

Abstract
We used the Illumina reversible-short sequencing technology to obtain 17-fold average depth (s.d. ~8) of ~94% of the euchromatic genome and ~1–5% of the heterochromatin sequence of the Drosophila melanogaster isogenic strain w1118; iso-2; iso-3. We show that this strain has a ~9 kb deletion that uncovers the first exon of the white (w) gene, ~4 kb of downstream promoter sequences, and most of the first intron, thus demonstrating that whole-genome sequencing can be used for mutation characterization. We chose this strain because there are thousands of transposon insertion lines and hundreds of isogenic deficiency lines available with this genetic background, such as the Exelixis, Inc., and the DrosDEL collections. We compared our sequence to Release 5 of the finished reference genome sequence which was made from the isogenic strain y1; cn1 bw1 sp1 and identified 356,614 candidate SNPs in the ~117 Mb unique sequence genome, which represents a substitution rate of ~1/305 nucleotides (~0.30%). The distribution of SNPs is not uniform, but rather there is a ~2-fold increase in SNPs on the autosome arms compared with the X chromosome and a ~7-fold increase when compared to the small 4th chromosome. This is consistent with previous analyses that demonstrated a correlation between recombination frequency and SNP frequency. An unexpected finding was a SNP hotpot in a ~20 Mb central region of the 4th chromosome, which might indicate higher than expected recombination frequency in this region of this chromosome. Interestingly, genes involved in sensory perception are enriched in SNP hotspots and genes encoding developmental genes are enriched in SNP coldspots, which suggests that recombination frequencies might be proportional to the evolutionary selection coefficient. There are currently 12 Drosophila species sequenced, and this represents one of many isogenic Drosophila melanogaster genome sequences that are in progress. Because of the dramatic increase in power in using isogenic lines rather than outbred individuals, the SNP information should be valuable as a test bed for understanding genotype-by-environment interactions in human population studies.
Keywords: personal genomes, Drosophila melanogaster, whole-genome SNP analysis
Introduction
Whole-genome sequences of humans and several model organisms have revolutionized genetics research in the past decade. Drosophila has long been a testing ground for developing genetics and genomics techniques that have later been applied to humans. For example, the shotgun sequencing technique of an entire eukaryotic genome was done as a proof-of-concept by Celera Genomics, Inc., and the International Drosophila Genome Project Consortium in 2000 with the D. melanogaster strain y1; cn1 bw1 sp1.1,2 This was four years before the near completion of the human genome sequence by Celera Genomics, Inc., and the International Human Genome Sequencing Consortium.3
D. melanogaster isogenic strain sequences will help in the development of personalized medicine.4 The central problem in personalized medicine is to identify single nucleotide polymorphisms (SNPs) that correlate with susceptibility to diseases and the efficacy of drugs.5 However, the characterization of SNPs in human genomes, because of its larger size and heterozygosity at many alleles, is at least 10-fold more expensive than D. melanogaster. Also, in humans but much less so in D. melanogaster, linkage disequilibrium between polymorphic markers often leads to false associations between sequence polymorphisms and disease states (International-HapMap-Consortium, 2005).6 Having additional D. melanogaster genome sequences will allow the development of novel statistical techniques that will improve the accuracy of human associative mapping studies and quantitative genetics. D. melanogaster has the advantages of over 100 years of genetics in which the functions of many genes are known.7,8 D. melanogaster also has known sequence polymorphisms that affect quantitative traits,9 inter-laboratory reproducibility, and multiple replications with genetically identical strains allows for more rapid computations in genome association studies.
We have used the Illumina massively parallel short-sequencing technology to obtain the genome sequence of D. melanogaster isogenic strain w1118; iso-2; iso-3.10 We chose this strain because there are thousands of transposon insertion stocks11 and hundreds of deficiencies12,13 available with this genetic background. Other laboratories have shown that relatively inexpensive short-sequencing technology of entire genomes, such as C. elegans, is orders of magnitude less expensive and more rapid than dideoxy sequencing.14
We compared the w1118; iso-2; iso-3 sequence to the reference genome sequence from the isogenic strain y1; cn1 bw1 sp1.1,2 We identified 356,614 candidate SNPs in the ~117 Mb euchromatin portion of the ~180 Mb D. melanogaster genome, representing a substitution rate of ~1/305 nucleotides. The alignment was done against the Release 5 finished genomic sequence with the R5.3 FlyBase gene annotation (www.flybase.org). This study shows a non-homogenous SNP distribution in the D. melanogaster genome, with hotspots enriched in sensory perception genes and coldspots located near developmental genes, chromosome ends, and the X and 4th chromosomes. This finding is consistent with earlier studies and population genetics theories that show a correlation between recombination rates and SNP frequencies in Drosophila melanogaster.15,16
The SNP information should be valuable not only to Drosophila researchers but, because of novel bioinformatics and experimental techniques developed in simple model organisms, to anyone interested in personal genomes and personalized medicine. While there are 12 Drosophila species sequenced,17 this is one of several isogenic D. melanogaster genomes that are currently being sequenced.
Results
Sequencing with the Illumina platform
The Illumina platform that we used produced reads of 36 nt. Current Illumina technology allows reads of over 100 nt with improved accuracy; and the quality, read lengths, and numbers of reads are steadily gaining ground. We analyzed sequences of 25–36 nt depending on the quality of the lanes (see Materials and Methods). The primary errors in these short sequences are substitutions, which are highest near the 3′ ends of the reads. The largest problem with short sequence read data is the difficulty in mapping these sequences to the reference genome, especially in the presence of the very sequence polymorphisms that we are attempting to identify. This results in poor coverage of polymorphic and repetitive regions of the genome.
The Illumina flow cells that we used contained 8 channels and generated as many as 5.2 million short-sequence reads per channel (~225 million nt/channel; current technology allows over 18 M reads per channel). Following the manufacturer’s instructions, at least one channel contained a control DNA library made from the bacteriophage φX174 for quality control purposes (this can also be done by mixing a small amount of phage DNA library to the fly library in one lane to increase the efficiency and cost effectiveness). We added DNA from a short-sequence library made from the D. melanogaster strain w1118; iso-2; iso-3 to up to 7 lanes (see Materials and Methods). This was repeated for a total of 3 flow cells of D. melanogaster sequences (~4.7 Gb). An example of the sequencing data is shown in Table 1. The full sequence data is available in Supplementary Tables S4–S15, which is available upon request from Douglas M. Ruden).
Table 1.
Comparison of sequence from BDGP with illumuna data
| X_position | Number | A | C | G | T | Call | Reference |
|---|---|---|---|---|---|---|---|
| 2680000 | 18 | 0 | 1 | 13 | 4 | G | G |
| 2680001 | 16 | 12 | 0 | 0 | 4 | A | A |
| 2680002 | 16 | 0 | 12 | 4 | 0 | C | C |
| 2680003 | 17 | 5 | 12 | 0 | 0 | C | C |
| 2680004 | 17 | 12 | 0 | 5 | 0 | A | A |
X_position, nucleotide position from D. melanogaster sequence (Release 5.3). Number, the number of times this nucleotide position was sequenced. A, C, G, T, the number of times these sequences were obtained. Call, the majority sequence from Illumina data. Reference, the reference genome sequence (y1; cn1 bw1 sp1).
