but how this contributes to the increased turbidity observed in the presence of this super-stoichiometric concentration of AJ is not yet known (Fig. 2). Together, these data show that although AJ is not efficient at initiating microtubule polymerization, it can enhance the rate and extent of tubulin polymerization in a manner that is markedly different from either PTX or LAU.
Figure 3.

Electron microscopy of microtubules. Electron micrograph images of microtubules formed in the presence of 2 mg/ml tubulin with 10 μM PTX, LAU, AJ or vehicle 4, 11, 30 or 60 min after tubulin reactions were warmed to 37°C, after chilling the reactions at -20°C for 30 min or after repolymerization at 37°C for 30 min after chilling. All images were acquired at a magnification of 2,000×.
Differential effects of microtubule stabilizers on microtubule stability
Microtubules are depolymerized by cold temperatures, as indicated by the decrease in turbidity following incubation of vehicle-treated microtubules at -20°C for 30 min (Fig. 4A, dashed line). Although PTX and LAU-initiated microtubules are somewhat cold stable, this stringent drop in temperature caused almost complete depolymerization of microtubules formed in the presence of either 10 μM PTX or LAU (Fig. 4B, C). However, once the vehicle, PTX or LAU-treated tubulin reactions were rewarmed to 37° C, microtubule re-polymerization occurred rapidly with kinetics similar to the initial polymerization. In contrast, microtubules formed in the presence of 10 μM AJ were insensitive to cold-induced depolymerization and once the reaction was rewarmed, a further increase in turbidity was observed (Fig. 4D). The differential cold stability of microtubules in the presence of PTX, LAU, AJ or vehicle was confirmed by electron microscopy at low (Fig. 3) and high (Supplementary Fig. S3) magnification. These findings demonstrate adramatic cold stability of AJ-induced microtubules and a propensity for increased microtubule polymerization upon rewarming. Electron micrographs show that after rewarming, the microtubule structures induced by AJ appear longer and that the microtubule density is greater than that observed after the original polymerization (Fig 3. bottom row, Supplementary Fig. S4). Similar effects were observed for taccalonolide AF-induced microtubules (data not shown), suggesting resistance to cold-induced depolymerization is a general property of the taccalonolides.
Figure 4.
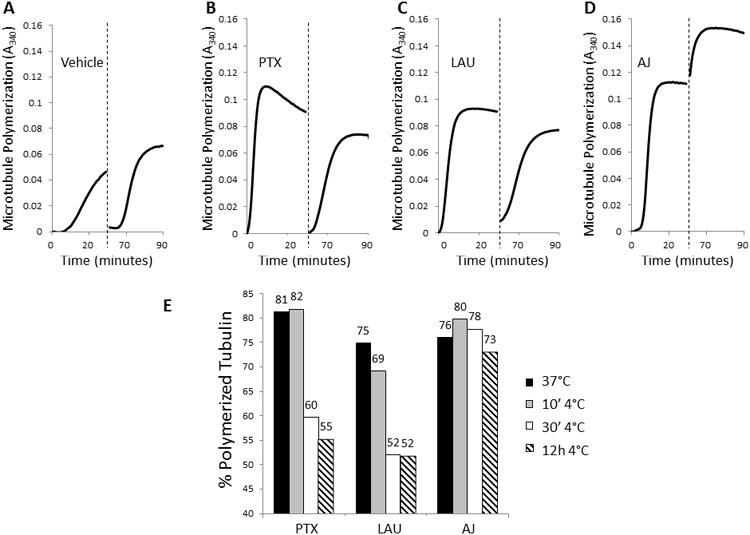
Cold stability of microtubule polymers in the presence of microtubule stabilizers. Polymerization of 2 mg/ml purified tubulin in the presence of vehicle (A), 10 μMPTX (B), 10 μM LAU (C) or 10 μM AJ (D) was monitored turbidimetrically for 30 min at 37°C. Samples were then cooled at -20°C for 30 min to depolymerize cold sensitive microtubules (dashed line) and then the plate was returned to 37°C and turbidity monitored for an additional 30 min. (E) The percentage of tubulin in the polymerized form in the presence of PTX, LAU or AJ after initial polymerization (black bars) or the indicated times after transfer to 4°C was determined by centrifugation.
To further explore the cold sensitivity of drug-treated microtubules, less stringent conditions were employed that caused only partial depolymerization of PTX or LAU-induced microtubules. Additionally, the extent of microtubule polymerization was monitored by separation of soluble tubulin from the microtubule pellet by centrifugation to more directly determine the distribution of polymerized and soluble tubulin. Consistent with the turbidimetric assays described above, at 10 μM each drug initiated a similar degree (75-81%) of tubulin polymerization after 30 min at 37°C (Fig. 4E, black bars). After an additional 10 min incubation at 4°C, little microtubule depolymerization was observed in the presence of any microtubule stabilizer (Fig. 4E, grey bars), but, as expected, vehicle-treated microtubules were completely depolymerized (data not shown). After a longer, 30 min incubation at 4°C, there was a 21-23% loss of PTX and LAU-induced polymer but no decrease in AJ-induced polymer (Fig. 4E, white bars). Extended cold exposure did not cause substantial further loss of polymer for any condition (Fig. 4E, hatched bars). While these results confirm reports that PTX and LAU impart some cold stability to microtubules, they further demonstrate that the cold stability initiated by AJ is more robust.
