

Abstract
Many malignant human tumors, including melanomas are auxotrophic for arginine due to reduced expression of argininosuccinate synthetase1 (ASS1), the rate-limiting enzyme for arginine biosynthesis. Pegylated arginine deiminase (ADI-PEG20), which degrades extracellular arginine resulting in arginine deprivation, has shown favorable results in clinical trials for treating arginine-auxotrophic tumors. Drug resistance is the major obstacle for effective ADI-PEG20 usage. To elucidate mechanisms of resistance, we established several ADI-PEG20-resistant (ADIR) variants from A2058 and SK-Mel-2 melanoma cells. Compared with the parental lines, these ADIR variants showed the following characteristics: (i) all ADIR cell lines showed elevated ASS1 expression resulting from the constitutive binding of the transcription factor c-Myc on the ASS1 promoter, suggesting that elevated ASS1 is the major mechanism of resistance; (ii) the ADIR cell lines exhibited enhanced AKT signaling and were preferentially sensitive to PI3K/AKT inhibitors, but reduced mTOR signaling and preferentially resistant to mTOR inhibitor; (iii) these variants showed enhanced expression of glucose transporter 1 and lactate dehydrogenase-A, reduced expression of pyruvate dehydrogenase, and elevated sensitivity to the glycolytic inhibitors, 2-deoxy-glucose and 3-bromopyruvate, consistent with the enhanced glycolytical pathway (the Warburg effect); (iv) the resistant cells showed higher glutamine dehydrogenase and glutaminase expression and were preferentially vulnerable to glutamine inhibitors. We demonstrated that c-Myc, not elevated ASS1 expression, is involved in upregulation of many of these enzymes because knockdown of c-Myc reduced their expression; whereas overexpressed ASS1 by transfection reduced their expression. This study identified multiple targets for overcoming ADI-PEG resistance in cancer chemotherapy using recombinant arginine-degrading enzymes.
Keywords: melanoma, argininosuccinate synthetase, Arginine Deiminase, glycolysis, glutamine metabolism
Introduction
The amino acid L-arginine has many physiological functions because it is a precursor for the biosyntheses of proteins, nitric oxide, polyamines, proline, glutamate, creatine and agmatine. It also plays important roles in immune response to antigen by impaired cytokine production and an arrest in T cell proliferation (1, 2). Arginine is primarily synthesized by 2 sequential enzymatic reactions in the urea cycle: argininosuccinate synthetase -1 (ASS1), which catalyzes the reaction of L-citrulline and aspartic acid to argininosuccinate, and argininosuccinate lyase (ASL) which recycles argininosuccinate back to arginine with the production of fumaric acid, an intermediate in the tricarboxylic acid (TCA) cycle (Fig. 1).
Figure 1.

The metabolic interrelationships of glycolysis, the TCA cycle, the urea cycle, and glutaminolysis. Upward arrows in the boxes denote that the expression is increased in the ADIR cell lines or can be induced by ADI-PEG20; downward arrows in the boxes denote downregulation. Abbreviations are: ACC=acetyl CoA carboxylase, ADI=arginine deiminase, ASL=argininosuccinate lyase, ASS1=argininosuccinate synthetase 1, FAS=fatty acid synthase, Glu-6-P=glucose-6-phosphate; GDH=glutamine dehydrogenase, Glut1=glucose transporter 1, GLS1=kidney-type glutaminase, LDH-A=lactate dehydrogenase-A, NOS=nitroxide synthetase, OTC=ornithine carbamoyltransferase, PDH=pyruvate dehydrogenase, PHD=prolyl hydroxylation, TCA= tricaboxylic acid cycle,
Many advanced human tumors, including melanomas, renal cell carcinomas, hepatocellular carcinomas, mesotheliomas, prostate cancers, and small cell lung cancers do not express ASS1 (3). ASS1-negative tumor cells therefore require arginine from the circulation for survival, whereas normal cells that synthesize arginine endogenously will survive under arginine deprivation conditions. Arginine deiminase (ADI) (4–7) and human arginase I (4, 8, 9) two arginine-depleting recombinant proteins (Fig. 1), have been under clinical development for treating arginine-auxotrophic tumors. ADI is produced by Mycoplasma spp. and humans do not produce this enzyme. Like most recombinant bacterial proteins, it is immunogenic and has a short half-life in vivo. Pegylated ADI (ADI-PEG20) has been formulated for clinical use. Clinical trials using ADI-PEG20 for treating various cancers including melanoma have produced favorable results (5, 6).
Drug resistance remains an important contributor for the treatment failure, owing to re-expression of the once-silenced ASS1. Using a cultured melanoma cell system, we previously demonstrated that transcriptional regulation of ASS1 by ADI-PEG20 involves a switch of promoter binding: c-Myc which functions as a positive regulator replaces hypoxia-inducible factor HIF-1α which functions as a negative regulator (10, 11).