Since the entire D. melanogaster genome is ~180 Mb (euchromatin ~117 million nt), ~4.7 Gb of sequencing data represents a ~26-fold coverage per nt. However, since much of the D. melanogaster genome consists of repeat sequences, which cannot be uniquely aligned against with current short sequencing technologies, and because of the higher error rates in short-sequencing methods, we have only been able to sequence (i.e., unambiguously align) ~90–95% of the euchromatin portion to a depth of ~17-fold (Table 2; see column “reads per base”). Because most of the heterochromatin is not unique, we were only able to align less than 5% of the heterochromatin genome. Consequently, the average reads per base for the heterochromatin sequence ranged from 1.4 for Y heterochromatin (Yhet) to 6.5 for 3L heterochromatin (3Lhet; Table 2). It is important to differentiate between “sequencing” and “aligning.” We likely sequenced the entire nuclear genome to a similar depth (~17-fold), but we were more successful in aligning the euchromatic portion. We chose not to randomly position ambiguous reads as this would impact the sensitivity of SNP identification. The mitochondrial genome was covered ~373-fold per nt, suggesting that there are ~22 mitochondrial genomes per cell (i.e., 373/17 ~22), which is consistent with other studies.18 However, this is from a mixed sex population of whole adult flies, and mitochondria often contain multiple genomes, so this represents a mean of a large number of tissues and the significance is uncertain.
Table 2.
Coverage of BDGP sequence with illumuna data
| Chromosome | % Bases covered | Av reads per base | ||
|---|---|---|---|---|
| Size (nt) | SNPs | (SNP frequency) | ||
| 2L | 23,011,550 | 94.20% | 80,318 | 17.1 (0.35%) |
| 2L Het | 368,900 | 38.90% | 146 | 4.5 (0.04%) |
| 2R | 21,146,750 | 94.10% | 77,147 | 17.2 (0.36%) |
| 2R Het | 3,288,800 | 44.50% | 236 | 5.6 (0.007%) |
| 3L | 24,543,600 | 93.90% | 82,420 | 17.1 (0.34%) |
| 3L Het | 2,555,500 | 53.0% | 114 | 6.5 (0.004%) |
| 3R | 27,905,100 | 96.40% | 72,551 | 17.9 (0.26%) |
| 3R Het | 2,517,550 | 51.20% | 146 | 5.1 (0.006%) |
| 4 | 1,351,900 | 89.76% | 620 | 16.5 (0.05%) |
| X | 22,422,850 | 93.70% | 41,551 | 13.8 (0.19%) |
| Yhet | 347,050 | 43.50% | 9 | 1.4 (0.003%) |
| Mitoch | 19,550 | 90.20% | 27 | 373.2 (0.14%) |
| U | 10,049,050 | 26.4% | 468 | 2.1 (0.005%) |
| Uextra | 29,004,700 | 13.3% | 860 | 1.2 (0.003%) |
| Total | 168,532,850 | 356,614 | ||
| Genome size | ~180,000,000 | |||
Chromosomes are broken down into arms (2L, 2R, 3L, 3R, 4 and X), centric heterochromatin (2L Het, 2R Het, 3R Het, Yhet) and mitochondria (Mitoch), un-annotated (U), un-annotated-extra (Uextra). % Bases Covered, the percentage of each region that has been resequenced. Average (Av) Reads per base, the average number of times each base is read. SNP frequency, is the number of SNPs divided by the size of the chromosome region (SNPs/Size).
A limitation of short sequence alignment is that repetitive sequences are much less likely to uniquely align to the reference genome. This is illustrated in the sequence coverage of the small 4th chromosome, which is only ~1.4 Mb in sizes and contains a large amount of repeats (Fig. 1). Figure 2 shows a region between 690 kb to 710 kb that contains a ~8 kb retrotransposon flanked by long terminal repeats (LTRs). Notice that the region of the repeat has a complete absence of coverage, presumably because these sequences are present hundreds of times in the D. melanogaster genome. Approximately 8% of the annotated 4th chromosome was not covered by short sequences, which is comparable to the 5–10% of the euchromatin portion of the X chromome and autosomes that were not covered (Supplementary Tables S4–S15; see “N’s” in called sequence column, Column G).
Figure 1.
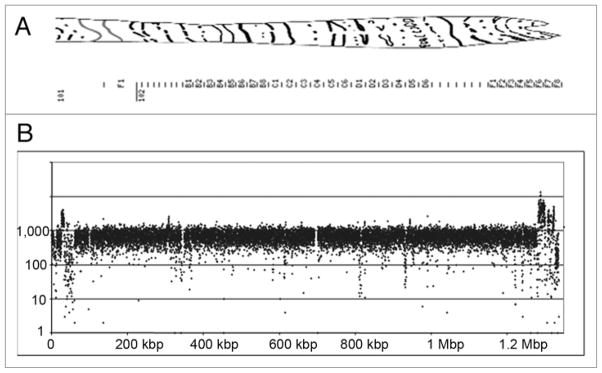
Sequence coverage of 4th Chromosome. (A) Polytene chromosome map of 4th chromosome.38 (B) Coverage of 4th chromosome in 100 nt increments.
Figure 2.

Repetitive Region of 4th Chromosome That is Not Sequenced. (A) Sequence coverage of region from 690 kb to 710 kb on the 4th chromosome in 100 nt increments. (B) Sequence coverage of same region in 100 nt increments. (C) Genome map shows that this region contains a rover retrotransposon flanked by long terminal repeats (LTR). Notice that there is no sequence coverage of the repetitive sequence.
Characterization of the w1118 allele
It has been reported to FlyBase that the w1118 mutation was caused by a small deletion that removes the first exon on the white (w) gene, but the precise breakpoints of the deletion have not yet been mapped (www.Flybase.org). We analyzed the Illumina data around the white gene and found a ~9 kb region that includes the entire first exon of this gene, ~4 kb of the promoter region and most of the first intron, and no other annotated gene (Fig. 3). Further analysis of the borders of the ~9 kb deletion reveals that coverage depth decreases in the region immediately adjacent to the deleted region in the genome (Fig. 4).
Figure 3.

Sequence of region on X chromosome near the white gene. (A) Notice that there is a ~9 kb region of no sequence coverage (double arrow). (B) genome map shows that the first exon of the white gene is deleted in the w1118; iso-2; iso-3 strain.
Figure 4.

Sequence coverage in region immediately flanking the w1118 deletion breakpoints. (A) 5′ end of ~9 kb deletion of the first exon of the white gene (See Fig. 3). Shown is the last sequenced nucleotide with a perfect match (arrow). (B) 3′ end of ~9 kb deletion. Shown is the first sequenced nucleotide with a perfect match (arrow). Notice that the sequence coverage decrease starting ~35 nt from the deficiency breakpoint (see text).
