

Abstract
Non-small cell lung carcinoma (NSCLC) is among the deadliest of human cancers. The CDKN2A locus, which houses the INK4a and ARF tumor suppressor genes, is frequently altered in NSCLC. However, the specific role of ARF in pulmonary tumorigenesis remains unclear. KRAS and other oncogenes induce the expression of ARF, thus stabilizing p53 activity and arresting cell proliferation. To address the role of ARF in Kras-driven NSCLC, we compared the susceptibility of NIH/Ola strain wild-type and Arf knockout mice to urethane-induced lung carcinogenesis. Lung tumor size, malignancy, and associated morbidity were significantly increased in Arf−/− compared to Arf+/+ animals at 25 weeks post-induction. Pulmonary tumors from Arf knockout mice exhibited increased cell proliferation and DNA damage compared to wild-type. A subgroup of tumors in Arf−/− animals presented as dedifferentiated and metastatic, with many characteristics of pulmonary sarcomatoid carcinoma, a neoplasm previously undocumented in mouse models. Our finding of a role for ARF in NSCLC is consistent with the observation that benign adenomas from Arf+/+ mice robustly expressed ARF, while ARF expression was markedly reduced in malignant adenocarcinomas. ARF expression also frequently co-localized with expression of p21CIP1, a transcriptional target of p53, arguing that ARF induces the p53 checkpoint to arrest cell proliferation in vivo. Together, these findings demonstrate that induction of ARF is an early response in lung tumorigenesis that mounts a strong barrier against tumor growth and malignant progression.
Keywords: p19Arf, p14ARF, ethyl carbamate, metastasis
Introduction
Non-small cell lung carcinoma (NSCLC) is the leading cause of cancer-related death in the United States and the world, with a five-year relative survival rate of only 17%.1 Lung adenocarcinoma, the predominant histological subtype of NSCLC, commonly harbors genetic alterations in KRAS (38%), CDKN2A (15%), and TP53 (36%).2 However, the timing, impact, and clinical significance of these changes are not yet fully understood.
The CDKN2A locus houses both the ARF (p14ARF in humans, p19Arf in mice) and INK4a (p16INK4a) tumor suppressor genes. ARF, so named because it is transcribed from an alternate reading frame, shares no homology with INK4a and is controlled by a separate promoter.3 Both proteins induce cell cycle arrest, albeit by regulating different signaling pathways. Under conditions of cellular stress, INK4a promotes G1 arrest by inhibiting Cyclin D-CDK4/6 complexes and preventing hyperphosphorylation of RB.4 In contrast, ARF triggers arrest at the G1 and G2 checkpoints,3 in part by antagonizing the MDM2 ubiquitin ligase and stabilizing cellular p53 levels.4 Although p53-independent tumor suppression by ARF has been demonstrated,5 these activities are not as well elucidated as the canonical ARF-MDM2-p53 signaling axis.
Several recent studies examining tumor suppression by ARF in NSCLC have yielded different results in different contexts. While ARF expression was found to be elevated in pre-malignant lung lesions in KrasG12V animals,6 expression of ARF was primarily restricted to high-grade adenocarcinomas in two mouse models of KrasG12D-driven lung cancer.7, 8 In addition, Arf loss had only a modest impact on lung tumor growth or progression in KrasG12D knock-in animals.9 These latter results are noteworthy, given the established relationship between RAS, ARF, and p53 (ref. 5) and the observation that Trp53 loss of function markedly enhanced the growth and malignant progression of Kras-driven lung tumors.10, 11 Moreover, loss of ARF expression has been observed in lung tumors in mice12 and in humans,13–15 and restoration of ARF expression arrests the growth of patient-derived lung cancer cell lines.16, 17 As ARF status may have important implications for NSCLC patient prognosis and clinical management, further examination of the role of ARF in lung tumorigenesis is warranted. Herein we identify ARF as a major suppressor of the growth and malignant progression of carcinogen-induced, Kras-driven NSCLC.
Results
Arf deficiency leads to increased lung tumor size and associated morbidity
To examine the role of ARF in lung tumorigenesis, we injected cohorts of Arf+/+, Arf+/− and Arf−/− mice with the pulmonary carcinogen urethane. Kaplan-Meier analyses indicated that urethane-exposed Arf−/− animals experienced significantly shortened overall survival (Supplementary Fig. 1A) and increased lung-tumor associated morbidity (Fig. 1A) compared to their Arf+/+, Arf+/− and unexposed Arf−/− littermates (logrank test for trend, P < 0.0001). By 25 weeks post-injection, 68.2% of Arf−/− mice exposed to urethane succumbed to lung tumors, whereas all Arf+/+, Arf+/− and unexposed Arf−/− mice remained viable (Fig. 1A). Arf−/− mice also presented with lymphomas and sarcomas (data not shown) at a rate consistent with prior reports.18 As described previously, two-thirds of Arf−/− animals also developed hepatic vascular lesions after urethane exposure.19 These lesions were typically small and had a limited impact on overall survival.
Figure 1. Arf loss increases lung tumor growth, malignancy, and morbidity in urethane-exposed mice.