The current investigation was initiated to elucidate resistance mechanisms of ADI-PEG20 in melanoma cells. We developed several stable ADI-resistant (ADIR) cell lines and found that compared with their parental counterparts, these ADIR variants exhibited elevated glycolytic metabolic activities and were preferentially sensitive to the killing by glycolytic inhibitors. Moreover, these ADIR cells also exhibited elevated expression of the kidney-type glutaminase (GLS1) and glutamine dehydrogenase (GDH), key enzymes for glutaminolysis, and were preferentially sensitive to glutamine inhibitors. These results demonstrate that ADI resistance in arginine-auxotrophic melanoma cells is associated with metabolic reprogramming that provides potential targets for overcoming the resistance.
Materials and Methods
Reagents, Antibodies and shRNA
Reagents were obtained from the following sources: ADI-PEG20 (specific activity, 5–10 IU/mg) from Polaris Pharmaceuticals, Inc. (San Diego, CA); LY294002, 2-deoxy-D-glucose (2-DG), 3-bromopyruvate (3-BP), 6-diazo-5-oxo-L-norleucine (DON), azaserine, deferoxamine mesylate (DFX), and CoCl2, from Sigma-Aldridge (St. Louis, MO); perifosine from Cayman Chemical (Ann Arbor, MI); temsirolimus (CCI-779) from Selleckchem (Houston, TX). Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) was prepared according to a previous report (12).
Antibodies were obtained from the following sources: mouse anti-human ASS1 monoclonal antibody from Polaris Pharmaceuticals; rabbit anti-argininosuccinate lyase (ASL) and rabbit anti-c-Myc (N262), rabbit anti-MCT1 and MCT4 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); rabbit anti-glucose transporter 1 (Glut-1) from FabGennix (Frisco, TX); antibodies against lactate dehydrogenase A (LDH-A), pyruvate dehydrogenase (PDH), acetyl CoA carboxylase (ACC), fatty acid synthase (FAS), mTORC1, p70S6 Kinase, p-p70S6 Kinase (Thr389), 4E-BP, eIF2α, p-eIF2 α (Ser51), ASCT2 (SLC15), AMPKα, and p-AMPK α (Thr172) from Cell Signaling (Danvers, MA); rabbit anti-kidney type glutaminase (GLS1) and rabbit anti-glutamine dehydrogenase (GDH) from Abcam (Cambridge, MA); and rabbit anti-α-tubulin antibodies from Sigma-Aldrich (St Louis, MO).
Recombinant shRNA for c-Myc were purchased from Sigma-Aldridge.
Cell culture and establishment of ADIR Cells
A2058, SK-Mel-2, and A375 melanoma cells were purchased from American Type Culture Collection Center without further authentication. All cell cultures were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum in 5% CO2 atmosphere.
ADIR cell lines were established by selecting the surviving population from A2058 and SK-Mel-2 cells that were continuously exposed to ADI-PEG20 with stepwise increases in concentrations, starting from 0.1 μg/mL and 0.05 μg/mL for A2058 and SK-Mel-2, respectively. Independent ADIR cell lines were established from single cells using 96-well-plate. Increased resistance in these clones were further developed by increased drug concentrations with 0.025 μg/mL increments for over 6 months, at which time the resistant cells were able to survive at 0.9 μg/mL of ADI-PEG20. Cells were routinely maintained at 0.3 μg/mL of ADI-PEG20.
Glucose uptake
Glucose transport was measured according to the procedure described (13). In brief, the experiments were started by adding 1 μCi of 2-deoxy-[H3]-D-glucose (2-DG) (5–10 Ci/mmol) into exponentially growing A2058 cells (5 × 105 in 6-wall, Corning). Cultured cells were maintained at 37°C for 20 min. Cells were harvested, washed 3 times with phosphate-buffered saline, and solubilized in 0.1N NaOH and the radioactivity was measured in a liquid scintillation counter.
Establishment of stable ASS1-overexpressing cell lines
The plasmid pCMV6-ASS1 carrying human full-length ASS1 cDNA sequence (GenBank, NM_000050) (Origene, Rockville, MD) and empty vector (pCMV6) were transfected into A2058 and A375 cells using lipofectamine and positive clones were selected with G418.
Other procedures
Western blotting, chromatin immunoprecipitation (ChIP), cytotoxicity assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylthiazolium bromide (MTT) were performed as described previously (10, 11).
Statistical Analyses
The IC50 values were obtained from nonlinear regression analysis of concentration-effect curves by the GraphPad Prism Software and represented by mean ± standard deviation of three independent experiments. For analysis with only two-groups (parental and ADIR cells), Student’s t test was used. The IC50 values were plotted in reference to that of A2058 which was set at 1.0. Significance was determined when p < 0.05.
Results
Elevated ASS1 expression in ADIR cell lines
We established 5 independent ADIR variants from A2058 (A2058-R1 to A2058-R5) and 4 SK-Mel-2 (SK-Mel-2-R1 to SK-Mel-2-R4) lines. Cytotoxicity tests showed that all these cell lines were more resistant to ADI-PEG20 than were their respective parental cell lines. More than 95% of all ADIR-A2058 cells could survive at 0.9 μg/mL ADI-PEG20 for 48 hr treatment, whereas < 25% of the parental cells survived (Fig. 2A). Likewise, treating SK-Mel-2 cells with 0.9 μg/ml ADI-PEG20 almost completely killed the cells, whereas >70% of ADIR SK-Mel-2 cells were able to survive under the same treatment (Fig. 2B).