An expected observation is that the coverage depth starts decreasing in a roughly linear manner starting ~36 nt from the deficiency breakpoints (Fig. 4, arrows). The likely reason for the decrease in coverage depth is that short sequences, which are also 36 nt, would contain chimeric sequences if they were less than 35 nt from the deletion breakpoint. These chimeric sequences would not adequately match the reference genome sequence and consequently would not be aligned by the alignment software. We confirmed this by sequencing the junction fragment after PCR amplification (data not shown).
Coverage considerations for SNP identification
In order to determine the optimal coverage required for accurately calling SNPs in our genome sequence data for w1118; iso-2; iso-3, we compared our sequence data with the SNPs previously identified in this strain as part of the original Drosophila genome sequencing project.10 The sequence polymorphisms were identified in this strain by sequencing PCR amplification products from genomic DNA with standard dideoxy sequencing, which is considered the “gold standard” for genome sequencing studies because SNPs are not introduced when directly sequencing PCR products. Hoskins and colleagues aligned each sequence to the Release 1 version of the D. melanogaster reference genome sequence and identified sequence polymorphisms.19 They identified 279 polymorphisms, of which 225 are single nucleotide substitutions. They also identified 17 small insertion/deletions, 8 dinucleotide substitutions and 29 more complex substitutions. We were not able to confirm these more complex nucleotide sequences with the short-sequencing approach presented here.
We compared 20 of the SNPs identified by Hoskins and colleagues with our sequence data and found that 18 of the SNPs are also found in our data (‘Agree Hoskins’, Table 2), but that two of our sequences agree with the Release 5 reference genome sequence and not with the Hoskins’ sequence (‘Agree Genomic’, Table 2; our two sequences also agree with the Release 1 reference genome sequence that Hoskins et al. used). The two sequences in which Hoskin’s data and our data disagree, were sequenced by the GA2 instrument 15 times and 12 times, respectively, with 100% of the reads agreeing with the reference genome sequence. This suggests the unlikely possibility that the 2 SNPs in question were misidentified as SNPs by Hoskins and colleagues (unlikely because Hoskins used the “gold-standard” technology) and correctly identified in our analyses (with an error-prone short-sequencing technology). We believe that a more likely possibility is that Hoskins et al. identified the SNPs correctly, whereas, our short-sequencing technique missed them (i.e., they are “false negatives”; see Discussion). The 18 sequences in which Hoskins’ data and our data agree varied from 6-fold to 18-fold depth and 71% to 100% agreement (Table 3). Interestingly, two of the SNPs that were identified by Hoskins and verified by our data have the genomic sequence as the minor allele in the Illumina data (Table 3, asterisks). Since some of the reference sequence is still apparently present, this might indicate that the SNPs appeared after the isogenization of the w1118 strain, but we think that this is unlikely (see Discussion).
Table 3.
Comparison of SNPs from BDGP with solexa data
| SNP | Call | Location | Chrom | Genome | Hoskins | Solexa | Nreads | A | C | G | T | %call |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DM3247 | Agree Hoskins | 4516312 | X | T | C | C | 14 | 0 | 14 | 0 | 0 | 100% |
| Agree Hoskins | 4516333 | X | C | G | G | 10 | 2 | 0 | 8 | 0 | 80% | |
| Agree Hoskins | 4516339 | X | C | T | T | 8 | 0 | 1 | 1 | 6 | 75%* | |
| Agree Hoskins | 4516471 | X | C | T | T | 8 | 0 | 1 | 0 | 7 | 88%* | |
| DM1954 | Agree Hoskins | 4839202 | X | T | A | A | 16 | 13 | 3 | 0 | 0 | 81% |
| DM1849 | Agree Genomic | 7999994 | X | A | T | A | 15 | 15 | 0 | 0 | 0 | 100% |
| DM3746 | Agree Hoskins | 11850082 | X | A | C | C | 8 | 0 | 6 | 0 | 2 | 75% |
| Agree Hoskins | 11850083 | X | A | G | G | 8 | 0 | 0 | 6 | 2 | 75% | |
| Agree Hoskins | 11850025 | X | T | G | G | 6 | 0 | 0 | 6 | 0 | 100% | |
| DM0478 | Agree Hoskins | 14944274 | X | A | G | G | 11 | 3 | 0 | 8 | 0 | 73%* |
| Agree Hoskins | 14944199 | X | A | C | C | 18 | 0 | 17 | 1 | 0 | 94% | |
| DM3491 | Agree Hoskins | 15468101 | X | T | C | C | 8 | 0 | 8 | 0 | 0 | 100% |
| DM3790 | Agree Hoskins | 16717058 | X | G | A | A | 21 | 21 | 0 | 0 | 0 | 100% |
| DM0509 | Agree Genomic | 17965763 | X | T | G | T | 12 | 0 | 0 | 0 | 12 | 100% |
| DM1846 | Agree Hoskins | 18088860 | X | C | A | A | 15 | 12 | 1 | 0 | 2 | 80%* |
| DM0529 | Agree Hoskins | 21932699 | X | G | T | T | 17 | 1 | 0 | 0 | 16 | 94% |
| DM0375 | Agree Hoskins | 1147996 | 2L | A | G | G | 14 | 1 | 0 | 10 | 3 | 71%* |
| Agree Hoskins | 1148062 | 2L | C | A | A | 15 | 15 | 0 | 0 | 0 | 100% | |
| DM2641 | Agree Hoskins | 2373268 | 2L | G | A | A | 10 | 7 | 3 | 0 | 0 | 70% |
| Agree Hoskins | 2373333 | 2L | A | G | G | 22 | 2 | 0 | 20 | 0 | 91%* |
SNP, SNP from Hoskins et al. (2001).10 Call, agrees with BDGP (Agree Hoskins) or agrees with y1; cn1 bw1 sp1 (Agree Genomic). Location, Drosophila Reference 5.3 genome. Chromosome, X, 2nd or 3rd. Genome, sequence in y1; cn1 bw1 sp1. Hoskins, sequence in.10 Solexa, consensus sequence from our data. Nreads, the number of short sequence reads in our data. A, C, G, T, the number of reads for each of the four bases. % call for base called, the frequency of the Solexa base.
At least one minor allele agrees with the genomic sequence.
While characterizing the short-sequence library for this study, we also identified 10 additional candidate SNPs by sequencing 96 library clones with dideoxy sequencing (data not shown). We found that the average insert size is ~85 nt and that 10 of the 96 sequenced clones contained a putative SNP. Eight of the dideoxy-identified SNPs correspond with the Illumina short sequence data, but two of them were apparently misidentified and agree with the reference genome sequence. This could be caused by errors introduced in the library during construction or amplification. Since the Hoskins’ SNPs were identified by PCR amplification of genomic DNA and dideoxy-sequencing, we believe that they are of greater accuracy than the SNPs in our library, which involved more steps (i.e., subcloning and library amplification). Therefore, based on the SNPs identified by Hoskins and colleagues, we found that at least 6-fold coverage at 70% agreement is sufficient for accurately calling a SNP.