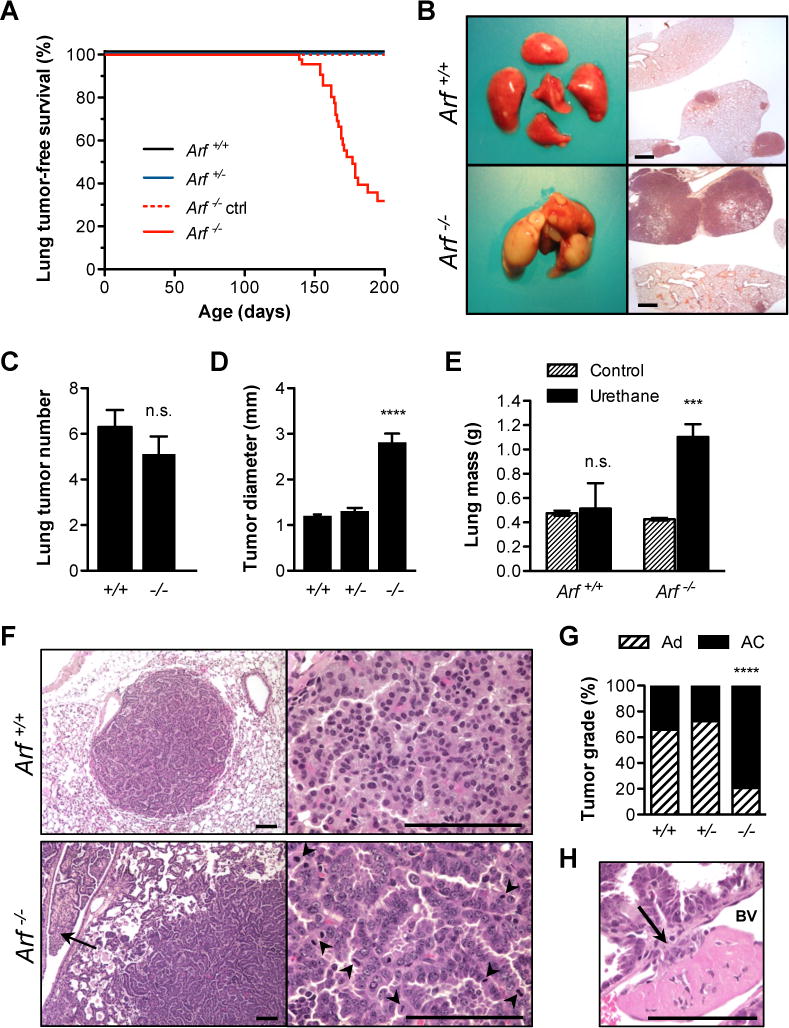
[A] Arf loss led to accelerated lung tumor-associated morbidity after urethane exposure (logrank test for trend, P < 0.0001). [B] At necropsy, Arf−/− mice (bottom) bore massive lung tumors compared to Arf+/+ mice (top). H&E images shown on right; scale bar 1 mm. [C] At 17 weeks post-exposure, an equivalent number of tumors were visible on the surface of Arf+/+ (6.300 ± 0.7461) and Arf−/− (5.111 ± 0.7718) lungs (P = 0.2840; n = 10 Arf+/+ and 9 Arf−/−). [D] Mean tumor diameter. Tumors were measured with calipers; largest diameter was used for analysis. Mean values differed significantly between genotypes (**** P < 0.0001; n = 351 Arf+/+ (1.205 ± 0.02899 mm), 196 Arf+/− (1.306 ± 0.07118 mm), and 211 Arf−/− tumors (2.815 ± 0.1883 mm)). [E] Mass of lungs from non-tumor bearing control and urethane-exposed male mice. Arf−/− mice (1.103 ± 0.106 g) carried a significantly greater lung tumor burden than Arf+/+ (0.514 ± 0.209 g) (*** P = 0.0007; n ≥ 4 animals each condition). [F] Adenomas occurred primarily in Arf+/+ mice (top panel). Tumors had discrete borders and were composed of a uniform cell population with small, round nuclei and moderate amounts of eosinophilic cytoplasm. Mitotic figures were rare. Adenocarcinomas predominated in Arf−/− mice (bottom panel). Invasion (arrow) and mitotic figures (arrowheads) were frequent. [G] Incidence of lung adenomas (Ad) and adenocarcinomas (AC) across three genotypes (**** P < 0.0001; n = 44 Arf+/+, 40 Arf+/− and 39 Arf−/− tumors examined). [H] Intravasation of Arf−/− adenocarcinoma. Tumor cells have broken through the basement membrane (arrow) to invade the neighboring blood vessel (BV). An endothelialized thrombus, in pink, has formed at the site. Scale bars 100 μm.
At necropsy, Arf−/− animals presented with substantial pulmonary tumor burden (Fig. 1B). Consistent with previous urethane carcinogenesis studies,20 all exposed animals developed at least one lung tumor nodule. However, although tumor multiplicity did not vary between genotypes (Fig. 1C), tumor size was greatly increased in Arf−/− compared to Arf+/+ mice (P <0.0001; Fig. 1D). Notably, while Arf+/+ and Arf+/− mice rarely developed tumors exceeding 3 mm in diameter, Arf−/− mice routinely developed tumors between 4 and 17 mm diameter (Supplementary Fig. 1B). This marked increase in tumor size corresponded with a nearly threefold increase in lung mass in Arf−/− mice (Fig. 1E). In summary, while homozygous loss of Arf did not affect lung tumor initiation after carcinogen exposure, it greatly increased the rate of lung tumor growth and associated morbidity and mortality.
Loss of Arf accelerates tumor invasion and metastasis
The urethane model of mouse lung carcinogenesis captures a tumor development spectrum that proceeds from hyperplasia to benign adenomas and, infrequently, to malignant adenocarcinomas.21 At 25 weeks post-urethane, the majority of tumors present in Arf+/+ (65.9%) and Arf+/− (72.5%) animals were identified as pulmonary adenomas (Fig. 1F, top, G). The incidence of adenocarcinomas was greatly increased (79.5%) in Arf−/− animals (P < 0.0001; Fig. 1G). These pulmonary adenocarcinomas were characterized by pleomorphic nuclei, abundant mitotic activity, and invasion into adjacent parenchyma and airways (Fig. 1F, bottom). Intravasation of adenocarcinoma cells was also observed (Fig. 1H).
A subset of adenocarcinomas in Arf−/− mice exhibited exceptionally poor differentiation (Fig. 2A, B) and metastasized to both intra- (Fig. 2D, bottom) and extra-thoracic sites (Fig. 2C, E). These tumors displayed histopathological characteristics typical of pulmonary sarcomatoid carcinoma,22 an aggressive tumor type that, to our knowledge, has not previously been observed in mice. Although the metastatic lesions most closely resembled undifferentiated spindle cell tumors, regions of the metastases that retained epithelial differentiation expressed pro-surfactant protein C (Fig. 2D), consistent with lung epithelial origin. Immunohistochemistry further illustrated that the mesenchymal marker vimentin was absent from the epithelioid compartments but stained strongly in the spindle regions of the metastatic lesions (Fig. 2E). Cytokeratin 8, a marker of epithelium, was expressed in both cell compartments. In summary, the sarcomatoid metastases exhibited a mixture of mesenchymal and epithelial markers. These unusual tumors were never observed in Arf+/+ or Arf+/− animals, but were found in 50% of Arf−/− mice examined (Table 1; n = 10, each genotype). Coincidental metastases to neighboring lymph nodes, the chest wall, or the peritoneal cavity were similarly identified in 40 to 50% of Arf-deficient animals.
Figure 2. Poorly differentiated, sarcomatoid lung tumors develop in Arf-deficient animals.