Figure 2.

Survivability test and protein expression of ADIR variants. A, A2058 and its ADIR variants (A2058-R1 to A2058-R5) and B, SK-Mel-2 and its ADIR variants (SK-R1 to SK-R4) were treated with different concentrations of ADI-PEG20 for 3 days. Viability of the treated cells was determined by MTT assay. C, Western blotting analyses of ASS1, c-Myc, HIF-1α, and α-tubulin expression in A2058 (denoted by A) and its ADIR variants (R1 to R5). D, Western blotting of protein expression in SK-Mel-2 (SK) and its ADIR lines (R1 to R4).
All the ADIR-A2058 cells exhibited increased ASS1 expression (about 15-fold by densitometry, not shown) as compared with that in the drug-sensitive counterpart (Fig. 2C). Likewise, all the ADIR-SK-Mel-2 cells had higher ASS1 expression levels than did the parental cell lines, but the SK-Mel-2-R1 and SK-Mel-2-R2 cell lines had lower ASS1 levels than did the SK-Mel-2-R3 and SK-Mel-2-R4 lines (Fig. 2D). Reduced expression of ASS1 in the SK-Mel-2-R1 and SK-Mel-2-R2 cell lines was correlated with less resistance to ADI-PEG20 (Fig. 2B). These observations, together with our previous finding that modulation of ASS1 levels affects cell sensitivity to ADI-PEG20 (4, 10), strongly support that ASS1 expression is the predominant mechanism for acquired resistance to ADI-PEG20 in cultured melanoma cells.
Roles of c-Myc and HIF-1α in the regulation of ASS1 expression in ADIR cell lines
We previously demonstrated that induction of ASS1 expression by ADI-PEG20 in melanoma cells was associated with the up-regulation of c-Myc which functions as a positive regulator, and the down-regulation of HIF-1α which functions as a negative regulator (10, 11). Figs. 2C and 2D show that c-Myc expression levels were similar between all ADIR variants and their respective parental cell lines; whereas reduced HIF-1α expression levels were seen in A2058-R2, A2058-R4, A2058-R5, and all ADIR-SK-Mel-2 lines, but not in A2058-R1 and A2058-R3 cells. The observations that no elevated expression of c-Myc and no reduction of HIF-1α expression in all ADIR cells prompted us to investigate whether c-Myc and HIF-1α remain important for regulating ASS1 expression in these ADIR cell lines.
We chose 4 representative ADIR cell lines (A2058-R1 which expressed higher HIF-1α levels than the control, and A2058-R2, SK-Mel-2-R1 and SK-Mel-2-R3 which did not express detectable levels of HIF-1α). To investigate whether accumulation of HIF-1α would suppress ASS1 expression in the ADIR cell lines, we treated these cells and their drug sensitive cells (as control) with hypoxic mimics, DFX and CoCl2 (14). Treating A2058 or SK-Mel-2 cells with DFX or CoCl2 suppressed ADI-PEG20-induced ASS1 expression (Fig. 3A, left). However, no suppression of ASS1 expression was found in A2058-R1, A2058-R2 and SK-Mel-2-R3, except SK-Mel-2-R1 cells (Fig. 3A, right). The suppression of ASS1 expression in SK-Mel-2-R1 cells by elevated HIF-1α, but not in the other ADIR cell lines may have been due to the lower expression levels of ASS1 in SK-Mel-2-R1 cells as compared with those in the other cell lines (Fig. 2D). These results suggest that the extent of ASS1 suppression by over-expressed HIF-1α may depend upon the intrinsic ASS1 expression levels.
Figure 3.

Roles of c-Myc and HIF-1α in the expression of ASS1 in ADIR cell lines and their parental cell lines. A, Effects of HIF-1α accumulation by hypoxia mimics, DFX (50 μM) or CoCl2, (100 μM) on the expression of ASS1 in A2058, SK-Mel2 induced by ADI-P EG 20 (0.3 μg/mL) and their ADIR cell lines. Note that accumulation of HIF-1α suppresses ASS1 expression in SK-R1 but not in SK-R3. B, knockdown of c-Myc by shRNA (Sh1 and Sh2) suppressed ASS1 expression in the indicated ADIR cell lines. C, ChIP assay of HIF-1α and c-Myc interactions with the ASS1 promoter in A2058 (denote by A, lane 1), A2058-R1 (lane 2) and A2058-R2 (lane 3). Top, the antibodies used for ChIP and control IgG. Input, genomic DNA prior to immunoprecipitation. D, similar experiment was performed in SK-Mel-2 (SK) and its ADIR cell lines (R1 and R3). ASS1 promoter sequences from ChIP were quantified by PCR assay.
To investigate the role of c-Myc in regulating ASS1 expression in ADIR variants, we used c-Myc shRNA strategy. Knockdown of c-Myc expression by 2 independent shRNA in lentiviral vectors downregulated ASS1 expression (Fig. 3B), supporting the positive role of c-Myc in the regulation of ASS1 in the ADIR cells.