Since AJ-induced microtubules were markedly cold stable, their ability to resist mechanical disruption was also evaluated. Under conditions of rigorous pipetting where vehicle, PTX and LAU-induced microtubules were sheared into numerous short microtubule polymers as observed by electron microscopy, AJ-induced microtubules were not affected (data not shown). These studies demonstrate that the taccalonolides impart robust stability to microtubules which differentiate them from PTX and LAU-stabilized microtubules.
Synergism and displacement studies
Synergism studies have been useful in identifying compounds with non-overlapping binding sites [34-36]. The ability of AF to cause synergistic antiproliferative effects in combination with PTX or LAU was evaluated. Low antiproliferative concentrations of AF in combination with PTX or LAU caused synergistic effects as determined by isobologram analysis and calculation of combination indices (CI) [30]. Combination indices of 0.65 to 0.84 were found with AF and LAU, indicating the two drugs act synergistically (Fig. 5A). A lesser degree of synergism was detected with the combination of AF and PTX, with CI values ranging from 0.84 to 0.95 (Fig. 5B). These finding suggested the possibility that the taccalonolides might bind to a site that is pharmacologically distinct from the two major stabilizer binding sites on tubulin and biochemical studies were initiated to test this possibility
Figure 5.
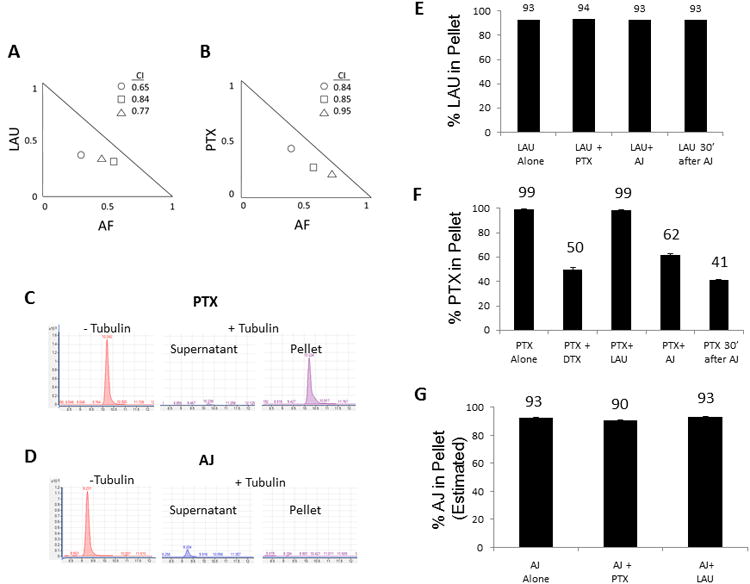
Synergism and displacement studies of the taccalonolides and other stabilizers. Isobolograms depicting synergy of (A) AF with LAU (○: 0.5 nM LAU + 10 nM AF;□: 0.5 nM LAU + 20 nM AF;Δ: 0.25 LAU + 10 nM AF) or (B) AF with PTX (○: 0.5 nM PTX + 10 nM AF; □: 0.5 nM PTX + 20 nM AF; Δ: 0.5 PTX + 30 nM AF). The combination indexes calculated for each point on the isobologram are listed. LC/MS traces of PTX (C) or AJ (D) in the absence of microtubules or in the supernatant or microtubule pellet after incubation with purified tubulin for 30 min. Quantitation of the percentage of LAU (E), PTX (F) or AJ (G) present in the microtubule pellet after incubation alone or in combination with other stabilizers. The percentage of AJ in the pellet was estimated based on the amount of drug that was depleted from the supernatant fraction.
Displacement studies were conducted to determine if the taccalonolides could compete with PTX or LAU binding to microtubules. High resolution mass spectrometry was used to detect drugs in the microtubule-containing pellet or residual supernatant after incubation with purified tubulin and compared to the total amount of drug detected in the absence of tubulin. As expected, when 10 μM PTX was incubated with 10 μM α/β-tubulin, essentially no drug was detected in the supernatant and 99% was extracted from the microtubule pellet (Fig. 5C, F). In contrast, AJ was undetectable in the microtubule pellet under the same conditions, even though only 7% of the drug remained in the supernatant (Fig. 5D). We hypothesized that the inability to detect significant levels of AJ in either fraction was a result of a tight interaction between AJ and microtubules that was not amenable to extraction with organic solvents. More stringent extractions of the microtubule pellet with a variety of detergents, proteases and heat were unable to liberate AJ from the microtubule pellet, indicating the possibility of a covalent interaction of AJ with microtubules. Due to its inability to be extracted from the microtubule pellet, the percentage of bound AJ to microtubules was estimated based on depletion of AJ from the supernatant using a standard curve (Fig 5G).