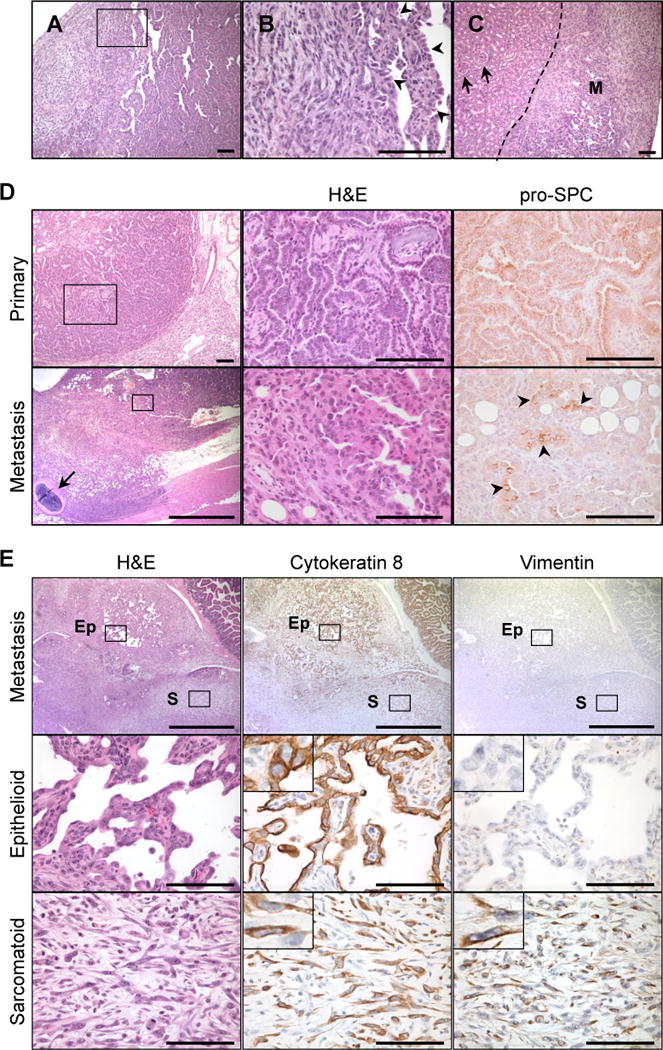
[A] Pulmonary sarcomatoid carcinoma-like lesions formed in the lung. [B] Magnification of boxed region in [A]. Arrowheads point to well-differentiated, epithelial regions. [C] Renal metastasis (M) of mixed epithelial and sarcomatoid components. Arrows indicate normal kidney glomeruli. [D] Lung adenocarcinomas (top) and sarcomatoid metastases (bottom) from the same mouse exhibited immunoreactivity for pro-surfactant protein C (pro-SPC), a marker of lung epithelial cells. Bottom panel displays invasion of the pleura and chest wall. Arrow indicates rib. The metastatic tumor maintained regions of epithelial differentiation (boxed region, magnified in middle panel) that expressed pro-SPC (arrowheads). [E] Metastasis of sarcomatoid carcinoma to peritoneal cavity. Whereas both the sarcomatoid (box S) and epithelioid (box Ep) compartments were positive for cytokeratin 8, vimentin expression was restricted to the sarcomatoid regions. Scale bars 100 μm, except panel [D, bottom left] (1 mm).
Table 1.
Spectrum of disease in urethane-exposed mice1
| Genotype | Primary PSC | Metastasis | ||
|---|---|---|---|---|
| LN | Pleura | PER | ||
| Arf+/+ | 0/10 (0) | 0/10 (0) | 0/10 (0) | 0/10 (0) |
| Arf+/− | 0/10 (0) | 0/10 (0) | 0/10 (0) | 0/10 (0) |
| Arf−/− | 5/10 (50) | 4/10 (40) | 5/10 (50) | 5/10 (50) |
Number of mice presenting with ≥ 1 indicated lesion (%). A subset of lung samples from urethane-exposed male mice of each genotype was selected at random for scoring. H&E stained sections were diagnosed blind to genotype. PSC, pulmonary sarcomatoid carcinoma; LN, lymph node; PER, peritoneal cavity.
ARF suppresses cell proliferation and protects against DNA damage
Based on the abundance of mitotic figures in Arf−/− tumors (Fig. 1F), we speculated that Arf-deficiency might accelerate tumor growth by conferring a proliferative advantage to lung cancer cells. To address this hypothesis, we examined the expression of the mitotic marker phosphorylated histone H3 in tumors from Arf+/+ and Arf−/− mice. Although no statistically significant difference in phospho-H3+ cells was observed in early stage tumors, adenocarcinomas from Arf-deficient animals exhibited elevated expression of phospho-H3 compared to Arf+/+ adenocarcinomas (Fig. 3A). A similar trend toward increased proliferation was identified with a second proliferation marker, Ki67 (Supplementary Fig. 2). The hyperproliferative effect of Arf deficiency was accompanied by a substantial increase in DNA damage. Marked elevation in the expression of phosphorylated histone H2A.X, a sensitive indicator of DNA damage, was observed in both Arf−/− adenomas and adenocarcinomas compared to wild-type (Fig. 3B). Notably, however, the apoptotic marker cleaved caspase 3 was largely undetectable at all stages of tumor development in both genotypes (Supplementary Fig. 3), suggesting that apoptosis plays little role in suppressing urethane-induced lung carcinogenesis.
Figure 3. RAS pathway signaling, cell proliferation, and DNA damage.
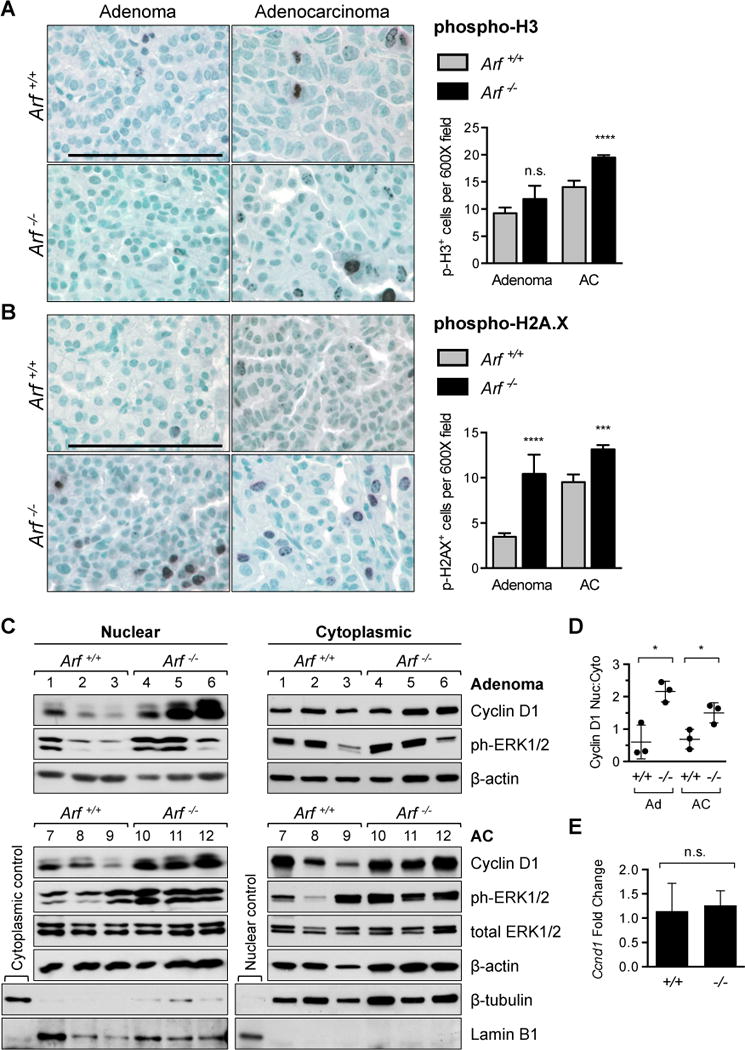
[A] Arf−/− adenocarcinomas (AC) displayed a higher proliferation index than tumors from Arf+/+ littermates, as shown by phosphorylated histone H3 staining (**** P < 0.0001; n = 119 adenoma and 220 AC fields counted from Arf+/+; n = 22 adenoma and 829 AC fields from Arf−/− mice). [B] Phosphorylated histone H2A.X staining illustrates a marked increase in DNA damage in tumors from Arf-deficient mice (**** P < 0.0001; *** P = 0.0001; n = 85 adenoma and 307 AC fields counted from Arf+/+; n = 31 adenoma and 788 AC fields from Arf−/− mice). In addition to H2A.X+ cells occurring more frequently per field, damage foci were also larger and more numerous per cell in Arf−/− mice compared to Arf+/+. Note that the paucity of adenomas in Arf−/− animals prevented inclusion of additional fields in the statistical analysis of phospho-H3 and phospho-H2A.X staining. [C] Nuclear cyclin D1 and phospho-ERK1/2 expression were strongly upregulated in adenomas (top) and adenocarcinomas (bottom) from individual Arf-deficient mice (lanes 4–6 and 10–12) compared to Arf+/+ mice (lanes 1–3 and 7–9). β-actin is provided as loading control. β-tubulin and lamin B1 are included as markers of cytoplasmic and nuclear compartments, respectively. [D] Relative intensities of nuclear versus cytoplasmic cyclin D1 bands from [C] compared between genotypes at each tumor stage. All samples were normalized to β-actin (* P = 0.0114 adenomas, * P = 0.0308 ACs). [E] Expression of Ccnd1 mRNA is equivalent in adenocarcinomas from Arf+/+ and Arf−/− animals. Gapdh was used as endogenous control. Data is plotted as fold change compared to mean Arf+/+ value (n = 6 tumors per genotype; P = 0.6600).