We also performed ChIP assay to determine the occupancy of HIF-1α and c-Myc at the ASS1 promoter. Consistent with our previous results (10), we found that HIF-1α, but not c-Myc, binds to the ASS1 promoter in the parental A2058 (Fig. 3C, lane 1) and SK-Mel-2 cells (Fig. 3D, lane 1). In contrast, in all the ADIR variants investigated, c-Myc, not HIF-1α, binds to the ASS1 promoter (Figs. 3C & 3D, lanes 2 & 3). These results taken together, demonstrate that HIF-1α and c-Myc remain important in transcriptional regulation of ASS1 expression in ADIR variants.
ADIR cell lines are sensitive to PI3K/AKT inhibitors
We previously observed that A2058 and SK-Mel-2 cells treated with ADI-PEG20 activated the Ras/PI3K/AKT-1 signaling cascade, resulting in c-Myc stabilization for ASS1 upregulation (11). Moreover, several recent reports suggested that PI3K/AKT play important roles in regulating c-Myc expression (15–17). Activation of PI3K/AKT-1 signal is elicited by phosphorylation of AKT-1 at threonine 308. We performed Western blotting analyses of 5 ADIR-A2058 cell lines using anti-phosphorylated AKT antibody and found that all these cells contained activated AKT-1 signal (Fig. 4A). Activation of AKT-1 in these ADIR cells was reflected by increased sensitivities to the killing by LY294002 and perifosine which inhibit PI3K and AKT1 activities, respectively (Fig. 4B).
Figure 4.

Analyses of protein expression in A2058 and its 5 ADIR cells. A, activated phosphorylation AKT (p-AKT) signaling; B, enhanced sensitivity of ADIR cells to the PI3K inhibitor (LY294002) and AKT (perifosine) as compared with their parental line. C, Western blotting analyses of the mTOR signaling, AMPKα, and eIF1α. D, Sensitivity of A2058 and its ADIR cells to mTOR inhibitor CCI-779. Cells were incubated with CCI-779 at concentrations ranging from 1 to 100 μM for 72 h and their viabilities were determined by MTT assay.
Downregulated mTOR signaling in ADIR Cell Lines
Three well-characterized sensing mechanisms for cellular amino acid and/or nutritional deprivation in eukaryotic cells include the mammalian target of rapamycin complex 1 (mTORC1), general amino acid control nonderepressible 2 (GCN2) and AMP-dependent protein kinase (AMPK) (18, 19). mTORC1 phosphorylates and activates protein kinase p70S6 kinase 1 (S6K1) which subsequently activates mRNA cap binding by phosphorylating eIF-4B for the formation of translational initiation complex. GCN2 is the only kinase for eukaryotic initiation factor 2α (eIF2α) that senses the absence of one or more amino acids by virtual of direct binding to uncharged cognate tRNA (20). Phosphorylation of elF2α (p-eIF2α) by activated GCN2 prevents the assembly of eIF2-GTP-Met-tRNAMet translation initiation complex, resulting in reduction of general protein synthesis (21, 22). AMPK is a metabolic sensor of energy crisis resulting from nutritional deprivation (23). We investigated the expression of these signals in 5 ADIR-A2058 cells. Fig. 4C shows that expression levels of mTOR and p-mTOR were drastically reduced in all 5 ADIR cell lines. Likewise, the downstream signal for mTOR, p70S6K was also downregulated. Downregulation of these signals in these ADIR cells is reflected by virtue of their resistance to clinically approved mTOR inhibitor, temsirolimus (CCI-779) (24) (Fig. 4D). Expression levels of AMPKα are variable among the 5 ADIR cell lines (Fig. 4C) and levels of phosphorylated AMPKα were too low to be detected in all cells (not shown). No apparent difference in eIF2α levels was observed between ADIR-A2058 cells and their parental cells, but levels of phosphorylated elF2α (p-eIF2α) were low in all the cell lines investigated, using cell lysate prepared from A2058 cells treated with ADI-PEG20 for 24 hr as a positive control (Fig. 4C). These results demonstrated that mTOR is the only signal that is consistently downregulated in the ADIR cells.
ADIR cells are associated with enhanced expression of ASL and glycolytic activities (the Warburg effect)
We then investigated whether ADI-PEG20 resistance would alter the metabolic programs. Because ASL is directly downstream from ASS1 in the urea cycle (Fig. 1), we performed Western blotting of its expression in 5 ADIR-A2058 lines. We found that ASL expression levels were elevated in all 5 ADIR cell lines (Fig. 5A).
Figure 5.

ADIR cell lines exhibited elevated glycolytic and glutaminolytic metabolism and increased sensitivity to glucose and glutaminase inhibitors A, Western blots of glucolytic enzymes in A2058 and its ADIR cells; B, enhanced uptake of 2-DG in ADIR cell lines as compared with A2058 cells; C, enhanced sensitivity of ADIR cell lines to glucolysis inhibitors 2-DG and 3-BP as compared with A2058 cells; D, expression levels of GLS1 and GDH in A2058 and its ADIR cell lines. E, enhanced sensitivity of ADIR cells to glutamine inhibitors, DON, azerserine and BPTES, as compared with A2058 cells.