SNPs are non-homogenously distributed in the euchromatin
Using the SNP cut off described in the previous section, i.e., at least 6-fold coverage at 70% agreement, we identified 356,614 SNPs in the w1118; iso-2; iso-3 strain compared with the y1; cn1 bw1 sp1 reference genome sequence. Since the euchromatin size is ~117 Mb, this SNP density corresponds to ~1 SNP/305 nt. Table 4 shows an example of the complete DMEL_SNP table, which is in Supplementary Table S1.
Table 4.
Table of D. melanogaster SNPs
| Chr | Position | Ref. | Seq (%) | # Seq | A | C | G | T |
|---|---|---|---|---|---|---|---|---|
| 2LHet | 20035 | C | G (88.9) | 9 | 0 | 0 | 8 | 1 |
| 2L | 16149 | A | T (88.2) | 17 | 0 | 2 | 0 | 15 |
| 2RHet | 19519 | G | T (85.71) | 14 | 0 | 0 | 2 | 12 |
| 2R | 22744 | C | A (81.25) | 16 | 13 | 0 | 3 | 0 |
| 3LHet | 114047 | G | A (80) | 10 | 8 | 0 | 2 | 0 |
| 3L | 57760 | C | A (81.48) | 27 | 22 | 4 | 1 | 0 |
| 3RHet | 39553 | G | T (77.8) | 9 | 0 | 0 | 2 | 7 |
| 3R | 7971 | A | C (75) | 8 | 2 | 6 | 0 | 0 |
| 4 | 22103 | T | A (75) | 8 | 6 | 0 | 0 | 2 |
| dmel_mit | 1077 | A | G (93.1) | 101 | 3 | 0 | 94 | 4 |
| Uextra | 8625 | G | C (83.3) | 12 | 0 | 10 | 2 | 0 |
| U | 45631 | G | A (75) | 8 | 6 | 0 | 2 | 0 |
| X | 8296 | C | T (88.9) | 36 | 0 | 3 | 1 | 32 |
| YHet | 44048 | T | A (76.5) | 17 | 13 | 0 | 0 | 4 |
This is a partial table of the much larger tables in the online Supplementary materials (Suppl. Tables S4–S15). Chr, chromosome on which the SNP is located. Total Number, the number of SNPs in the indicated chromosome region; Position, sequence location based on D. melanogaster Release 5.3, Ref, the sequence from the reference genome (y1; cn1 bw1 sp1). Seq(%), the percentage of SNP called in the w1118; iso-2; iso-3 sequence. #Seq, the number of times this nucleotide was sequenced. A, C, G, T, the number of times this sequence was called for all 4 bases.
As expected from previous studies,15,20 the SNPs are not homogenously distributed in the euchromatin. The SNPs on the autosome arms (2L, 2R, 3L and 3R) are almost twice as frequent as the SNPs on the X (0.35% versus 0.19%, respectively). Even more dramatic is the finding that the SNP frequency on the small 4th chromosome is only 0.05%, which is 7 times less than on the autosome arms (Table 2). Figure 5 shows the distribution of SNPs along the chromosome arms. The most obvious finding is that SNP frequency decreases near the ends of the chromosomes and near the centric heterochromatin (Fig. 5). There are also several large regions of SNP cold spots in the central regions of the chromosome arms (Fig. 5). The small 4th chromosome has a SNP hotspot, relative to the rest of the chromosome, from ~800–1,000 kb near the right end of the chromosome (Fig. 5). Since the non-sequenced (i.e., unaligned) regions of the 4th chromosome and the euchromatic portions of the X chromosome and autosomes are all within the same range (5–10%), correcting for unaligned sequences would not significantly change the SNP frequency calculations (Suppl. Tables S4–S15; see “N” in the “called sequence” column, Column G).
Figure 5.

SNP distribution in the D. melanogaster genome. (A–F) SNPs/10 kb on the 2nd (2L, 2R), 3rd (3L, 3R), X, and 4th chromosomes arranged in order along the chromosomes. Notice the dips near the ends of the chromosomes.
We also found that there is a non-homogenous distribution of SNPs in genes. Table 5 shows a small portion of the ‘SNPs-in-genes’ list and Supplementary Table S2 shows the entire list. There are a total of 215,808 SNPs in 9,738 genes, which represents 60.5% of the SNPs in the transcribed regions of ~72% of the ~13,500 annotated genes (Table 5). The average SNP-SNP distance in the genes with SNPs is 311 nt, but the median SNP-SNP distance in genes is 773 nt because there is a long tail of extremely long SNP-SNP distances (Fig. 6).
Table 5.
Table of D. melanogaster SNPs in genes
| Gene name | Gene beginning | Gene end | #SNPs | Distance between SNPs (nt) | Amino acid change | SNP type | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Obp57d | 16436127 | 16436603 | 9 | 52.88889 | |||||||
| SNP | Chr arm | SNP loc | Ref | Seq(%) | #Seq | A | C | G | T | ||
| SNP + 147 | 2R | 16436274 | C | T (76.5) | K > K | Syn | 17 | 1 | 3 | 0 | 13 |
| SNP + 152 | 2R | 16436279 | C | T (100) | D > N | NS | 17 | 0 | 0 | 0 | 17 |
| SNP + 173 | 2R | 16436300 | C | T (76.9) | A > T | NS | 13 | 0 | 3 | 0 | 10 |
| SNP + 201 | 2R | 16436328 | A | G (88.9) | C > C | Syn | 18 | 1 | 0 | 16 | 1 |
| SNP + 238 | 2R | 16436365 | G | A (78.9) | T > I | NS | 19 | 15 | 4 | 0 | 0 |
| SNP + 361 | 2R | 16436488 | A | G (73.7) | IVS | IVS | 19 | 2 | 0 | 14 | 3 |
| SNP + 408 | 2R | 16436535 | T | C (81.8) | Q > Q | SYN | 22 | 2 | 18 | 1 | 1 |
| SNP + 449 | 2R | 16436576 | C | T (94.1) | V > I | NS | 17 | 0 | 1 | 0 | 16 |
| SNP + 450 | 2R | 16436577 | A | G (100) | L > L | Syn | 19 | 0 | 0 | 19 | 0 |
This is a partial table of the larger table in the online Supplementary materials (Suppl. Table S2). The first two rows show the gene name, the beginning and end of the gene (Release 5.3), the number of SNPs in the gene, the average distance between SNPs (nt), the amino acid change (e.g., K > K is lysine (reference genome) to lysine (w1118)), the SNP type is synonymous (Syn) or non-synonymous (NS), intron (IVS). Not shown are 5′UTR and 3′UTR, which are also included in the “gene beginning” and “gene end” region.
Figure 6.

SNP-SNP Distances. (A) Distance between SNPs (nt) versus the number of genes. The first bar represents the number of genes with an average SNP-SNP distance between 33 nt and 100 nt (0 nt < SNP-SNp < 100 nt), the second bar represents 100 nt < SNP-SNp < 200 nt, etc. (B) Genes with SNP-SNP distances between 4 kb and 100 kb. The first bar represents 4 kb < SNP-SNp < 5 kb, etc.