To elucidate the anti-proliferative properties of ARF, we examined mitogenic signaling in wildtype and Arf-deficient lung tumors. Consistent with prior reports on urethane carcinogenesis,23 activating mutations in the Kras oncogene were identified in all lung tumors examined (n = 4 animals, each genotype; Supplementary Fig. 4A). However, expression of phosphorylated ERK1/2, a downstream mediator of oncogenic KRAS, was further elevated in tumors from Arf−/− compared to Arf+/+ mice (Fig. 3C). The greatest increase in phospho-ERK1/2 expression occurred in the nuclear fraction of Arf−/− tumor cells (Supplementary Fig. 4B, C), a pattern linked to mitogenic stimulation and cell cycle entry.24 KRAS-ERK pathway signaling promotes expression of cyclin D1, a key component of the cell cycle machinery that is essential for transformation by oncogenic RAS.25, 26 Immunoblot analysis of cyclin D1 expression revealed upregulation of the cyclin D1 protein in adenomas and adenocarcinomas from Arf−/− compared to Arf+/+ mice (Fig. 3C). Similar to our observations of phospho-ERK1/2 localization, the gains in cyclin D1 expression were largely restricted to the nuclear compartment (Fig. 3D). Quantitative real-time polymerase chain reaction (QPCR) analysis found no significant difference in the expression of Ccnd1 transcript between adenocarcinomas harvested from Arf−/− and Arf+/+ mice (Fig. 3E), implying that the misregulation occurs post-transcriptionally.
Early induction of ARF by mutant KRAS activates a p53-dependent anti-proliferative response
Many of the tumor suppressive properties previously ascribed to ARF are linked to its role in stabilizing p53 levels in cells stressed by oncogenic RAS.5, 27 Having found that homozygous Arf loss facilitates urethane carcinogenesis, we hypothesized that ARF may act as an in vivo barrier to Kras-driven lung tumor growth and progression. We therefore examined ARF expression in lung tumors from wild-type mice. Early stage adenomas exhibited prominent ARF expression (Fig. 4A, top), while expression of ARF in adenocarcinomas was restricted to small, sporadic foci (Fig. 4A, bottom). Consequently, the number of ARF+ cells per field was significantly reduced in malignant (17.39 ± 1.59) compared to benign (35.07 ± 3.12) tumors (P < 0.0001; Fig. 4B). Normal lung parenchyma did not exhibit ARF expression (Supplementary Fig. 5A). ARF expression was thus triggered early and silenced late during Kras-driven pulmonary tumorigenesis.
Figure 4. The ARF-p53 signaling pathway.
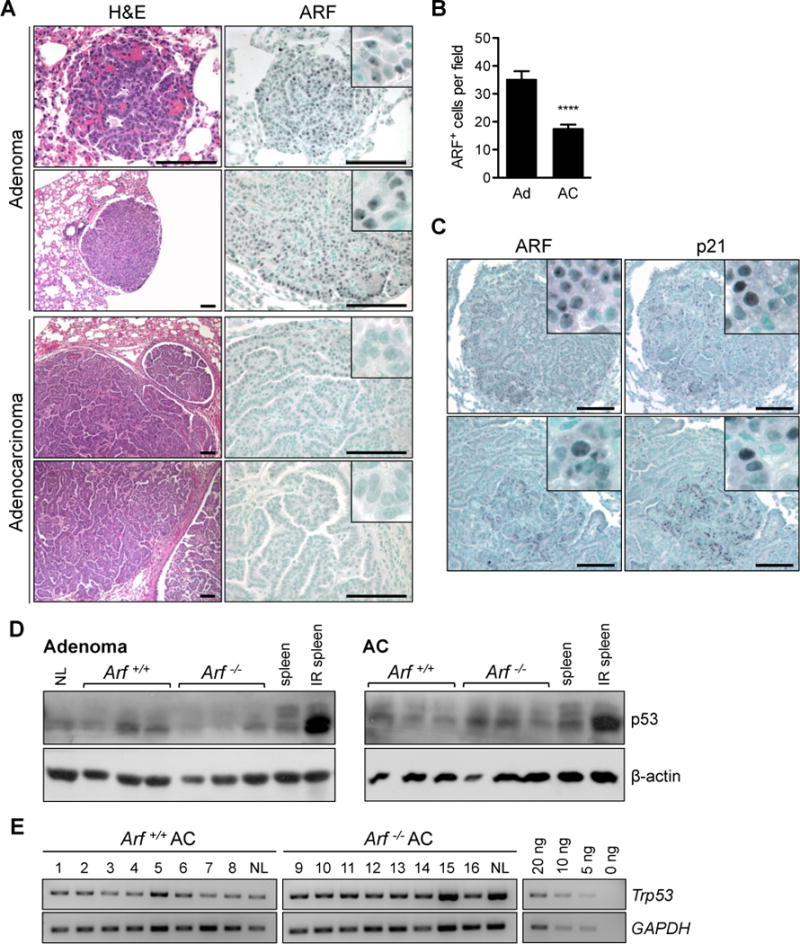
[A] Adenomas (Ad) from Arf+/+ mice robustly expressed ARF, but adenocarcinomas (AC) frequently lost ARF expression. Inset shows nucleolar staining of ARF in adenomas. [B] Quantification of ARF positive cells (**** P < 0.0001; n = 46 Ad and 145 AC fields examined). [C] ARF and p21 proteins colocalize by IHC in adenomas (top) and regions of low-grade adenocarcinomas (bottom) in wild-type mice. All scale bars 100 μm. [D] Western blot analysis of p53 reveals expression of p53 protein in adenomas and adenocarcinomas isolated from both genotypes. Irradiated spleen is positive control. β-actin is provided as loading control. [E] PCR amplification of genomic DNA shows that the Trp53 locus has not been deleted in adenocarcinomas from either genotype. GAPDH is provided as loading control. A dilution series is included to demonstrate that the PCR was performed in the exponential range. NL = normal lung.