Next, we investigated the expression of several key players involved in the glycolytic pathway in the ADIR cell lines. We first investigated Glut1 which is the major transporter for glucose and found that all 5 ADIR-A2058 cells showed about 8-fold higher Glut1 levels than those in A2058 parental cells. Elevated Glut1 expression in these the ADIR cell lines was correlated with the enhanced transport of [3H]-2-DG, a non-metabolizable form of glucose (Fig. 5B). Compared with the parental cell lines, the ADIR variants showed higher levels of LDH-A which catalyzes the conversion of pyruvate into lactate (Fig. 1), but lower levels of PDH (Fig. 5A). PDH is the key enzyme that catalyzes the conversion of pyruvate into acetyl CoA, an important metabolic intermediate in the TCA cycle (Fig. 1).
Monocarboxylate transporter family (MCT, SLC16) catalyzes the rapid transport across the plasma membrane of many monocarboxylates including lactate (25). Since these ADIR cells show elevated expression of LDH-A, we examined the expression levels of MCT1 and MCT4 in ADIR A2058 cells. Results showed that levels of MCT1 were increased in A2058-R1, A2058-R2, A2058-R4, and A2058-R5, but reduced in A2058-R3; whereas levels of MCT4 were increased in A2058-R2, A2058-R3, and A2058-R5, but reduced in A2058-R4, and no change in A2058-R1 (data not shown). Thus, the expression levels of MCT1 and MCT4 are heterogeneous among the 5 ADIR-A2058 cells. Their roles in ADI resistance are not certain.
ADIR cell lines are preferentially sensitive to glycolytic inhibitors
Since ADIR cells exhibit elevated expression of Glut1 and enhanced glucose uptake, known as the Warburg effect, we asked whether these cells would be preferentially killed by 2-DG, a commonly used inhibitor of glycolysis (26–28). We found that, indeed, all the 5 ADIR-A2058 variants exhibited enhanced sensitivity to 2-DG (Fig. 5C). Moreover, we found that these 5 ADIR cells were also vulnerable to another glucolytic inhibitor 3-BP, a lactate/pyruvate analogue (29, 30) (Fig. 5C), demonstrating that these ADIR cells are preferentially sensitive to glycolytic inhibitors as compared with their parental cell lines.
ADIR cells exhibit elevated glutaminolytic metabolism and increased sensitivity to glutaminase Inhibitors
Two key enzymes in glutaminolytic metabolism, glutaminase (GLS) and glutamine dehydrogenase (GDH), catalyze the 2-step sequential conversion of glutamine into α-ketoglutarate via glutamate intermediate (Fig. 1). Mammals express 2 GLS isoforms, kidney-type (or GLS1) and liver-type (GLS2). GLS1 is expressed in a broad spectrum of cell lines. We demonstrated elevated expression of GLS1 and GDH in all 5 ADIR-A2058 cell lines as compared with those in the parental control, indicating the increased glutaminolytic activities in these ADIR variants (Fig. 5D). These results were supported by the demonstration that all the 5 ADIR cell lines were associated by preferential sensitivity to GLS inhibitors, DON, azaserine, and BPTES (Fig. 5E). Despite increased glutaminolytic enzymes in these ADIR cell lines, we found only A2058-R3 exhibited elevated ASCT2 glutamine transporter among the 5 ADIR A2058 cells (data not shown).
C-Myc, but not elevated ASS1, contributes to the enhanced glycolytic and glutaminolytic metabolisms in ADIR cell lines
To investigate the kinetics of induction of these glycolytic and glutaminolytic enzymes by ADI-PEG20 in the ADIR cells, we performed an ADI-PEG20 time-course treatment of A2058 cells from 8 to 72 hrs. We found that ADI-PEG20 induced not only ASS1 and c-Myc, but also Glut1, LDH-A, GLS1 (but not GDH) within this time frame. These results indicate that induction of ASS1, Glut1, LDH-A, occurred very early during the development of ADI resistance, and elevation of GDH levels occurred subsequently (Fig. 6A).
Figure 6.
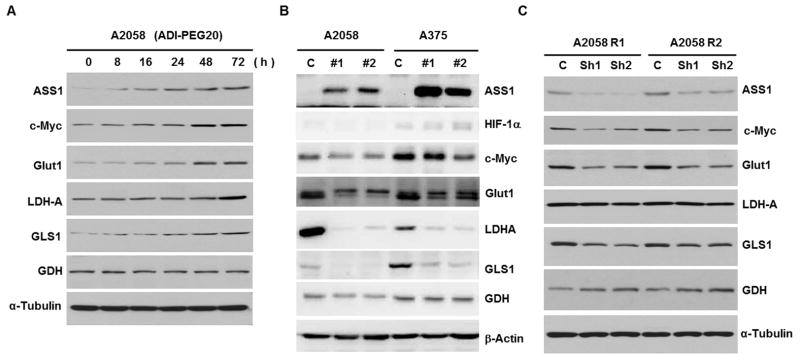
Western blotting analyses of protein expression. A, time-course induction of various enzymes by ADI-PEG20 treatment (0.3 μg/mL) in A2058 cells; B, effects of ASS1-overexpression in A2058 and A375 cell lines by ASS1 transfection (2 independent cell lines each) on the expression of several proteins as indicated. C, effects of c-Myc knockdown on the expression of proteins as indicated in two ADIR-A2058 cell lines.