Gene Ontology (GO) analyses indicate that certain categories of genes are enriched in the genes with SNP hotspots and cold spots. GO enrichment was determined with the program DAVID (Database for Annotation, Visualization and Integrated Discovery).21,22 Among the genes with SNPs, there are 441 genes with SNP-SNP distances greater than 25 kb (i.e., ‘SNP cold-spots’) and 436 genes with a SNP-SNP distance of <100 nt (i.e., ‘SNP hotspots’). The genes with SNP coldspots are 4.84-fold enriched in the GO category ‘transcription regulation’ (FDR = 1.3E-08) and 3.64-fold enriched in ‘developmental protein’ (FDR = 2.1E-06; Table 6). The genes with SNP hotspots are 6.0-fold enriched in the category “sensory perception of chemical perception” (FDR = 3.5E-07; Table 6).
Table 6.
GO enrichment of genes with SNPs
| Description | GO term | Count | % | Genes | Fold enrichment | FDR |
|---|---|---|---|---|---|---|
| Genes with SNP-SNP distance >25 kb (441 genes) | Transcription regulation | 29 | 6.68% | cnc, grn, Poxm, Hira, Rga, sv, pan, zfh2, lolal, topi, pho, Hnf4, ewg, ab, pros, stc, spen, Topors, salm, fkh, cic, ash1, cas, Hr4, dsx, MED25, su(w[a]), H, ct, | 4.84 | 1.3E-08 |
| Genes with SNP-SNP distance >25 kb (441 genes) | Developmental protein | 31 | 7.14% | cnc, dpld, Poxm, l(2)gl, DIP2, ph-p, sv, pcx, Appl, Crk, pan, rno, lolal, topi, pho, hdc, ab, pros, spen, d, jar, salm, fkh, cic, ash1, cas, fog, sdk, H, ct | 3.64 | 2.1E-06 |
| Genes with SNP-SNP distance <100 nt (436 genes) | sensory perception of chemical stimulus | 21 | 5.02% | Gr23a, Gr58b, Or67c, Or67d, Or30a, Gr22c, Gr93c, Gr64f, Gr59f, Or67a, Gr61a, Gr64a, Gr64b, Or92a, Gr22a, Obp58c*, Or46a, Or49b, Gr92a, Obp57d*, Gr93d* | 6.0 | 3.5E-07 |
GO analyses were done with 441 genes with the largest SNP-SNP distance (Genes with SNP-SNP distance >25 kb) and with 436 genes with the smallest SNP-SNP distance (Genes with SNP-SNP distance <100 nt). GO enrichment was determined with the program DAVID (Database for Annotation, Visualization and Integrated Discovery) which is available free online (http://david.abcc.ncifcrf.gov/home.jsp).21,22
These are the only genes in this group that are not G-protein coupled receptors (Gpcrs).
Further examination of genes in the SNP coldspots and hotspots suggests that they are surrounded by genes with similar SNP distributions. The two sensory perception genes Gr23a and Or67c have over 10 SNPs per kb, and several surrounding genes in unrelated gene families have similarly high SNP frequencies (Fig. 7A and B). The two developmental genes cnc and rno have fewer than 1 SNP/25 kb, and they are surrounded by genes with similar SNP deficits (Fig. 7C and D). The gene rno is near the 2R telomere, and it is know that telomeres are recombination and SNP coldspots.10 As an example of a gene in a SNP hotspot, the sensory perception gene Obp57d has a 759 nt transcript and 9 SNPs (which corresponds to ~11.9 SNPs/kb). We show in Figure 8 that 4 SNPs are synonymous mutations (i.e., they do not change the amino acid sequence), 4 SNPs are non-synonymous mutations (i.e., they change the amino acid), and one SNP is in the short intron.
Figure 7.
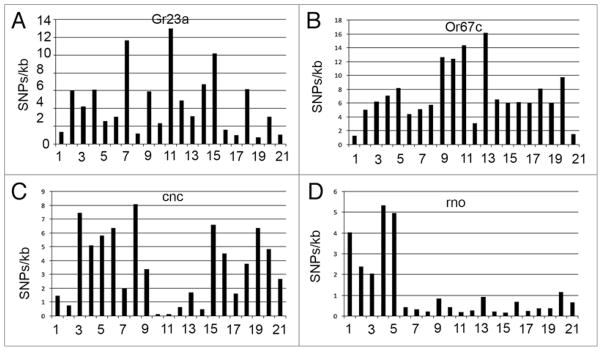
SNP distribution near SNP hotspot and coldspot genes. (A and B) SNP hotspot genes Gr23a and Or67c, which are both involved in sensory transduction, and 10 genes on either side are shown. Notice the peak near the hotspot genes. (C and D) SNP coldspot genes cnc and rno, which are both developmental genes, and 10 genes on either side are shown. Notice the trough near the coldspot genes.
Figure 8.

SNPs in the gene Obp57d and the number of SNPs per probeset. (A) Location of 9 SNPs in Obp57d. Synonymous mutations do not change the amino acid (L, Q, C, K) and non-synonymous mutations are indicated with an asterisk (I*, I*, T*, N*). There is also a t > c SNP in the intron (lower case letters).
SNPs in affymetrix probes
The Mackay laboratory in an extensive collaboration with members of the Nutrition and Genomics Laboratory at JM-USDA HNRCA (Jene Mayer-USDA Human Nutrition Research Center on Aging) at Tufts University, recently developed a technique that they call “Speed-Mapping” quantitative trait loci (QTL) that genotypes F2 progeny made by crossing the isogenic strains Oregon R to Russian 2b and then mass-crossing the F1 progeny (F1xF1). They genotyped the surviving F2 flies in a longevity experiment after ~90% of the flies died of old age. The genotyping was done en masse and was based on hybridization the bulk-purified DNA to an Affymetrix Dros2 gene expression array.23 Speed-mapping is based on the changes of gene frequency of 2,326 single-feature polymorphisms (SFPs) to identify QTLs affecting lifespan.23 The SFPs were identified, not based on direct sequence data, but rather indirectly by looking for outliers in probesets that do not correspond to the other probes in the probeset.
We thought that we could improve the Speed-Mapping technique by utilizing the SNPs that we identified in this study. Each probeset, which was designed to measure the relative steady state RNA levels for each of the ~13,500 D. melanogaster genes, has ~12 “perfect-match” and ~12 “mismatch” probes. The probes are 25mers and mismatch probe has a transversion (G > C, C > G, A > T or T > A) in the central (12th) position. In the Dros2 array, there are approximately ~225,000 match and ~225,000 mismatch probes for a total of ~450,000 probes at ~225,000 locations.
When we compared the ~225,000 perfect-match probe sequences, which were based on the reference genome sequence, with the w1118; iso-2; iso-3 sequence, we identified 12,685 SNPs in the perfect-match probe sequences (i.e., ~5.6% of the perfect match probes) in 7,265 genes (i.e., ~54% of the genes). The majority of the genes (4,000) have a single probe with a SNP, but 1,872 have two probes with SNPs and 14 have 7 probes with SNPs (Fig. 8). This is especially remarkable because the Affymetrix Dros2 array has non-overlapping probes in each probeset. Interestingly, 84 of the probes have a SNP that causes the mismatch probe to become a perfect match probe (i.e., it is a transversion at the 12th position). A short version of the Affymetrix SNP table is shown in Table 7 and the complete data is in Supplementary Table S3.