To investigate whether ARF acts through a p53-dependent pathway to suppress tumor development, we examined expression of the prototypical p53 transcriptional target p21CIP1 (i.e. p21). ARF frequently co-localized with p21 in adenomas and mixed-grade tumors from wildtype mice (Fig. 4C). In accord with Figure 3B, these ARF+p21+ tumor regions were negative for the DNA damage marker phospho-histone H2A.X (Supplementary Fig. 5B, top). QPCR analysis of two additional p53 transcriptional targets, Mdm2 (Supplementary Fig. 5C) and Bax (Supplementary Fig. 5D), further identified a trend toward decreased expression in Arf-deficient compared to Arf+/+ tumors. Together, these data suggest that expression of ARF triggers an anti-proliferative response through p53 that occurs in the absence of persistent DNA damage signaling.
We attempted to confirm the p53-dependence of pulmonary tumor suppression by ARF by repeating the urethane carcinogenesis study in Trp53−/− mice. Unfortunately, all animals died from lymphoma by 15 weeks post exposure, precluding analysis of lung tumor development (data not shown). An examination of lung tumors isolated from Arf+/+ and Arf−/− animals revealed similar low-level expression of p53 protein throughout tumor development (Fig. 4D). Notably, p53 exhibited no phosphorylation on the Serine 15 residue, implying that the DNA damage response did not contribute to p53 stabilization (Supplementary Fig. 5D). Genetic analysis of adenocarcinomas from Arf+/+ and Arf−/− mice found that the Trp53 locus remained intact in both genotypes (Fig. 4E; n = 8 each). However, sequencing of Trp53 exons 2 through 11 identified 3 missense mutations in wild-type adenocarcinomas and 1 frameshift and 2 missense mutations in Arf-deficient adenocarcinomas (Supplementary Table 1), demonstrating that selection for Trp53 mutation occurs even in the absence of Arf. It is also important to note that we observed continued p21CIP1 expression in adenocarcinomas from Arf−/− mice, although the pattern of immunoreactivity differed appreciably. In Arf-deficient tumors, unlike in wildtype, p21 expression was closely associated with that of phospho-H2A.X (Supplementary Fig. 5B, bottom). Induction of p21 by DNA damage signaling has been previously demonstrated and can occur in the absence of p53.28 Regardless of the initiating signal, it is clear that remnant p21 expression is insufficient to restrain lung tumorigenesis in an Arf-null environment.
Discussion
Herein we tested the hypothesis that ARF suppresses carcinogen-induced, Kras-driven lung adenocarcinoma. Germline Arf deletion resulted in the development of large, poorly differentiated, and metastatic lung tumors. Increased proliferation and DNA damage in tumors from Arf-deficient mice, accompanied by elevated expression of nuclear cyclin D1 and phospho-ERK1/2, indicate that ARF functions to restrain hyperproliferation and maintain genomic integrity in a Kras-mutant lung environment. In wild-type mice ARF expression was typically confined to benign adenomas and co-localized with p21CIP1, suggesting that tumor suppression by ARF is an early response in lung carcinogenesis that proceeds through a p53-dependent pathway.
The Cdkn2a locus, which houses both Arf and Ink4a, has been proposed as a candidate for the Papg-1 (i.e. pulmonary adenoma progression 1) susceptibility locus in mice. The locus maps to mouse Chromosome 4 and was so named because of its significant association with lung tumor size.29 A previous study investigating this connection employed the double knockout Arf−/−;Ink4a−/− mouse, precluding direct analysis of the contributions of each gene to pulmonary tumor suppression in vivo.30 Based on strain-specific variation in INK4a function, but not in ARF, the authors deduced that Ink4a is the major lung tumor suppressor gene encoded from Cdkn2a. Indeed, there is strong evidence that Ink4a deficiency contributes to pulmonary tumorigenesis.17, 31 However, our study clearly demonstrates that Arf loss is sufficient to facilitate the malignant progression of pulmonary neoplasia. Parsing the tumor suppressive activities of ARF and INK4a has proven difficult, both in mice and humans. Selective inactivation of ARF does occur in human cancers, but silencing of the entire CDKN2A locus is a far more common event.32 The findings described herein are highly suggestive of an important role for both ARF and INK4a in suppressing lung adenocarcinoma development.
Arf-deficient mice frequently presented with lung tumors that were markedly dedifferentiated and, at times, sarcomatoid in appearance. These latter lesions exhibited many of the characteristics of pulmonary sarcomatoid carcinoma (PSC), a rare neoplasm in humans defined as poorly differentiated non-small cell lung carcinomas containing abundant sarcomatoid or spindle cell elements.22 These lesions have not previously been described in rodents.33 PSC is believed to be a “transitional” tumor type, with the sarcomatoid elements of the lesion deriving from epithelial carcinoma cells through the activation of an epithelial-mesenchymal transition (EMT) program.34 The development of PSC lesions in Arf-deficient animals argues that ARF may regulate the in vivo differentiation of cancer cells. In support of this, loss of heterozygosity at Chromosome 9p21 (i.e. CDKN2A) has been reported in human PSC cases,35 and Arf-deficiency has been shown to promote EMT in a breast cancer model.36 Furthermore, the spindle cell morphology and dual staining of cytokeratin 8 and vimentin observed in these lesions fit the proposed diagnostic criteria for EMT tumors in the mouse.37 Additional investigation of the contribution of ARF to the EMT program in carcinogenesis, as well as the clinical significance of these unusual lesions, is clearly warranted. Our model will provide a unique platform on which to conduct future examinations of PSC pathogenesis.
Our study further revealed that Arf loss engenders the nuclear accumulation of cyclin D1, a crucial regulator of CDK4/6 kinase activity during the G1 phase of the cell cycle.38 ARF has been shown to inhibit transcription of cyclin D1,39, 40 but we found no difference in mRNA levels of Ccnd1 between Arf+/+ and Arf−/− tumors, suggesting the misregulation occurs post-transcriptionally. Although the precise mechanistic link between ARF and cyclin D1 remains to be determined, previous studies have demonstrated that persistent nuclear localization of cyclin D1 promotes DNA re-replication and genomic instability.41, 42 For this reason, increased nuclear cyclin D1 is a poor prognostic indicator in NSCLC.43 It is tempting to speculate that the high levels of DNA damage we observed in Arf-deficient animals result from misregulation of cyclin D1, but additional pathways connecting ARF to DNA damage signaling and repair have been proposed.44–46 Regardless, as recent works have described,47, 48 Arf loss may have deleterious consequences for genomic stability.
The timing and functional importance of ARF expression during lung tumorigenesis are likely context dependent.6–9 Notably, studies identifying ARF expression as a late event in tumor development were performed in the context of Trp53 nullizygosity. It is perhaps not surprising that regulation of ARF expression is altered in a Trp53-deficient environment, given that p53 represses Arf transcription.49 Our data clearly demonstrate that, in a pulmonary environment without ab initio alteration of tumor suppressor pathways, induction of ARF expression is an early event in neoplastic development. As benign adenomas progress to malignant adenocarcinomas, ARF protein expression decreases significantly, a finding consistent with the increased incidence of adenocarcinomas in Arf-deficient animals. All told, our results bear many similarities to previously published accounts of lung tumorigenesis in Kras-mutant, p53-deficient animals, including increased lung tumor growth, accelerated malignancy, and enhanced metastatic capability.10, 11, 50 These commonalities strongly suggest that lung tumor suppression by ARF is mediated, at least in part, through p53.