To measure drug displacement, equimolar combinations of LAU, PTX and AJ were incubated with tubulin and the percentage of each drug present in the microtubule pellet was calculated. When LAU was incubated with tubulin alone, 93% was bound to microtubules (Fig. 5E). As expected, the percentage of LAU bound to the pellet was not decreased with PTX co-incubation since they bind tonon-overlapping sites. AJ was unable to displace LAU whether it was added simultaneously or prior to LAU (Fig. 5E) and conversely LAU was unable to displace AJ (Fig. 5G), indicating that these two drugs do not compete for binding to microtubules.
Similarly, PTX was unable to be displaced from microtubules by LAU, but DTX was able to inhibit PTX binding by 50%, consistent with their ability to competitively bind to the taxane site (Fig. 5F). When AJ was added simultaneously with PTX in equimolar concentrations, the percentage of PTX associated with the microtubule pellet decreased by 38%, indicating that AJ inhibited PTX binding, although to a lesser extent than DTX (Fig. 5F). Given the inhibition of PTX binding by AJ, it was surprising to find that the converse was not true; the distribution of AJ was not affected by PTX (Fig. 5G).
Based on the assumption that the binding of AJ to microtubules was irreversible and that this property may contribute to its inability to be displaced by PTX, the effect of the order of drug addition on binding was tested. When AJ was added to tubulin 30 min before PTX, the percentage of PTX associated with the microtubule pellet decreased by 59%(Fig. 5F). Together, these results demonstrate that AJ inhibits PTX binding, but not LAU binding and that neither PTX nor LAU affected AJ binding. Although these data are consistent with some competition for microtubule binding between PTX and AJ, the fact that the timing of AJ addition impacts the extent to which it is able to displace PTX combined with the inability of PTX to displace AJ suggests the two drugs interact with tubulin in different ways.
Characterization of taccalonolide binding by mass spectrometry
Mass spectrometry was employed to aid in characterizing the interaction between the taccalonolides with tubulin/microtubules and determine their impact on microtubule stability. Peptic fragments of tubulin were generated in the presence of AJ or DTX and detected by LC-MS using ion extraction chromatography to identify peptides by their predicted mass. Strikingly, the peptide containing residues 212-230 of β-tubulin was observed in the presence of DTX (Fig. 6A, blue trace) but not with AJ (Fig. 6A, red trace). Since our binding experiments suggested AJ may covalently interact with tubulin, the generation of peptide(s) conjugated to AJ was explored. Indeed, a peptide with the predicted mass of β212-230 linked to AJ was detected in the presence of AJ (Fig. 6B, red trace) but not DTX (Fig. 6B, blue trace), confirming that AJ covalently binds to this peptide on β-tubulin. In addition to β212-230, AJ was also found to bind directly to another, slightly shorter peptic fragment, β213-230 (Supplementary Fig. S5). The peptides to which AJ binds contain both the T218 and N226 cyclostreptin binding residues and are distinct but partially overlapping with the taxane site (Fig. 6C, D), consistent with our displacement data (Fig. 5F). Therefore, AJ may bind to one or both of the residues that interact with cyclostreptin or another nearby residue. Unfortunately, the labeled residue(s) could not be localized in this study because tandem mass spectrometry using collisionally-induced dissociation only induced the removal of AJ as a neutral loss fragment, whereas electron-transfer dissociation did not generate an informative peptide sequence, only charge reduction. More complex follow-up studies to map the residue(s) of β-tubulin that directly bind to AJ and model drug binding are ongoing.
Figure 6.