Table 7.
Table of D. melanogaster SNPs in affymetrix probes
| Affy_Probe | Chr | 5′ end | 3′ end | SNP loc | SNP pos | #seq | Ref | Seq(%) | A | C | G | T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1627446_at_1899 | 2L | 82730 | 82754 | 82745 | 15 | 20 | C | T(80) | 4 | 0 | 0 | 16 |
| 1634783_s_at_213 | 2L | 155674 | 155698 | 155691 | 17 | 15 | C | G(86.7) | 0 | 1 | 13 | 1 |
| 1625452_at_2387 | 2L | 264290 | 264314 | 264313 | 23 | 15 | T | G(100) | 0 | 0 | 15 | 0 |
| 1630529_at_619 | 2L | 293128 | 293152 | 293132 | 4 | 24 | C | A(100) | 24 | 0 | 0 | 0 |
This is a partial table of the much larger table in the online Supplementary materials (Suppl. Table S3). These are SNPs based on the probes in the Dros2 array from Affymetrix. Affy_Probe, Affymetrix probe ID; Chr, chromosome arm where SNP is located. 5′ end and 3′ end, the location of the ends of the probe. SNP loc, the location of the SNP in the Drosophila Reference 5.3 genome. SNP pos, the position in the 23mer probe that has the SNP. #seq, the number of times the nucleotide was sequenced. A, C, G, T, the number of times this position was called for the four bases.
Discussion
We have sequenced the D. melanogaster strain w1118; iso-2; iso-3 and have identified 356,614 candidate SNPs using the short sequence Illumina platform. This Illumina re-sequencing technique was also used to identify SNPs in C. elegans,14 in an Asian man,24 and in an African man.25 Recently, the Hobert laboratory re-sequenced the entire genome of C. elegans as a quick method to identify a mutation that produces a hard-to-map phenotype.26 We have characterize the w1118 deletion and thereby show that whole-genome sequencing is also a viable method for characterizing mutations in Drosophila.
What is the best way to re-sequence an organism? The Illumina platform currently produces reads of over 100 nt, and this is likely to be dramatically improved in the near future. The primary errors in these short sequences are substitutions, which are highest near the 3′ ends of the reads. The largest problem with short sequence read data is the difficulty in mapping these sequences to the reference genome, especially in the presence of the very sequence polymorphisms that we are attempting to identify. This results in poor coverage of polymorphic and repetitive regions of the genome. Fortunately, Illumina has developed dual-end read technology that allows short sequences to be read from both ends of the library clones.25 However, due to the short insert size in our D. melanogaster library (~85 nt), we were not able to fully take advantage of the paired-end technology in this study. Also, the software to reliably characterize short INDELs using short read sequencing is still in the early evaluation stages and cannot be considered reliable. Therefore, the raw sequence data has been archived in the NCBI’s trace archive for future analysis. A combination of short sequencing and long sequencing technologies, such as whole-genome pyrosequencing,27 are required to optimally obtain the INDEL and repeat sequences in a whole genome.
What are the false positive and false negative error rates with short sequencing technology? Two of the 20 SNPs identified by Hoskins and colleagues10 by dideoxy-sequencing (the gold standard) surprisingly had the reference strain sequences in our analyses. One possibility is that the isogenic w1118 sub-strain that was sequenced by Hoskins and colleagues had picked up new mutations, whereas our sub-strain had the reference genome sequence. We do not think that this is likely because the 2008 Whitepaper by Mackay and colleagues found only a few hundred SNPs after re-sequencing the reference strain (www.flybase.org), and it is unlikely that Hoskins and colleagues identified two such SNPs from a small number of random sequences. We note that the the isogenic w1118 sub-strain that we sequenced was the same sub-strain used by Hoskins (http://flystocks.bio.indiana.edu/Reports/5905.html). Another possibility, which we think is more likely, is that several of our short-read sequences, which have a high error frequency, aligned to the reference genome sequence by chance because perfect sequences (which were actually sequencing artifacts) are more likely aligned with the MAQ 2.1 software. If this were the case, the Hoskin SNPs are in fact correct and, therefore, “false negatives” in our analysis (i.e., our analysis might have a false negative rate as high as ~10%). The two SNPs in question can be re-examined by dideoxy sequencing after PCR amplification, but this is beyond the scope of this project. Instead, we are in the process of designing custom SNP chips to validate the SNPs that we observe in this study (Douglas M. Ruden, in progress).
Re-sequencing D. melanogaster yields an unequalled resource of genetic information. Quantitative trait nucleotide (QTN) analyses have been done in Drosophila, but they require a massive amount of sequencing analyses of dozens or hundreds of D. melanogaster strains.9 Trudy Mackay and colleagues recently wrote a 2008 Drosophila White Paper, that proposes sequencing 192 D. melanogaster strains from a single natural population from the Raleigh-Durham area of North Carolina that have been inbred to homozygosity. Several laboratories have conducted extensive complex trait analyses with these strains (www.FlyBase.org). According to the 2008 Drosophila White Paper, this database will create: (1) A Drosophila community resource for association mapping of QTL; (2) a Drosophila community resource of SNPs, which will be valuable for QTL mapping, population genetic analyses, and allele specific expression (ASE) studies; and (3) a “test bench” for statistical methods used in QTL association mapping in all organisms, including humans. Since the D. melanogaster genome size is ~5% that of the human genome size, the Drosophila genome sequences can be analyzed with much greater power and lower costs than human sequences.
There are practical and immediate uses of our SNP analyses. For example, one could more efficiently perform genotyping experiments without any additional sequencing, such as by using existing oligonucleotide microarrays or custom arrays. We identified 12,685 SNPs in the perfect-match probe sequences (i.e., ~5.6% of the perfect match probes) in 7,265 genes (i.e., ~54% of the genes) in the Dros2 gene expression array from Affymentrix, Inc. If one were to have mutations generated in on the two sequenced D. melanogaster strains, then one could relatively easily map the gene to the nearest SNP, such as by the method described in the Bellen laboratory.28
An expected outcome of this study was the finding that SNPs do not have a uniform distribution along the euchromatin. We found a ~2-fold higher rate of SNPs on the autosomes compared with the X chromosome and a ~7-fold higher rate of SNPs compared with the small ‘dot’ 4th chromosome (although this might partly reflect a slightly lower coverage of sequences on the 4th). The hemizygosity of the X chromosome in males might help explain the deficiency of SNPs on this chromosome because hemizygosity results in more effective selection against deleterious mutations,16 whereas, beneficial mutations are more readily fixed by selection on the X.29 The lack of recombination on the 4th chromosome and lower recombination frequencies in centromeric and telomeric regions might help explain the deficiency of SNPs in these regions because a similar inverse correlation between recombination rate and genetic divergence has previously been identified in D. melanogaster,15,16 and several years later in humans.30 Perhaps an error-prone DNA recombination repair process is the cause of this correlation.31 The relative hotspot for SNPs on the 4th chromosome might indicate a higher than expected recombination frequency in a restricted region of this chromosome.