Since elevated expression of ASS1 is the principal mechanism of ADI resistance, we investigated whether enhanced expression of these enzymes was the consequence of ASS1 upregulation in the ADIR cell lines. To this end, we used transfection to establish 2 independently overexpressed ASS1 cell lines in A2058 and A375 cells. While no apparent alteration of HIF-1α levels was discernible even using prolonged exposure of the Western blots (not shown). We found that expression of c-Myc, Glut1, LDH-A, GLS1 and GDH was reduced (Fig. 6B). This expression profile is directly opposite from that in the ADIR cell lines. These results demonstrate that elevated expression of these enzymes in ADIR cells was not likely due to the enhanced ASS1 expression.
We next investigated whether c-Myc plays a role in regulating the glycolytic and glutaminolytic enzymes in these ADIR cells, in light that c-Myc has been recognized as an important transcription regulator of 10% to 20% of total cellular genes through E-box interactions in mammalian cells (31, 32). A2058-R1 and A2058-R2 cells were treated with 2 independent c-Myc shRNA for 48 hr. Although the shRNA we used could only partially reduce c-Myc levels, we found reduced expression of Glut1 and GLS1 in these ADIR cells as compared with those in the scramble shRNA-treated controls, whereas levels of LDH-A and GDH were not affected. The inability of reducing LDH-A and GDH suggest that either other mechanisms are involved for the regulation of LDH-A and GDH or regulation of these enzymes by c-Myc is more stringent than Glut1 or GLS1. In any event, these results support that c-Myc plays a role in regulating metabolic rewiring during the development of ADI-PEG20 resistance.
Expression of enzymes involved in the lipid biosynthesis in the ADIR cell lines
We also determined the expression of enzymes involved in fatty acid biosynthesis in ADIR cell lines. Compared with their parental counterpart, all the ADIR A2058 cell lines, except A2058-R4, had lower expression levels of ACC which catalyzes the conversion of acetyl-CoA into malonyl-CoA (Fig. 1) (data not shown), and all the ADIR A2058 cell lines, except A2058-R2, had lower levels of FAS which converts malonyl-CoA into palmitoyl-CoA (data not shown). These results suggest that fatty acid metabolism may also be reduced in the ADIR cell lines compared with their parental counterpart.
Discussion
The discovery that many human malignant tumors do not express ASS1 and are arginine-auxotrophic provides a mechanistic basis for using recombinant arginine-depleting enzymes in targeted therapy of these diseases (4). In this communication, we demonstrate that in a panel of 9 independently established ADIR cell lines all expressed elevated levels of ASS1, further strengthening the role of ASS1 in ADI resistance. These ADIR cell lines serve as a valuable resource for investigating the metabolic reprogramming associated with ADI resistance as summarized in Fig 1. Several important findings are discussed below.
We found that these ADIR variants exhibited elevated AKT signaling and are preferentially sensitive to PI3K/AKT inhibitors. These findings support the important role of this pathway in ADI-PEG20 resistance (11). We also observed that expression of mTOR and its downstream signal p70S6K was downregulated in all ADIR cells. While the canonical pathway of mTOR regulation is through the PI3K/AKT pathway (33), however, downregulation of mTOR independent of this pathway has also been reported (34). The molecular mechanism for downregulation of mTOR signal in ADI resistance remains to be investigated.
We further demonstrated that ASL was elevated in these ADIR lines, suggesting that the ADIR cells may require robust ASS1/ASL activities in the urea cycle to overcome ADI-PEG20-induced autophagy and apoptosis in the otherwise arginine-auxotrophic cancer cells (11, 35). Although ASS1 has often been considered as the rate-limiting enzyme for the biosynthesis of arginine (36), our results suggest that ADIR cells may require additional enzymes to replenish arginine under prolong arginine deprivation conditions. Our results are consistent with recent findings that several enzymes in the urea cycle may be co-regulated (37), including ASS1, ASL, nitric oxide synthetase, and cationic amino acid transporter 1 and may form a supermolecular complex to perform important physiological function (38).
We found that these ADIR cells have altered metabolic program toward aerobic glycolysis by enhancing Glut1 and LDH-A expression but reducing PDH expression, exhibiting the Warburg phenomenon. Since Warburg’s work in 1929, it has been evidenced that cancer cells, even under aerobic conditions, frequently upregulate glucose metabolism leading to a high uptake and use of glucose, but a moderate rate of mitochondrial respiration (39, 40). This characteristic has led to the development of 2-[18F]fluoro-2-deoxy-D-glucose (FDG) in positron emission tomography (PET) for the imaging of primary and metastatic tumors in patients (41). Our observations that ADIR cells exhibited elevated Warburg phenomenon suggest that it may be possible to use FDG-PET for monitoring the development of ADI resistance during cancer chemotherapy. In clinical setting, melanoma is known to be strongly PET positive and usually become less FDG avid when tumor response to treatment. Once tumors become resistant to ADI-PEG20, melanoma will again become FDG avid. This was also observed clinically when patient failed ADI-PEG20.