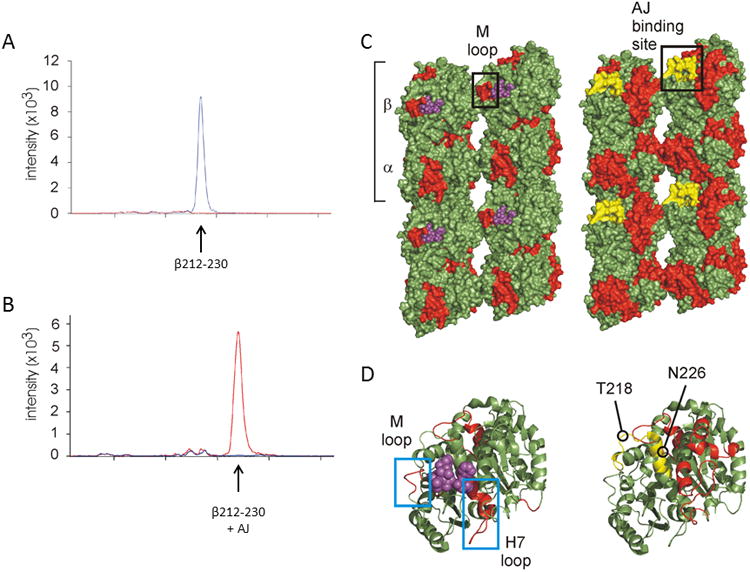
Mapping the interaction of AJ with microtubules by structural mass spectrometric analysis.(A) Chromatogram of the β212-230 peptide [m/z 544.7995 (+4) ] showing the peak intensity for DTX (blue) or AJ (red) treated microtubules. (B) Chromatogram of the AJ-bound form of the β212-230 peptide [m/z 713.87 (4+) ] showing the peak intensity for DTX (blue) or AJ (red) treated microtubules. (C) Lumen-to-exterior view of two parallel protofilaments in surface rendering and (D) a zoomed-in view of β-tubulin in cartoon (same orientation as C). Structures on left represent DTX-treated GMPCPP-microtubules and structures on right represent taccalonolide-treated GMPCPP-microtubules. Red highlights depict reductions in deuterium labeling induced by drug binding. DTX is shown as purple spheres, and the peptide covalently-bound to AJ (β213-230) is shown in yellow. An estimation of the binding region for AJ is marked with a black box in (C). The M-loop involved in taxane-binding and interprotofilament stabilization is highlighted (black box in C, left blue box in D). Notice the M-loop is unaffected in AJ-treated microtubules as indicated by green coloring. The H7 helix and loop, involved in taxane-binding and intraprotofilament stabilization, is also highlighted (right blue box in D) and unaffected by AJ. The residues to which cyclostreptin covalently binds, T218 and N226, are marked for reference.
In the absence of the ability to precisely map AJ binding to tubulin, we employed hydrogen-deuterium exchange mass spectrometry to further probe the allosteric effects elicited by this covalent interaction as has been done for many other microtubule binding drugs [9, 19, 20, 22]. A decreased rate of hydrogen exchange demonstrates a lower accessibility of residues to solvent, which is used to identify less flexible and more stable regions of a protein. This method was used to demonstrate the effect of AJ or DTX on the stability of microtubule peptides as compared to GMPCPP-stabilized microtubules, which allows for differentiation of drug specific effects from those associated with general microtubule stabilization. Decreases in accessibility to hydrogen deuterium exchange on tubulin residues in the presence of AJ are depicted in red on a β-tubulin monomer (Fig. 6D, right side) and in the context of a larger microtubule fragment to show wider allosteric effects (Fig. 6C, right side). Since we could not determine whether the covalent binding of AJ to β213-230 directly or indirectly altered the exchange properties of this peptide, it was excluded from our analysis. The changes observed with AJ binding were compared to those elicited by the binding of DTX to microtubules (Fig. 6C, D, left side). Although some of the regions on tubulin that showed decreased exchange upon drug binding are similar for the two drugs, there are notable differences. The binding of DTX results in stabilization and therefore decreased hydrogen deuterium exchange in the M-loop of β-tubulin as depicted in by its representation in red (Fig. 6C, D, left side), which is also the case for most other microtubule stabilizers [19-21]. In contrast, the M-loop is not stabilized upon AJ binding, as shown by the lack of red in this area (Fig. 6C, D, right side). The stabilization of the intradimer interface, involving helix H7 on β-tubulin, is also observed with DTX and other stabilizers [37] but not AJ (Fig. 6D). Rather, AJ induced a markedly higher stabilization of the lateral inter-protofilament contacts centering on α-tubulin in a manner that does not appear to involve the M-loop (Fig. 6C, right side).
In vivo antitumor activity of taccalonolides AF and AJ
The distinct binding characteristics of the taccalonolides led us to evaluate the in vivo antitumor effects of AF and AJ in the MDA-MB-231 breast cancer xenograft model. Initial dose tolerance tests indicated that doses of 2-2.5 mg/kg AF administered twice a week caused a dose dependent, but generally recoverable weight loss. For these antitumor trials, the positive control, PTX, was administered at 10 mg/kg on days 1, 3, 5 and 8 for a cumulative dose of 40 mg/kg (Fig. 7, filled squares). AF was found to have potent dose-dependent antitumor effects with inhibition of tumor growth almost identical to that of PTX when the lower, 2 mg/ml, dose was administered on days 1, 4 and 8 for a cumulative dose of 6 mg/kg (Fig. 7, open triangles). When a higher, 2.5 mg/kg dose, was administered on days 1 and 5 for a cumulative dose of 5 mg/kg, complete inhibition of tumor growth and even measureable reductions in tumor size were observed (Fig. 7, open circles). This is significant because tumor shrinkage is rarely observed in this model. However, it is important to note that this dosing of AF also caused significant weight loss and represented the LD20 with one mouse succumbing to toxicity on day 15, which was 10 days after the last dose was administered. These data indicate that AF has antitumor activity in vivo with the potential to reduce tumor mass, albeit with a narrow therapeutic window, consistent with the effects of less potent taccalonolides [6, 7].