For evolutionary studies, one should be cautious that the polymorphisms identified so far are between two isogenic laboratory strains. Thus, the rate of polymorphism identified here only reflects the density of SNPs between two these two isogenic lines. These findings may not reflect the mutation rate/polymorphisms of D. melanogaster in the natural population, nor should one necessarily apply our findings to general laboratory stocks.
With these caveats in mind, the most intriguing observation in this study is that certain genes, especially those involved in sensory perception such as certain G protein coupled receptors, are enriched in SNP hotspots. Genes involved in sensory perception have been found to have very high ‘positive selection’ rates in humans, chimps and mice.32–35 Other genes with high rates of positive selection in humans involve immune-response genes and sperm-function genes. We see a 12.5-fold enrichment in immune-response genes in the 436 genes in the largest SNP hotspots (Dro, IM2, IM1, AttC, IM4 and IM23), but this does not have a significant FDR (FDR~0.1). In contrast to what is observed when comparing humans and chimpanzees,33 the ‘SNP hotspots’ we see in D. melanogaster have generally not undergone ‘positive selection’ because ‘SNP hotspots’ have dN/dS ~ 1, whereas, ‘positive selection’ involves regions that have an excess of nonsynonymous mutations compared to synonymous mutations (dN/dS > 1).36 A complete analysis of synonymous and nonsynonymous SNPs is beyond the scope of this paper and will be presented elsewhere.
Previous experiments and standard evolutionary models that show a correlation between recombination frequency and mutation rate might explain the non-homogenous SNP distribution that we observe.10,15,20 Alternatively, an undefined hyper-mutation mechanism (such as via recombination hotspots) might account for SNP hotspots in sensory perception genes. For example, there might be an epigenetic mechanism that increases the random mutation frequency (via increasing recombination frequency) in evolutionarily-important genes, such as we propose in the Epigenetic Directed Genetic Error (EDGE) hypothesis.37 Mutation hotspots might also explain why there are similar numbers of nonsynonymous and synonymous SNPs in sensory perception genes from individual humans (i.e., dN/dS ~ 1), but, as mentioned above, dN/dS ≫ 1 for many of the same genes when they are compared between humans and chimpanzees.33 In other words, a hyper-mutated region would initially have dN/dS ~ 1 within a species, but after a long period of positive selection, this could change to dN/dS ≫ 1 for the same genes in closely related species. This is presumably because there are powerful selective pressures for the new genotypes that were better suited to the novel environments.
One possibility, which we cannot rule out in this study, is that the high rate of SNPs in sensory perception genes that we report here is an artifact caused by mis-aligning the error-prone short-sequencing reads to the incorrect members of the gene families. This can be addressed by longer sequencing reads, paired-end reads, and by custom oligonucleotide arrays (SNP-chips). However, we do not believe that this is the case because the regions of elevated mutation rates encompass not only sensory perception genes but also neighboring genes in unrelated families (Fig. 7). Perhaps a higher recombination rate (and therefore higher mutation rate) is tolerated near a gene that is plastic in evolution, such as a sensory perception gene.
In conclusion, we have provided proof-of-concept that Illumina short-read sequencing can identify hundreds of thousands of SNPs in D. melanogaster at a cost and speed that is a small fraction of that of the first genome sequence of this organism. By characterizing the w1118 deletion, we also show that this technique can also be used for rapid characterization of the molecular basis of a mutant strain. By making recombinant inbred lines or F2 hybrid lines between the two sequenced D. melanogaster strains, our data will facilitate the mapping of QTLs or to do traditional mapping of mutant alleles that were generated in one of the two sequenced strains. Our SNP analyses will enable the entire Drosophila community to more efficiently perform genotyping experiments without any additional sequencing, such as by using existing oligonucleotide microarrays or custom arrays. Finally, evolutionary implications are implied by the non-homogenous distribution of SNPs in highly polymorphic genes involved in sensory perception.
Methods
Preparing genomic DNA library for paired-end sequencing
Drosophila genomic DNA from the strain w1118; iso-2; iso-3 was prepared using an AutoPure LS (Qiagen). A genomic DNA library was prepared from 5 μg purified Drosophila DNA according to the standard protocol using a Paired-End Sample Prep Kit for the GA2 (Illumina). The library was validated by cloning an aliquot into pCR4Blunt-TOPO using Zero Blunt TOPO PCR Cloning Kit for Sequencing (Invitrogen). Single colonies were selected and amplified by PCR, purified, sequenced with dideoxy terminator sequencing (AB 3730), and verified (Sequencher, BLAST) for adapters and inserts. The DNA library was then used for cluster generation and sequencing analysis using the Genome Analyzer II using Illumina standard protocols.
Alignment of short DNA sequence reads
Methods for DNA manipulation, including sample preparation, formation of single-molecule arrays, cluster growth and sequencing were all done by following the standard protocols from Illumina, Inc. All sequencing was performed on a 2008 Illumina Genome Aanalyzer version 2 (GA2) that was equipped with a one-megapixel camera. All purity filtered read data are available for download from the Short Read Archive at NCBI. In total 7 lanes were run in paired-end sequencing mode and 18 lanes run in single end sequencing mode to generate the short sequence archive for alignment.
Analysis software
Image analysis software and the ELAND aligner were provided as part of the Genome Analyzer analysis pipeline and configured for fully automatic parameter selection. For paired end data ELAND was run in ELAND_PAIR mode and for single-end data ELAND_EXTENDED was used. Paired end reads were 36 × 2 bases in total length and single end reads 36 bases in length. Short reads from the GA2 are highly reliable but error rates increase markedly after the 25th base. Base masking to select only the most reliable read data was determined on a lane by lane basis relative to the reported gross alignment error rate. Where the gross alignment errors were high in the terminal base reads these bases were masked from the analysis. Base masking ranged from no bases masked (36 nucleotide reads) to the masking of the terminal eleven nucleotides of each read (25 nucleotide reads). In order to allow SNP detection some difference between the reference genome and aligned sequence must be allowed. ELAND U0, U1 and U2 alignments were accepted, permitting read alignment to a single unique location with at most 2 base mismatches between the read and reference genome. The analyzed reads were aligned to the D. melanogaster reference genome (Release 5 finished genomic sequence with the R5.3 FlyBase gene annotation) using ELAND and cross validated by realigning with the open source short read alignment tool MAQ 2.1 (http://maq.sourceforge.net/maq-man.shtml). Aggregation of short aligned reads with the reference assembly was achieved with a custom VB application to report for each base in the reference genome the distribution of bases generated from aligned reads.
