

Abstract
We describe an approach for engineering peptide–lipid nanoparticles that function similarly to high-density lipoprotein (HDL). Branched, multivalent constructs, bearing multiple 23- or 16-amino-acid peptides, were designed, synthesized and combined with phospholipids to produce nanometer-scale discoidal HDL-like particles. A variety of biophysical techniques were employed to characterize the constructs, including size exclusion chromatography, analytical ultracentrifuge sedimentation, circular dichroism, transmission electron microscopy, and fluorescence spectroscopy. The nanoparticles functioned in vitro (human and mouse plasma) and in vivo (mice) to rapidly remodel large native HDLs into small lipid-poor HDL particles, which are key acceptors of cholesterol in reverse cholesterol transport. Fluorescent labeling studies showed that the constituents of the nanoparticles readily distributed into native HDLs, such that the peptide constructs coexisted with apolipoprotein A-I, the main structural protein in HDLs. Importantly, nanolipid particles containing multivalent peptides promoted efficient cellular cholesterol efflux and were functionally superior to those derived from monomeric peptides. The multivalent peptide-lipid nanoparticles were also remarkably stable toward enzymatic digestion in vitro and displayed long half-lives and desirable pharmacokinetic profiles in mice, providing a real practical advantage over previously studied linear or tandem helical peptides. Encouragingly, a two-week exploratory efficacy study in a widely used animal model for atherosclerosis research (LDLr-null mice) using nanoparticles constructed from a trimeric peptide demonstrated an exceptional 50% reduction in the plasma total cholesterol levels compared to the control group. Altogether, the studies reported here point to an attractive avenue for designing synthetic, HDL-like nanoparticles, with potential for treating atherosclerosis.
INTRODUCTION
An important goal in advancing nanomedicine is to devise novel strategies that allow the fabrication of robust, well-defined nanoparticles with useful functional properties. Lipoprotein nanoparticles, as complexes between phospholipids and apolipoproteins, can effectively serve in his context. Indeed, synthetic lipid nanoparticles of tunable composition, size, and morphology would be of considerable interest for understanding the molecular basis of protein–lipid interactions and for developing new therapeutic agents.1
High-density lipoprotein (HDL) is a structurally and functionally fascinating class of lipoproteins that encompass nanoparticles with distinct sizes (6–13 nm) and shapes (discoidal and spherical).2 HDL plays a crucial role in protecting against cardiovascular disease through a number of mechanisms that depend on the particular particle composition. The small, dense, discoidal particles are especially important in absorbing and transporting cholesterol. The antiatherogenic effects of HDL derive in part from its involvement in reverse cholesterol transport (RCT), whereby excess cholesterol and other lipids are translocated from peripheral tissues to the liver for elimination (Figure 1).3 A major goal in developing HDL-targeted therapeutics is to enhance RCT by promoting cellular cholesterol efflux and by remodeling HDL to increase the levels of lipid-poor species, which have the greatest capacity to absorb cellular cholesterol. In this vein, the functional mimicry of HDLs with synthetic lipid nanoparticles has garnered intense interest as a route to potential agents for managing atherosclerosis.4
Figure 1.
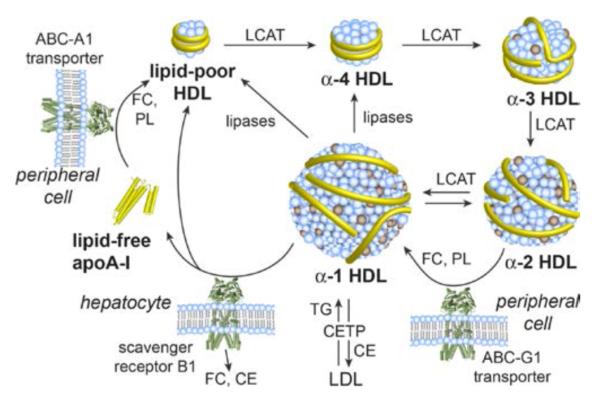
Major pathways in the formation and dynamic remodeling of HDL, a heterogeneous mixture of interconverting particles that undergo constant remodeling mediated by various transporters, receptors, and enzymes. The process by which HDL removes cholesterol from peripheral tissues and transport it to the liver for elimination is known as reverse cholesterol transport (RCT). ApoA-I is shown as yellow tubes. Abbreviations: ABC, ATP-binding cassette transporter protein; CE, cholesterol ester; CETP, cholesteryl ester transfer protein; FC, free cholesterol; LCAT, lecithin–cholesterol acyltransferase; LDL, low-density lipoprotein; PL, phospholipid; TG, triglyceride.
Apolipoprotein A-I (apoA-I), the primary structural protein within HDL particles, has been mimicked by many short, amphiphilic, α-helical peptides.5 However, some of the structural roles of apoA-I in supporting nanolipid particles may not be fully replicated by monomeric peptides, as apoA-I contains 10 amphiphilic α-helices. For example, apoA-I adapts to various particle sizes and morphologies during the course of HDL maturation from small, cholesterol-poor discoidal particles to larger, cholesterol-rich spherical particles, and plays a central role in the remodeling of HDL (Figure 1). This remodeling is a constant dynamic process in which proteins and enzymes mediate the influx, efflux, or modification of constituent lipids, cholesterol, and small-molecule components, greatly impacting HDL particle composition and function.2 Thus, we supposed that multimeric α-helical constructs (linear or branched), by virtue of their multiple interacting helices, would afford improved scaffolding and conformational adaptability to support the HDL remodeling process. Although previous studies of helix multivalency have employed linear truncation or deletion variants of apoA-I,6 and linear or branched synthetic constructs,7 the role of multivalency in apoA-I function has not been systematically investigated. As a design perspective, we planned to use branched peptide constructs with different numbers of attached α-helices, which would present a distinctly different topology from the linear arrangement of α-helices in the native protein (Figure 1). These novel, branched constructs might supply the morphological plasticity to support the range of HDL particle sizes and shapes to advance the chemical biology of HDL and serve as potential therapeutic agents. Our multivalent approach included the use of two different lengths of peptide building blocks, 23-mers and 16-mers, to examine the functional effects of helix multimerization more thoroughly. Herein, we report on the remarkable properties of these multivalent peptide constructs in generating functional, synthetic, HDL-like nanolipid particles.
RESULTS
Design and Synthesis of Multivalent Constructs
We endeavored to systematically assess the potential of multivalent peptide constructs in generating functional nanolipid particles. A convergent strategy involving two distinct families of monomer, dimer, trimer, and tetramer peptide constructs with different lengths for the amphiphilic α-helical peptide segments was adopted (Figure 2). For synthetic efficiency we utilized native chemical ligation (NCL)8 to attach multiple copies of the selected peptide to simple small-molecule scaffolds that differed in the number of reactive arms (Figure 3).9 The scaffolds were designed to be flexible and uncharged (Figure 3b), so as not to unduly influence the biophysical properties of the constructs, and to be synthesized in a few steps from commercial starting materials (Figure S1).
Figure 2.