Figure 7.
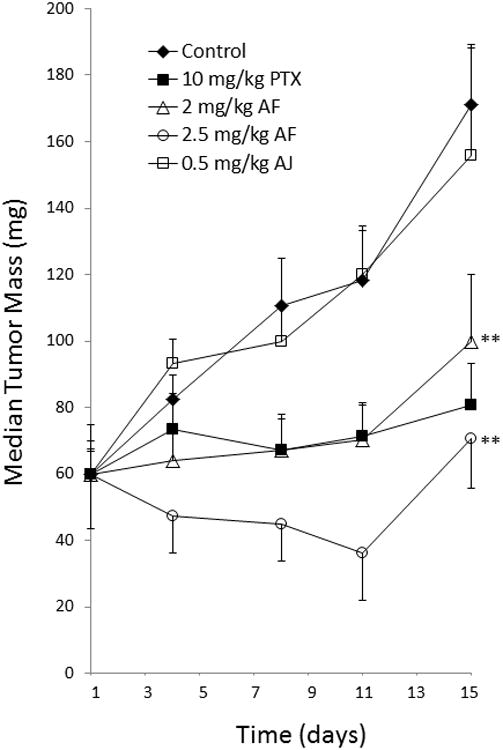
Antitumor effects of AF and AJ. Mice bilaterally implanted with MDA-MB-231 breast cancer cells were treated with 2.0 mg/kg of AF on days 1, 4 and 8, 2.5 mg/kg AF on days 1 and 5 or 0.5 mg/k AJ on days 1, 3, 5 and 8. PTX was administered at a dose of 10 mg/kg on days 1, 3, 5 and 8 as a positive control. Median tumor volumes with standard error of the mean (n = 10) are graphically represented. **p < 0.01.
Following initial dose tolerance testing, antitumor studies were also performed with 0.5 mg/kg AJ dosed on days 1, 3, 5 and 8 for a cumulative dose of 2 mg/kg. This intense schedule of AJ showed no indication of antitumor effects (Fig. 7, open squares) in spite of the fact that an average weight loss of greater than 10% was observed and two mice succumbed to toxicity on days 11 and 12. The fact that no measureable antitumor activities were observed at the LD40 demonstrates that AJ does not have a therapeutic window for antitumor activity. Follow-up studies to optimize the dose and schedule of AJ confirmed this as lower dosing regimens that minimized toxicity also produced no antitumor effects. Modest antitumor effects were observed with a dosing regimen of 0.85 mg/kg on days 1, 4 and 8 albeit with unacceptable toxicity that led to the LD80. Although AJ was highly potent, causing notable weight loss at doses 20-fold lower than paclitaxel, the lack of antitumor efficacy at doses lower than the LD80 demonstrated the absence of any therapeutic window.
The fact that a small but notable therapeutic window was identified for AF but not AJ prompted an investigation into differences in chemical stability that might relate to in vivo metabolism. Analysis of the chemical breakdown of both AF and AJ showed very different chemical stability in aqueous conditions; AJ showed marked stability in pH7 PBS of over 20 h while AF had a t1/2 of 9h. Analysis of the degradation products of AF indicated that AJ was the major breakdown product. Given the high toxicity observed for AJ in vivo and the fact that AF is rapidly broken down into AJ in aqueous solution at physiological pH, it is possible that the toxicity associated with AF administration in vivo could be due at least in part to its conversion into AJ. We hypothesize that the identification of taccalonolides that do not have this liability of hydrolysis at C15 might facilitate the discovery of a taccalonolide with a better therapeutic window.
Discussion
All microtubule stabilizing agents in clinical use bind within the classical taxane pocket on microtubules. Laulimalide site agents, which show synergism with taxane-binding drugs [24, 34, 36], are able to circumvent some clinically relevant forms of taxane resistance in vitro [38-40], but have so far failed to advance to clinical trials due in part to the lack of clear antitumor effects in murine models [38, 41]. We show that PTX and LAU have almost identical effects on microtubule polymerization and stability in spite of the fact that they bind to distinct sites on microtubules, which is consistent with previous reports [9, 24]. In contrast, AF and AJ have very different properties, including a slow rate of microtubule nucleation as evidenced by a persistent lag period for polymerization even at concentrations that eventually enhance the rate, extent and cold stability of microtubule polymerization as compared to other stabilizers. These results suggest that the taccalonolides do not stimulate microtubule nucleation; but that once they are formed they are remarkably stable. This finding is reminiscent of those obtained with cyclostreptin, which causes weak tubulin assembly reactions with a significant lag period but enhanced cold stability [27, 42]. The ability of cyclostreptin to bind to β-tubulin residue T218 deep within the pore of the microtubule has prompted references to cyclostreptin as a “gatekeeper” for agents that require transport through this pore to gain access to the taxane site on the interior of the microtubule [43]. Similar to zampanolide, cyclostreptin can also covalently modify the N226 residue in the taxane pocket on the inside of the microtubule if it is able to traverse the pore without becoming covalently attached [26]. In spite of the robust literature detailing the biochemical effects on tubulin [26-28, 42] the antitumor effects of cyclostreptin have not been well characterized [44].