SNP calls were made where the distribution of bases in the short read data was found to lie counter to that in the reference genome. Thresholds for SNP calling were determined relative to published capillary sequencing data. The set of candidate SNPs were mapped to Affymetrix Drosophila probe locations annotated at Flybase.org (ftp://ftp.flybase.net/genomes/dmel/dmel_r5.12_FB2008_09/gff/) and to the NCBI’s map of RefSeq and candidate Drosophila genes (ftp://ftp.ncbi.nih.gov/genomes/Drosophila_melanogaster/mapview/seq_gene.md.gz).
Data access
Sequence data are freely available from the NCBI Trace archive: http://www.ncbi.nlm.nih.gov/Traces/home/ (accession ID pending).
Supplementary Material
Acknowledgments
This work was supported by a Michigan Core Technology grant from the State of Michigan’s 21st Century Fund Program to the Wayne State University Applied Genomics Technology Center. This work was also supported by the Environmental Health Sciences Center in Molecular and Cellular Toxicology with Human Applications Grant P30 ES06639 at Wayne State University, NIH R01 grants (ES012933 and CA105349) to Douglas M. Ruden, and DK071073 to Xiangyi Lu.
Footnotes
Note
Supplementary Tables 1–3 can be found at:
www.landesbioscience.com/supplement/PlattsFLY3-3-Sup01.pdf
www.landesbioscience.com/supplement/PlattsFLY3-3-Sup02.pdf
www.landesbioscience.com/supplement/PlattsFLY3-3-Sup03.pdf
Supplementary Tables 4–17 are available upon request from the corresponding author.
References
- 1.International Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 2.Internaltional HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 4.Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–95. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 5.Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 6.Ruden DM. Personalized medicine and quantitative trait transcripts. Nat Genet. 2007;39:144–5. doi: 10.1038/ng0207-144. [DOI] [PubMed] [Google Scholar]
- 7.Lee SS, Mudaliar A. Medicine. Racing forward: the Genomics and Personalized Medicine Act. Science. 2009;323:342. doi: 10.1126/science.1165768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashburner M. Drosophila: A laboratory Handbook. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 10.Rubin GM, Lewis EB. A brief history of Drosophila’s contributions to genome research. Science. 2000;287:2216–8. doi: 10.1126/science.287.5461.2216. [DOI] [PubMed] [Google Scholar]
- 11.De Luca M, Roshina NV, Geiger-Thornsberry GL, Lyman RF, Pasyukova EG, Mackay TF. Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat Genet. 2003;34:429–33. doi: 10.1038/ng1218. [DOI] [PubMed] [Google Scholar]
- 12.Hoskins RA, Phan AC, Naeemuddin M, Mapa FA, Ruddy DA, Ryan JJ, et al. Single nucleotide polymorphism markers for genetic mapping in Drosophila melanogaster. Genome Res. 2001;11:1100–13. doi: 10.1101/gr.178001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thibault ST, Singer MA, Miyazaki WY, Milash B, Dompe NA, Singh CM, et al. A complementary transposon tool kit for Drosophila melanogaster using P and piggyBac. Nat Genet. 2004;36:283–7. doi: 10.1038/ng1314. [DOI] [PubMed] [Google Scholar]
- 14.Parks AL, Cook KR, Belvin M, Dompe NA, Fawcett R, Huppert K, et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat Genet. 2004;36:288–92. doi: 10.1038/ng1312. [DOI] [PubMed] [Google Scholar]
- 15.Ryder E, Ashburner M, Bautista-Llacer R, Drummond J, Webster J, Johnson G, et al. The DrosDel deletion collection: a Drosophila genomewide chromosomal deficiency resource. Genetics. 2007;177:615–29. doi: 10.1534/genetics.107.076216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5:183–8. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- 17.Begun DJ, Aquadro CF. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature. 1992;356:519–20. doi: 10.1038/356519a0. [DOI] [PubMed] [Google Scholar]
- 18.Charlesworth B, Coyne JA, Barton NH. The relative rates of evolution of sex chromosomes and autosomes. Am Naturalist. 1987;130:113–46. [Google Scholar]
- 19.Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–18. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Moreno MA, Farr CL, Kaguni LS, Garesse R. Drosophila melanogaster as a model system to study mitochondrial biology. Methods Mol Biol. 2007;372:33–49. doi: 10.1007/978-1-59745-365-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams JC. Characterization of a Drosophila melanogaster orthologue of muskelin. Gene. 2002;297:69–78. doi: 10.1016/s0378-1119(02)00887-9. [DOI] [PubMed] [Google Scholar]
- 22.Charlesworth B. Background selection and patterns of genetic diversity in Drosophila melanogaster. Genet Res. 1996;68:131–49. doi: 10.1017/s0016672300034029. [DOI] [PubMed] [Google Scholar]
- 23.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 24.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai CQ, Leips J, Zou W, Roberts JF, Wollenberg KR, Parnell LD, et al. Speed-mapping quantitative trait loci using microarrays. Nat Methods. 2007;4:839–41. doi: 10.1038/nmeth1084. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–5. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarin S, Prabhu S, O’Meara MM, Pe’er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods. 2008;5:865–7. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satkoski JA, Malhi R, Kanthaswamy S, Tito R, Malladi V, Smith D. Pyrosequencing as a method for SNP identification in the rhesus macaque (Macaca mulatta) BMC Genomics. 2008;9:256. doi: 10.1186/1471-2164-9-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci USA. 2003;100:10860–5. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torgerson DG, Singh RS. Sex-linked mammalian sperm proteins evolve faster than autosomal ones. Mol Biol Evol. 2003;20:1705–9. doi: 10.1093/molbev/msg193. [DOI] [PubMed] [Google Scholar]
- 32.Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet. 2003;72:1527–35. doi: 10.1086/375657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frankenberg-Schwager M, Becker M, Garg I, Pralle E, Wolf H, Frankenberg D. The role of nonhomologous DNA end joining, conservative homologous recombination, and single-strand annealing in the cell cycle-dependent repair of DNA double-strand breaks induced by H(2)O(2) in mammalian cells. Radiation research. 2008;170:784–93. doi: 10.1667/RR1375.1. [DOI] [PubMed] [Google Scholar]
- 34.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilad Y, Segre D, Skorecki K, Nachman MW, Lancet D, Sharon D. Dichotomy of single-nucleotide polymorphism haplotypes in olfactory receptor genes and pseudogenes. Nat Genet. 2000;26:221–4. doi: 10.1038/79957. [DOI] [PubMed] [Google Scholar]
- 37.Clark AG, Glanowski S, Nielsen R, Thomas PD, Kejariwal A, Todd MA, et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science. 2003;302:1960–3. doi: 10.1126/science.1088821. [DOI] [PubMed] [Google Scholar]
- 38.Hughes AL, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–70. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- 39.Ruden DM, Jamison DC, Zeeberg BR, Garfinkel MD, Weinstein JN, Rasouli P, Lu X. The EDGE hypothesis: Epigenetically directed genetic errors in repeat-containing proteins (RCPs) involved in evolution, neuroendocrine signaling, and cancer. Frontiers in neuroendocrinology. 2008;29:428–44. doi: 10.1016/j.yfrne.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindsley DL, Zimm G. The Genome of Drosophila melanogaster. San Diego: Academic Press; 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.