Metabolic reprogramming in ADIR cell lines also include elevated expression of GLS1 and GDH, another common phenomenon in cancer cells (42, 43). These enzymes are important for producing α-ketoglutarate, an important metabolite in the TCA cycle. Tumor cells fuel their metabolism with glucose and glutamine as important bioenergetic sources for cell proliferation. The dependence of these ADIR cells to glucose and glutamine for growth was demonstrated by the findings that these ADIR cells exhibited elevated sensitivity to the killing by glucose (2-DG and 3-BP) and glutamine inhibitors (DON, azaserine, and BPTES). While DON and azaserine have long been known to be toxic in human clinical evaluations (44), presumably due to the presence of chemically reactive diazo group. The increased sensitivity of these ADIR cells to BPTES demonstrated in this study may have promising therapeutic implication. New generation of GLS1 inhibitors such as BPTES derivatives which are devoid of diazo group found in DON and azaserine (12), as well as compound 968 and its derivatives, another structurally distinct series of GLS1 inhibitor (45, 46) that are currently under clinical evaluations, may pave the way for future clinical applications.
We also found that c-Myc plays important roles in the metabolic rewiring of ADI resistance. Previous studies have demonstrated that c-Myc regulates genes involved in the glycolytic pathway by directly binding to the E-box located at the promoters of these genes (47). C-Myc can also upregulates glutamine metabolism via transcriptional repression of miR-23a and miR-23b (48). Although levels of c-Myc were not elevated in these ADIR cells, however, our kinetic analysis demonstrated that expression of c-Myc was induced by ADI-PEG20 within the same time-frame when many of these enzymes were also induced. Elevated levels of glycolytic and glutaminolytic pathways apparently are needed for the maintenance of ADI resistance, because inhibitors to these pathways overcome the resistance. The observation that no elevated c-Myc levels in all the 9 ADIR cell lines, despite their early induction, suggests that persistently high-levels of c-Myc may be disadvantageous for cell growth during the development of ADI resistance.
We also discovered that elevated expression of ASS1 by transfection downregulates Glut1, LDH-A, GLS1, and GDH expression, demonstrating that modulating ASS1 levels can influence the expression of other enzymes in distance, i.e. glycolytic and glutaminolytic pathways. While the precise mechanism(s) remain to be investigated, downregulation of c-Myc may be responsible for the reduced expression of these enzymes in these ASS1-treansfected cells, although this remain to be elucidated. Moreover, the findings of reduced levels c-Myc levels in these ASS1-transfected cells may explain why ADIR cells did not show elevated c-Myc, as this may be due to feedback inhibition mechanism of c-Myc regulation once ASS1 is overly expression. These possible mechanisms would further support the important roles of c-Myc in the regulation of glycolytic and glutaminolytic metabolism in melanoma cells.
Finally, the identification of metabolic reprogramming associated with ADI resistance in the arginine-auxotrophic melanoma model bears important clinical implications for targeted therapy of arginine-auxotrophic tumors. Many anti-tumor small molecules targeting PI3K/AKT, glycolytic and glutaminolytic signalings have been approved for clinical use. Combination of these agents with arginine-degrading enzymes, such as ADI-PEG20 or pegylated human arginase I may improve the treatment efficacy of many human cancers that require arginine for survival.
Acknowledgments
Grant Support: Supported in part by ROI-CA149260 from the National Cancer Institute (M. T. Kuo and L. G. Feun) and R21NS074151 from the National Institute of Neurological Disorders and Stroke (T. Tsukamoto).
The authors thank Dr. Bor-Wen Wu (Polaris) for providing ADI-PEG20 and anti-ASS1 antibody.