The ability of AJ to bind covalently to residue(s) in the 213-230 peptide of β-tubulin is reminiscent of both cyclostreptin and zampanolide binding because this peptide encompasses both the T218 pore site and the N226 residue in the taxane pocket. Although the identification of the exact residue(s) of β-tubulin that are covalently modified by AJ was not determined in these studies due to technical limitations, the allosteric effects on microtubule stabilization afforded by AJ binding are striking (Fig. 6). It has been demonstrated that the covalent binding of zampanolide to microtubules increases the lateral interaction between protofilaments by promoting helical structuring of the M-loop on β-tubulin [21]. In contrast, the microtubule stabilization promoted by covalent binding of AJ appears to cause profound interprotofilament stability through a distinct mechanism that does not involve changes to the M-loop. Interestingly, it has also been proposed that the binding of cyclostreptin to T218 is not anticipated to affect M-loop stability or tubulin polymerization and that its ability to bind within the taxane pocket is what enhances microtubule stabilization [45]. The distinct interprotofilament interactions initiated by AJ indicate, for the first time, that a drug can covalently bind and stabilize microtubules independently of M-loop stabilization, demonstrating that it interacts with tubulin in a manner distinct from all other stabilizers that have been previously characterized. Preliminary data indicate that all taccalonolides that are sufficiently potent to enhance the polymerization of tubulin in biochemical extracts bind irreversibly to the resulting microtubule polymer, suggesting that covalent binding of the taccalonolides to microtubules is likely a general property of this class of microtubule stabilizers. Further biochemical analysis is warranted to fully characterize the interaction of the taccalonolides with tubulin.
The distinct allosteric effects imparted on microtubule stability by AJ shed insight into many unique properties of this compound class as compared to other microtubule stabilizers. For example, it may facilitate the ability of the taccalonolides to cause bundling of interphase microtubules at antiproliferative concentrations, which is important in light of recent evidence that suggests interphase effects play an important role in the antitumor properties of microtubule targeted agents [46]. The covalent binding of the taccalonolides to microtubules may also help to explain their potency in vivo. As observed for other taccalonolides [6, 7], AF and AJ were much more potent in vivo than would have been expected from their in vitro IC50s. For example, PTX has an in vitro IC50 10-fold lower than AF yet was administered at 5-fold higher concentrations than AF to observe similar antitumor effects. This represents an effective 50-fold increase in in vivo potency for AF as compared to PTX, which is likely at least partially attributable to the ability of taccalonolides to bind covalently to microtubules. This would not be immediately reflected in an in vitro IC50 where the cells are constantly bathed in the drug, but becomes evident in vivo where non-bound drug is actively cleared. The high cellular persistence of the taccalonolides in clonogenic assays after drug washout was the first indication that their cellular effects were irreversible [8].
The in vivo potency of AF and AJ, presumably initiated by their covalent binding, also has the advantage of allowing them to be administered in aqueous solvents without the need for Cremophor or polysorbate-80, which can cause hypersensitivity reactions. The covalent attachment of the taccalonolides to microtubules also likely explains their ability to circumvent drug resistance mediated by expression of the Pgp drug efflux pump, which limits the efficacy of many anticancer agents [4, 7]. Indeed, in addition to the taccalonolides, other microtubule stabilizers that covalently bind microtubules, including cyclostreptin and zampanolide, have shown the ability to circumvent Pgp-mediated drug resistance [23, 26, 28]. In spite of the potency of AF and AJ, it is possible that the lack of antitumor efficacy for AJ and the narrow therapeutic window of AF and other taccalonolides may also be due in part to irreversible binding. In addition to identifying new taccalonolides with a larger therapeutic window, an alternative strategy is to conjugate a taccalonolide to atumor-targeting antibody. This has been successful with the recent FDA approval of T-DM1 [47]. The prospects of employing this strategy to specifically load a drug that irreversibly binds to its target into cancer cells may be highly effective.
Supplementary Material
Acknowledgments
We thank Dr. Phil Crews for providing laulimalide (fijianolide B).
This work was supported by NCI CA121138 (SLM), DOD-CDMRP Postdoctoral Award BC087466 (ALR), COSTAR Program NIDCR DE 14318 (JL), NCI P30 CA054174 (SLM), the CTSA support grant (SLM), Remeditex Ventures (SLM), Alberta Ingenuity-Health Solutions 201000620 (DCS) and NSERC/CIHR CHRP 385880 (DCS). Support of the CTRC Mass Spectrometry Shared and Macromolecular Structure (NMR) Resources are gratefully acknowledged.