The oncogenic stress response acting through ARF-p53, rather than the short-term DNA damage response, has been proposed as the predominant tumor suppressive mechanism in vivo.51, 52 We have previously shown that ARF has both p53-dependent and p53-independent roles in the suppression of Hras-driven skin carcinogenesis.53 Our observation that ARF and p21 are frequently co-expressed in lung tumors, without concomitant DNA damage, similarly argues that the ARF-p53 signaling axis is activated by mutant KRAS and arrests pulmonary tumorigenesis. p53 protein was found to be present in both adenomas and adenocarcinomas from Arf+/+ and Arf−/− mice. However, regulation of p53 activity is an extremely complex process that involves many layers of post-translational modification and protein-protein interaction.54, 55 These complexities are difficulty to interrogate and were not fully addressed in the current study. We also observed that Arf-deficient adenocarcinomas developed Trp53 mutations at a similar frequency as wildtype adenocarcinomas. Interestingly, Trp53 mutations in tumors from Arf+/+ mice were found exclusively in the DNA binding domain, while the mutations in Arf−/− mice predominantly occurred in the transactivation domain. The significance of this segregation of mutations by genotype, along with the functionality of p53 in Arf-deficient tumors, will be the subject of future investigations. It is worth noting, however, that Arf and Trp53 loss are not necessarily mutually exclusive events in lung tumorigenesis,14, 15 and that mice with alterations in both Trp53 and Arf exhibit increased lung tumor multiplicity and accelerated progression compared to either alone.56 It therefore remains possible, and perhaps even probable, that ARF and p53 also have independent tumor suppressive functions in the lung.
Using the urethane lung carcinogenesis model we have identified a novel role for ARF as an important inhibitor of pulmonary tumor growth and invasion. Taken together with our previous report on tumor suppression by ARF in cutaneous squamous cell carcinoma,53 our results argue that induction of ARF is an early response to oncogenic signaling that erects a barrier against tumor growth and invasion. ARF status may therefore have important implications for lung cancer patient prognosis and clinical management.
Materials and Methods
Animal Model
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Fred Hutchinson Cancer Research Center. Arf-deficient mice57 were backcrossed onto the NIH/Ola strain (Harlan Olac, UK) for 20 generations. Arf+/− breeder mice were intercrossed and lung carcinogenesis studies performed on littermates of each genotype: Arf−/− (n = 59), Arf+/− (n = 33), and Arf+/+ (n = 56). DNA was isolated by digestion of ear tissue with proteinase K in InstaGene Matrix (Bio-Rad, Hercules, CA, USA), and each mouse was genotyped as described.57 Mice were subjected to a single intraperitoneal injection of urethane (Sigma, St. Louis, MO, USA) in a PBS vehicle (1 mg/g bodyweight) between 21 and 28 days of age. A cohort of uninjected Arf−/− (n = 39), Arf+/− (n = 14), and Arf+/+ (n = 33) mice was included for control. Animals were euthanized by CO2 asphyxiation at indicated time points or when moribund. Lungs were filled with fixative before excision. Tumors macroscopically visible on the pleural surface were counted, and microcalipers were used to measure tumor diameter. Normal and tumor tissues were snap-frozen in liquid nitrogen and/or fixed in neutral buffered formalin.
Immunohistochemistry and Histopathology
Formalin-fixed tissues were processed to paraffin. For analysis, 4 μm-thick sections were stained for either hematoxylin and eosin (H&E) or a specific protein. Immunostaining was performed using standard methods for the following proteins: phospho-histone H3 (Ser10), phospho-histone H2A.X (Ser139), cleaved caspase 3 (Asp175), phospho-p44/42 MAP kinase (Thr202/Tyr204), i.e. ERK1/2, (all from Cell Signaling, Danvers, MA, USA), p19Arf (Santa Cruz, Santa Cruz, CA, USA), p21 (BD Biosciences, San Jose, CA, USA), Ki67 (Vector Laboratories, Burlingame, CA, USA), pro-surfactant protein C (Millipore, Billerica, MA, USA), cytokeratin 8 (University of Iowa, Iowa City, IA, USA) and vimentin (Novus Biologicals, Littleton, CO, USA).
Serial sections stained with H&E were analyzed and diagnosed as described.58 In order to match the original stained slide, global adjustments to white balance, brightness, and/or color saturation were made to copies of some photomicrographs using Adobe Photoshop. To calculate labeling indices for phospho-H3, phospho-H2A.X, Ki67, and ARF, at least 10 fields at 600X magnification were counted for each individual tumor, where possible (n ≥ 3 mice for each genotype and/or stage). To control for differences in staining between edge and interior fields of large-diameter tumors, only immunoreactive cells within two fields of the tumor perimeter were counted for all samples.
Western Blot Analysis
Frozen lung tumor tissues were used to prepare nuclear and cytoplasmic protein lysates, as described.59 Buffers were supplemented with standard protease and phosphatase inhibitors. Proteins were quantified using the Bradford assay (Bio-Rad), diluted in loading buffer, run on SDS-PAGE gels, and transferred to PVDF membranes. Primary antibodies included cyclin D1, lamin B1, β-tubulin, total ERK1/2 and phospho-ERK1/2 (Cell Signaling), and β-actin (BioVision, Milpitas, CA, USA). HRP-conjugated secondary antibodies (Santa Cruz) were used together with SuperSignal West detection substrate (Thermo Scientific, Rockford, IL, USA) to visualize bands. Densitometry was performed in Adobe Photoshop.
Semi-Quantitative PCR
Genomic DNA was isolated from frozen Arf+/+ and Arf−/− lung adenocarcinomas and non-tumor bearing lung tissue using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) or Trizol (Invitrogen, Carlsbad, CA, USA) using the manufacturer’s protocol. DNA concentrations were measured in triplicate using the NanoDrop 1000 spectrophotometer (Thermo Scientific). Primers for Trp53 were used to amplify equivalent amounts (20 ng) of genomic DNA over a 30 cycle PCR. Amplification reactions for a second gene, Gapdh, were performed in separate tubes, and the products run jointly on an agarose gel. The primers used were as follows: Trp53 (5′-CTTCTTGTAGATGTGGCGCGGACACG-3′) and (5′-CGTGTGCTGTAGGAGCTGCTAGAGAC-3′), and Gapdh (5′-TTCCATCCTCCAGAAACCAG-3′) and (5′-CCCTCGAACTAAGGGGAAAG-3′).
RNA Isolation and Gene Expression Analysis
Total RNA was isolated from normal lung and lung adenocarcinomas using TRIzol reagent (Invitrogen) and subsequently purified with the RNeasy Mini Kit (Qiagen). Synthesis of cDNA was performed with SuperScript II Reverse Transcriptase using oligo(dT) or random hexamer primers (Invitrogen). The expression of Ccnd1, Bax, and Mdm2 were analyzed by Real-Time PCR using predesigned Taqman Gene Expression Assays (Life Technologies, Carlsbad, CA, USA). Ct values were normalized to the endogenous control Gapdh and compared to the mean wild-type Ct. Fold change was analyzed between genotypes.