Footnotes
Authors’ contributions:
Conception and design: M.T. Kuo, N. Savaraj, L.G. Feun
Development of Methodology: Y. Long, W.-B. Tsai, M. Wangpaichitr, T. Tsukamoto
Acquisition of data: Y. Long, W-B. Tsai, M. Wangpaichitr
Analysis and interpretation: Y. Long, W-B. Tsai, M. Wangpaichitr, N. Savaraj, M. T. Kuo
Writing and review: M.T. Kuo, N. Savaraj, L.G. Feun, T. Tsukamoto
Study supervision: M. T. Kuo, N. Savaraj
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest to disclose
References
- 1.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100:826–33. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- 4.Kuo MT, Savaraj N, Feun LG. Targeted cellular metabolism for cancer chemotherapy with recombinant arginine-degrading enzymes. Oncotarget. 2010;1:246–51. doi: 10.18632/oncotarget.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feun LG, Marini A, Walker G, Elgart G, Moffat F, Rodgers SE, et al. Negative argininosuccinate synthetase expression in melanoma tumours may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br J Cancer. 2012;106:1481–5. doi: 10.1038/bjc.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ott PA, Carvajal RD, Pandit-Taskar N, Jungbluth AA, Hoffman EW, Wu BW, et al. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs. 2013;31:425–34. doi: 10.1007/s10637-012-9862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang TS, Lu SN, Chao Y, Sheen IS, Lin CC, Wang TE, et al. A randomised phase II study of pegylated arginine deiminase (ADI-PEG 20) in Asian advanced hepatocellular carcinoma patients. Br J Cancer. 2010;103:954–60. doi: 10.1038/sj.bjc.6605856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mauldin JP, Zeinali I, Kleypas K, Woo JH, Blackwood RS, Jo CH, et al. Recombinant human arginase toxicity in mice is reduced by citrulline supplementation. Transl Oncol. 2012;5:26–31. doi: 10.1593/tlo.11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, et al. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2013;31:99–107. doi: 10.1007/s10637-012-9807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai WB, Aiba I, Lee SY, Feun L, Savaraj N, Kuo MT. Resistance to arginine deiminase treatment in melanoma cells is associated with induced argininosuccinate synthetase expression involving c-Myc/HIF-1alpha/Sp4. Mol Cancer Ther. 2009;8:3223–33. doi: 10.1158/1535-7163.MCT-09-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012;72:2622–33. doi: 10.1158/0008-5472.CAN-11-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. 2012;55:10551–63. doi: 10.1021/jm301191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan Y, Dickman KG, Zong WX. Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J Biol Chem. 2010;285:7324–33. doi: 10.1074/jbc.M109.035584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaelin WG., Jr Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb Symp Quant Biol. 2011;76:335–45. doi: 10.1101/sqb.2011.76.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilic N, Utermark T, Widlund HR, Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muellner MK, Uras IZ, Gapp BV, Kerzendorfer C, Smida M, Lechtermann H, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–93. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med. 2011;17:1116–20. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, et al. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ Res. 2012;110:1192–201. doi: 10.1161/CIRCRESAHA.111.259952. [DOI] [PubMed] [Google Scholar]
- 19.Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013;49:379–87. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 21.Kimball SR, Jefferson LS. Control of translation initiation through integration of signals generated by hormones, nutrients, and exercise. J Biol Chem. 2010;285:29027–32. doi: 10.1074/jbc.R110.137208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–27. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles? Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- 24.Stock C, Zaccagnini M, Schulze M, Teber D, Rassweiler JJ. Temsirolimus. Recent Results Cancer Res. 2010;184:189–97. doi: 10.1007/978-3-642-01222-8_13. [DOI] [PubMed] [Google Scholar]
- 25.Pinheiro C, Longatto-Filho A, Azevedo-Silva J, Casal M, Schmitt FC, Baltazar F. Role of monocarboxylate transporters in human cancers: state of the art. J Bioenerg Biomembr. 2012;44:127–39. doi: 10.1007/s10863-012-9428-1. [DOI] [PubMed] [Google Scholar]
- 26.Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, et al. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007;6:3049–58. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- 27.Pradelli LA, Beneteau M, Chauvin C, Jacquin MA, Marchetti S, Munoz-Pinedo C, et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene. 2010;29:1641–52. doi: 10.1038/onc.2009.448. [DOI] [PubMed] [Google Scholar]
- 28.Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer. 2002;87:805–12. doi: 10.1038/sj.bjc.6600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, et al. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun. 2004;324:269–75. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 30.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–9. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, et al. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–29. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–9. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YK, Park SY, Kim YM, Kim DC, Lee WS, Surh YJ, et al. Suppression of mTOR via Akt-dependent and -independent mechanisms in selenium-treated colon cancer cells: involvement of AMPKalpha1. Carcinogenesis. 2010;31:1092–9. doi: 10.1093/carcin/bgq040. [DOI] [PubMed] [Google Scholar]
- 35.Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69:700–8. doi: 10.1158/0008-5472.CAN-08-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 126:2762–72. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- 37.Oyadomari S, Gotoh T, Aoyagi K, Araki E, Shichiri M, Mori M. Coinduction of endothelial nitric oxide synthase and arginine recycling enzymes in aorta of diabetic rats. Nitric Oxide. 2001;5:252–60. doi: 10.1006/niox.2001.0344. [DOI] [PubMed] [Google Scholar]
- 38.Erez A, Nagamani SC, Shchelochkov OA, Premkumar MH, Campeau PM, Chen Y, et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med. 2011;17:1619–26. doi: 10.1038/nm.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dang CV, Hamaker M, Sun P, Le A, Gao P. Therapeutic targeting of cancer cell metabolism. J Mol Med (Berl) 2011;89:205–12. doi: 10.1007/s00109-011-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer. 2002;2:683–93. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 42.Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle. 2010;9:3884–6. doi: 10.4161/cc.9.19.13302. [DOI] [PubMed] [Google Scholar]
- 43.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magill GB, Myers WP, Reilly HC, Putnam RC, Magill JW, Sykes MP, et al. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer. 1957;10:1138–50. doi: 10.1002/1097-0142(195711/12)10:6<1138::aid-cncr2820100608>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 45.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–19. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katt WP, Ramachandran S, Erickson JW, Cerione RA. Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther. 2012;11:1269–78. doi: 10.1158/1535-7163.MCT-11-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–64. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 48.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–5. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]