Footnotes
Conflict of Interest: Authors Risinger, Li, Peng and Mooberry are inventors on a pending patent application on new taccalonolides that is assigned to the University of Texas system.
References
- 1.Fojo T, Menefee M. Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol. 2007;18(5):v3–8. doi: 10.1093/annonc/mdm172. [DOI] [PubMed] [Google Scholar]
- 2.Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(3):194–204. doi: 10.1038/nrc2803. [DOI] [PubMed] [Google Scholar]
- 3.Rowinsky EK, Donehower RC. Paclitaxel (taxol) N Engl J Med. 1995;332(15):1004–14. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 4.Tinley TL, et al. Taccalonolides E and A: Plant-derived steroids with microtubule-stabilizing activity. Cancer Res. 2003;63(12):3211–20. [PubMed] [Google Scholar]
- 5.Li J, et al. Potent taccalonolides, AF and AJ, inform significant structure-activity relationships and tubulin as the binding site of these microtubule stabilizers. J Am Chem Soc. 2011;133(47):19064–7. doi: 10.1021/ja209045k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng J, et al. Identification and biological activities of new taccalonolide microtubule stabilizers. J Med Chem. 2011;54(17):6117–24. doi: 10.1021/jm200757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Risinger AL, et al. The taccalonolides: microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res. 2008;68(21):8881–8. doi: 10.1158/0008-5472.CAN-08-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Risinger AL, Mooberry SL. Cellular studies reveal mechanistic differences between taccalonolide A and paclitaxel. Cell Cycle. 2011;10(13):2162–71. doi: 10.4161/cc.10.13.16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett MJ, et al. Discovery and characterization of the laulimalide-microtubule binding mode by mass shift perturbation mapping. Chem Biol. 2010;17(7):725–34. doi: 10.1016/j.chembiol.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 10.Nogales E, et al. Structure of tubulin at 6.5 A and location of the taxol-binding site. Nature. 1995;375(6530):424–7. doi: 10.1038/375424a0. [DOI] [PubMed] [Google Scholar]
- 11.Rao S, et al. Characterization of the Taxol binding site on the microtubule. Identification of Arg(282) in beta-tubulin as the site of photoincorporation of a 7-benzophenone analogue of Taxol. J Biol Chem. 1999;274(53):37990–4. doi: 10.1074/jbc.274.53.37990. [DOI] [PubMed] [Google Scholar]
- 12.Rao S, et al. 3′-(p-azidobenzamido)taxol photolabels the N-terminal 31 amino acids of beta-tubulin. J Biol Chem. 1994;269(5):3132–4. [PubMed] [Google Scholar]
- 13.Rao S, et al. Characterization of the taxol binding site on the microtubule. 2-(m-Azidobenzoyl)taxol photolabels a peptide (amino acids 217-231) of beta-tubulin. J Biol Chem. 1995;270(35):20235–8. doi: 10.1074/jbc.270.35.20235. [DOI] [PubMed] [Google Scholar]
- 14.Honore S, et al. Synergistic suppression of microtubule dynamics by discodermolide and paclitaxel in non-small cell lung carcinoma cells. Cancer Res. 2004;64(14):4957–64. doi: 10.1158/0008-5472.CAN-04-0693. [DOI] [PubMed] [Google Scholar]
- 15.Martello LA, et al. Taxol and discodermolide represent a synergistic drug combination in human carcinoma cell lines. Clin Cancer Res. 2000;6(5):1978–87. [PubMed] [Google Scholar]
- 16.Kowalski RJ, et al. The microtubule-stabilizing agent discodermolide competitively inhibits the binding of paclitaxel (Taxol) to tubulin polymers, enhances tubulin nucleation reactions more potently than paclitaxel, and inhibits the growth of paclitaxel-resistant cells. Mol Pharmacol. 1997;52(4):613–22. [PubMed] [Google Scholar]
- 17.Madiraju C, et al. Tubulin assembly, taxoid site binding, and cellular effects of the microtubule-stabilizing agent dictyostatin. Biochemistry. 2005;44(45):15053–63. doi: 10.1021/bi050685l. [DOI] [PubMed] [Google Scholar]
- 18.Kowalski RJ, Giannakakou P, Hamel E. Activities of the microtubule-stabilizing agents epothilones A and B with purified tubulin and in cells resistant to paclitaxel (Taxol(R)) J Biol Chem. 1997;272(4):2534–41. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 19.Khrapunovich-Baine M, et al. Hallmarks of molecular action of microtubule stabilizing agents: effects of epothilone B, ixabepilone, peloruside A, and laulimalide on microtubule conformation. J Biol Chem. 2011;286(13):11765–78. doi: 10.1074/jbc.M110.162214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao H, et al. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci U S A. 2006;103(27):10166–73. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prota AE, et al. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science. 2013;339(6119):587–90. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 22.Khrapunovich-Baine M, et al. Distinct pose of discodermolide in taxol binding pocket drives a complementary mode of microtubule stabilization. Biochemistry. 2009;48(49):11664–77. doi: 10.1021/bi901351q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Field JJ, et al. Zampanolide, a potent new microtubule-stabilizing agent, covalently reacts with the taxane luminal site in tubulin alpha, beta-heterodimers and microtubules. Chem Biol. 2012;19(6):686–98. doi: 10.1016/j.chembiol.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gapud EJ, et al. Laulimalide and paclitaxel: a comparison of their effects on tubulin assembly and their synergistic action when present simultaneously. Mol Pharmacol. 2004;66(1):113–21. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]