Kras and Trp53 Mutational Profiling
Amplification of Kras exons 1 and 2 from cDNA (prepared as described above) was performed using primers 5′-AGGCCTGCTGAAAATGACTG-3′ and 5′-CCAGGACCATAGGCACATCT-3′. The following primers were used to sequence Kras codons 12 and 13 (5′-CTCTATCGTAGGGTCGTAC-3′), and 61 (5′-GACTCCTACAGGAAACAAGT-3′). The Trp53 cDNA transcript spanning exons 2–11 was PCR amplified as previously described,60 and cloned into a TOPO-TA vector (Life Technologies). Top10 competent cells were transformed, and several colonies sequenced for mutations in the Trp53 gene using an ABI 3730xl DNA Analyzer with ABI BigDye terminator cycle sequencing method (PE Applied Biosystems, Foster City, CA, USA). Sequencing results were aligned to the Trp53 transcript VEGA OTTMUST00000013379 using Sequencher 4.10.1 (Gene Codes, Ann Arbor, MI, USA).
Statistical Methods
Time until development of pulmonary lesions causing death or requiring euthanasia was graphically summarized using the Kaplan-Meier method, and survival differences were analyzed for significance using the logrank test for trend. Animals euthanized for reasons other than the presentation of pulmonary lesions were included in the analysis of overall mortality but were considered censored observations in the second logrank analysis. The incidence of lung tumors by 17 weeks post urethane-injection was compared between genotypes; differences were analyzed for significance using the unpaired t test. Differences in mean tumor diameter and lung mass were analyzed using the unpaired t test. Animals who died before 20 weeks post-injection were excluded from analysis of tumor size. Individual tumor diameters were graphically summarized in a scatter plot, and variations between genotypes were statistically assessed using the Mann Whitney test. Incidence of benign and malignant lesions was compared between genotypes using the Fisher’s exact test. Nuclear-to-cytoplasmic ratios, relative gene expression, and immunoreactive cell counts were graphed and analyzed for significant differences between groups using the unpaired t test. Values given represent mean ± SEM. All P values were two-tailed, except for the statistical analysis of Ki67 cell counts, which used a one-sided P value. P values < 0.05 were considered significant. Statistical analyses were performed in Prism (GraphPad Software, La Jolla, CA, USA).
Supplementary Material
Acknowledgments
We are grateful to Sue Knoblaugh for her assistance in analyzing tumor histopathology, and to numerous colleagues for their helpful commentary. This work was supported by NCI MMHCC U01 CA141550, and by NIEHS 5 R01 ES020116. SEB was supported by PHS NRSA T32 GM007270 from NIGMS.
Grant Support: This work was supported by NCI MMHCC U01 CA141550, and by NIEHS 5 R01 ES020116. SEB was supported by PHS NRSA T32 GM007270 from NIGMS.
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.SEER Cancer Statistics Review, 1975–2008. Bethesda, MD: National Cancer Institute; Available from: http://seer.cancer.gov/csr/1975_2008/. [Google Scholar]
- 2.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 4.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 6.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 7.Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572–575. doi: 10.1038/nature09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young NP, Jacks T. Tissue-specific p19Arf regulation dictates the response to oncogenic K-ras. Proc Natl Acad Sci U S A. 2010;107:10184–10189. doi: 10.1073/pnas.1004796107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Zhang Z, Lubet RA, You M. A mouse model for tumor progression of lung cancer in ras and p53 transgenic mice. Oncogene. 2006;25:1277–1280. doi: 10.1038/sj.onc.1209182. [DOI] [PubMed] [Google Scholar]
- 12.Tam AS, Devereux TR, Patel AC, Foley JF, Maronpot RR, Massey TE. Perturbations of the Ink4a/Arf gene locus in aflatoxin B1-induced mouse lung tumors. Carcinogenesis. 2003;24:121–132. doi: 10.1093/carcin/24.1.121. [DOI] [PubMed] [Google Scholar]
- 13.Vonlanthen S, Heighway J, Tschan MP, Borner MM, Altermatt HJ, Kappeler A, et al. Expression of p16INK4a/p16alpha and p19ARF/p16beta is frequently altered in non-small cell lung cancer and correlates with p53 overexpression. Oncogene. 1998;17:2779–2785. doi: 10.1038/sj.onc.1202501. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Cespedes M, Reed AL, Buta M, Wu L, Westra WH, Herman JG, et al. Inactivation of the INK4A/ARF locus frequently coexists with TP53 mutations in non-small cell lung cancer. Oncogene. 1999;18:5843–5849. doi: 10.1038/sj.onc.1203003. [DOI] [PubMed] [Google Scholar]
- 15.Nicholson SA, Okby NT, Khan MA, Welsh JA, McMenamin MG, Travis WD, et al. Alterations of p14ARF, p53, and p73 genes involved in the E2F-1-mediated apoptotic pathways in non-small cell lung carcinoma. Cancer Res. 2001;61:5636–5643. [PubMed] [Google Scholar]
- 16.Gao N, Hu YD, Cao XY, Zhou J, Cao SL. The exogenous wild-type p14ARF gene induces growth arrest and promotes radiosensitivity in human lung cancer cell lines. J Cancer Res Clin Oncol. 2001;127:359–367. doi: 10.1007/s004320000184. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Zhu J, Bai J, Jiang H, Liu F, Liu A, et al. Comparison of the inhibitory effects of three transcriptional variants of CDKN2A in human lung cancer cell line A549. J Exp Clin Cancer Res. 2010;29:74. doi: 10.1186/1756-9966-29-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- 19.Busch SE, Gurley KE, Moser RD, Kemp CJ. ARF suppresses hepatic vascular neoplasia in a carcinogen-exposed murine model. J Pathol. 2012;227:298–305. doi: 10.1002/path.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogers S. Age of the host and other factors affecting the production with urethane of pulmonary adenomas in mice. J Exp Med. 1951;93:427–449. doi: 10.1084/jem.93.5.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tannenbaum A, Maltoni C. Neoplastic response of various tissues to the administration of urethan. Cancer Res. 1962;22:1105–1112. [PubMed] [Google Scholar]
- 22.Pelosi G, Sonzogni A, De Pas T, Galetta D, Veronesi G, Spaggiari L, et al. Review article: pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18:103–120. doi: 10.1177/1066896908330049. [DOI] [PubMed] [Google Scholar]
- 23.You M, Candrian U, Maronpot RR, Stoner GD, Anderson MW. Activation of the Ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc Natl Acad Sci U S A. 1989;86:3070–3074. doi: 10.1073/pnas.86.9.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouyssegur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18:664–674. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavoie JN, Rivard N, L’Allemain G, Pouyssegur J. A temporal and biochemical link between growth factor-activated MAP kinases, cyclin D1 induction and cell cycle entry. Prog Cell Cycle Res. 1996;2:49–58. doi: 10.1007/978-1-4615-5873-6_5. [DOI] [PubMed] [Google Scholar]