- 25.Barasoain I, et al. Probing the pore drug binding site of microtubules with fluorescent taxanes: evidence of two binding poses. Chem Biol. 2010;17(3):243–53. doi: 10.1016/j.chembiol.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Buey RM, et al. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat Chem Biol. 2007;3(2):117–25. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- 27.Bai R, et al. Interaction of a cyclostreptin analogue with the microtubule taxoid site: the covalent reaction rapidly follows binding. J Nat Prod. 2008;71(3):370–4. doi: 10.1021/np800056m. [DOI] [PubMed] [Google Scholar]
- 28.Calvo E, et al. Cyclostreptin derivatives specifically target cellular tubulin and further map the paclitaxel site. Biochemistry. 2012;51(1):329–41. doi: 10.1021/bi201380p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato B, et al. A new antimitotic substance, FR182877. II. The mechanism of action. J Antibiot (Tokyo) 2000;53(2):204–6. doi: 10.7164/antibiotics.53.204. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC. Talalay, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 31.Bennett MJ, et al. Structural mass spectrometry of the alpha beta-tubulin dimer supports a revised model of microtubule assembly. Biochemistry. 2009;48(22):4858–70. doi: 10.1021/bi900200q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slysz GW, et al. Hydra: software for tailored processing of H/D exchange data from MS or tandem MS analyses. BMC Bioinformatics. 2009;10:162. doi: 10.1186/1471-2105-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fourniol FJ, et al. Template-free 13-protofilament microtubule-MAP assembly visualized at 8 A resolution. J Cell Biol. 2010;191(3):463–70. doi: 10.1083/jcb.201007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark EA, et al. Laulimalide and synthetic laulimalide analogues are synergistic with paclitaxel and 2-methoxyestradiol. Mol Pharm. 2006;3(4):457–67. doi: 10.1021/mp060016h. [DOI] [PubMed] [Google Scholar]
- 35.Hamel E, et al. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol. 2006;70(5):1555–64. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]
- 36.Wilmes A, et al. Peloruside A synergizes with other microtubule stabilizing agents in cultured cancer cell lines. Mol Pharm. 2007;4(2):269–80. doi: 10.1021/mp060101p. [DOI] [PubMed] [Google Scholar]
- 37.Huzil JT, et al. A unique mode of microtubule stabilization induced by peloruside A. J Mol Biol. 2008;378(5):1016–30. doi: 10.1016/j.jmb.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, et al. In vitro and in vivo anticancer activities of synthetic (-)-laulimalide, a marine natural product microtubule stabilizing agent. Anticancer Res. 2007;27(3B):1509–18. [PubMed] [Google Scholar]
- 39.Mooberry SL, et al. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999;59(3):653–60. [PubMed] [Google Scholar]
- 40.Pryor DE, et al. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry. 2002;41(29):9109–15. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 41.Johnson TA, et al. Sponge-derived fijianolide polyketide class: further evaluation of their structural and cytotoxicity properties. J Med Chem. 2007;50(16):3795–803. doi: 10.1021/jm070410z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edler MC, et al. Cyclostreptin (FR182877), an antitumor tubulin-polymerizing agent deficient in enhancing tubulin assembly despite its high affinity for the taxoid site. Biochemistry. 2005;44(34):11525–38. doi: 10.1021/bi050660m. [DOI] [PubMed] [Google Scholar]
- 43.Snyder JP. The microtubule-pore gatekeeper. Nat Chem Biol. 2007;3(2):81–2. doi: 10.1038/nchembio0207-81. [DOI] [PubMed] [Google Scholar]
- 44.Sato B, et al. A new antimitotic substance, FR182877. II. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 2000;53(2):123–30. doi: 10.7164/antibiotics.53.123. [DOI] [PubMed] [Google Scholar]
- 45.Prussia AJ, et al. Cyclostreptin and microtubules: is a low-affinity binding site required? Chembiochem. 2010;11(1):101–9. doi: 10.1002/cbic.200900538. [DOI] [PubMed] [Google Scholar]
- 46.Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18(1):51–63. doi: 10.1158/1078-0432.CCR-11-0999. [DOI] [PubMed] [Google Scholar]
- 47.Boyraz B, et al. Trastuzumab emtansine (T-DM1) for HER2-positive breast cancer. Curr Med Res Opin. 2013;29(4):405–14. doi: 10.1185/03007995.2013.775113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.