- 26.Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–2474. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin AW, Lowe SW. Oncogenic ras activates the ARF-p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci U S A. 2001;98:5025–5030. doi: 10.1073/pnas.091100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 29.Manenti G, Gariboldi M, Fiorino A, Zanesi N, Pierotti MA, Dragani TA. Genetic mapping of lung cancer modifier loci specifically affecting tumor initiation and progression. Cancer Res. 1997;57:4164–4166. [PubMed] [Google Scholar]
- 30.Zhang Z, Wang Y, Herzog CR, Liu G, Lee HW, DePinho RA, et al. A strong candidate gene for the Papg1 locus on mouse chromosome 4 affecting lung tumor progression. Oncogene. 2002;21:5960–5966. doi: 10.1038/sj.onc.1205725. [DOI] [PubMed] [Google Scholar]
- 31.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 32.Saporita AJ, Maggi LB, Jr, Apicelli AJ, Weber JD. Therapeutic targets in the ARF tumor suppressor pathway. Curr Med Chem. 2007;14:1815–1827. doi: 10.2174/092986707781058869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–2316. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 34.Thompson L, Chang B, Barsky SH. Monoclonal origins of malignant mixed tumors (carcinosarcomas). Evidence for a divergent histogenesis. Am J Surg Pathol. 1996;20:277–285. doi: 10.1097/00000478-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Dacic S, Finkelstein SD, Sasatomi E, Swalsky PA, Yousem SA. Molecular pathogenesis of pulmonary carcinosarcoma as determined by microdissection-based allelotyping. Am J Surg Pathol. 2002;26:510–516. doi: 10.1097/00000478-200204000-00015. [DOI] [PubMed] [Google Scholar]
- 36.Debies MT, Gestl SA, Mathers JL, Mikse OR, Leonard TL, Moody SE, et al. Tumor escape in a Wnt1-dependent mouse breast cancer model is enabled by p19Arf/p53 pathway lesions but not p16 Ink4a loss. J Clin Invest. 2008;118:51–63. doi: 10.1172/JCI33320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cardiff RD. Epithelial to Mesenchymal Transition Tumors: Fallacious or Snail’s Pace? Clin Cancer Res. 2005;11:8534–8537. doi: 10.1158/1078-0432.CCR-05-2250. [DOI] [PubMed] [Google Scholar]
- 38.Quelle DE, Ashmun RA, Shurtleff SA, Kato JY, Bar-Sagi D, Roussel MF, et al. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- 39.D’Amico M, Wu K, Fu M, Rao M, Albanese C, Russell RG, et al. The inhibitor of cyclin-dependent kinase 4a/alternative reading frame (INK4a/ARF) locus encoded proteins p16INK4a and p19ARF repress cyclin D1 transcription through distinct cis elements. Cancer Res. 2004;64:4122–4130. doi: 10.1158/0008-5472.CAN-03-2519. [DOI] [PubMed] [Google Scholar]
- 40.Andrique L, Fauvin D, El Maassarani M, Colasson H, Vannier B, Seite P. ErbB3(80 kDa), a nuclear variant of the ErbB3 receptor, binds to the Cyclin D1 promoter to activate cell proliferation but is negatively controlled by p14ARF. Cell Signal. 2012;24:1074–1085. doi: 10.1016/j.cellsig.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 41.Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, et al. Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev. 2007;21:2908–2922. doi: 10.1101/gad.1586007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pontano LL, Diehl JA. Speeding through cell cycle roadblocks: Nuclear cyclin D1-dependent kinase and neoplastic transformation. Cell Div. 2008;3:12. doi: 10.1186/1747-1028-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. Cyclin D1 in non-small cell lung cancer: a key driver of malignant transformation. Lung Cancer. 2007;55:1–14. doi: 10.1016/j.lungcan.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 44.Normand G, Hemmati PG, Verdoodt B, von Haefen C, Wendt J, Guner D, et al. p14ARF induces G2 cell cycle arrest in p53- and p21-deficient cells by down-regulating p34cdc2 kinase activity. J Biol Chem. 2005;280:7118–7130. doi: 10.1074/jbc.M412330200. [DOI] [PubMed] [Google Scholar]
- 45.Eymin B, Claverie P, Salon C, Leduc C, Col E, Brambilla E, et al. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol. 2006;26:4339–4350. doi: 10.1128/MCB.02240-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J Biol Chem. 2005;280:18771–18781. doi: 10.1074/jbc.M413387200. [DOI] [PubMed] [Google Scholar]
- 47.di Tommaso A, Hagen J, Tompkins V, Muniz V, Dudakovic A, Kitzis A, et al. Residues in the alternative reading frame tumor suppressor that influence its stability and p53-independent activities. Exp Cell Res. 2009;315:1326–1335. doi: 10.1016/j.yexcr.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tompkins VS, Hagen J, Frazier AA, Lushnikova T, Fitzgerald MP, di Tommaso A, et al. A novel nuclear interactor of ARF and MDM2 (NIAM) that maintains chromosomal stability. J Biol Chem. 2007;282:1322–1333. doi: 10.1074/jbc.M609612200. [DOI] [PubMed] [Google Scholar]
- 49.Zeng Y, Kotake Y, Pei XH, Smith MD, Xiong Y. p53 binds to and is required for the repression of Arf tumor suppressor by HDAC and polycomb. Cancer Res. 2011;71:2781–2792. doi: 10.1158/0008-5472.CAN-10-3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng S, El-Naggar AK, Kim ES, Kurie JM, Lozano G. A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene. 2007;26:6896–6904. doi: 10.1038/sj.onc.1210493. [DOI] [PubMed] [Google Scholar]
- 51.Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature. 2006;443:214–217. doi: 10.1038/nature05077. [DOI] [PubMed] [Google Scholar]
- 52.Efeyan A, Garcia-Cao I, Herranz D, Velasco-Miguel S, Serrano M. Tumour biology: Policing of oncogene activity by p53. Nature. 2006;443:159. doi: 10.1038/443159a. [DOI] [PubMed] [Google Scholar]
- 53.Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS Biol. 2004;2:E242. doi: 10.1371/journal.pbio.0020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 55.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Zhang Z, Kastens E, Lubet RA, You M. Mice with alterations in both p53 and Ink4a/Arf display a striking increase in lung tumor multiplicity and progression: differential chemopreventive effect of budesonide in wild-type and mutant A/J mice. Cancer Res. 2003;63:4389–4395. [PubMed] [Google Scholar]
- 57.Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 58.Kelly-Spratt KS, Philipp-Staheli J, Gurley KE, Hoon-Kim K, Knoblaugh S, Kemp CJ. Inhibition of PI-3K restores nuclear p27Kip1 expression in a mouse model of Kras-driven lung cancer. Oncogene. 2009;28:3652–3662. doi: 10.1038/onc.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philipp-Staheli J, Kim KH, Payne SR, Gurley KE, Liggitt D, Longton G, et al. Pathway-specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell. 2002;1:355–368. doi: 10.1016/s1535-6108(02)00054-5. [DOI] [PubMed] [Google Scholar]
- 60.Wang YV, Leblanc M, Wade M, Jochemsen AG, Wahl GM. Increased radioresistance and accelerated B cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell. 2009;16:33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.