Multivalent peptide constructs used in preparing HDL-like peptide-lipid nanoparticles. (a) Schematic illustrations of the multivalent peptide constructs, which contain 2–4 copies of amphiphilic helical peptide ligated at the peptide N-terminus to small scaffolds via native chemical ligation. This study involves two series of constructs that differ in the length of the peptide subunit. (b) Helical-wheel diagrams for the peptide subunits used in this study, illustrating their amphiphilic nature. Color-coding in the helical wheels: yellow, hydrophobic; blue, cationic; red, anionic; green, polar uncharged.
Figure 3.

Synthesis of multivalent peptide constructs. (a) Representative synthetic scheme for preparing a dimeric construct. (b) Chemical structures of scaffolds used to prepare the multivalent constructs. (c) HPLC traces showing formation of the trimer23 peptide construct over time via native chemical ligation.
For the first series of peptides, we chose to use helix 10 of human apoA-I (residues 221–241), which is critical for lipid binding and cholesterol efflux in the native protein.10 The 21-amino-acid peptide was altered for our studies by introducing two conservative amino acid substitutions: Ala for Val-227 to increase the amphiphilicity and Trp for Tyr-236 to improve spectrophotometric analyses (Figure 2). We also appended Cys-Gly to the N-terminus to allow efficient NCL, giving a 23-amino acid monomeric peptide (denoted as monomer23). In designing the second family of constructs, we supposed that the structures, by virtue of their multivalent, amphiphilic, α-helical arrangements, should have stronger, more cooperative interactions with lipid membranes than typical monomeric apoA-I mimetic sequences, thus permitting the use of shorter helical modules. Therefore, the second series was constructed by using a 16-amino-acid peptide (denoted as monomer16), which is a truncated variant of the parent 23-mer peptide, with seven residues (one turn of an α-helix) removed from the N-terminus (Figure 2). The required 23-mer and 16-mer peptides were synthesized and purified according to standard solid-phase protocols. The multivalent constructs were synthesized (Figure 3) by mixing peptide (1.5 mol equiv) and scaffolds in neutral aqueous solution for 10–16 h, after which the cysteine side chains were capped by reaction with bond formation. Overall isolated yields of the constructs iodoacetamide to prevent complications from disulfide starting from the monomer were 60–85%. This excellent synthetic efficiency allowed us to smoothly produce sufficient trimeric construct (~2.0 g) for chronic antiatherosclerosis studies in mice, which are currently ongoing in our laboratory.
Biophysical Characterization of Lipid-Free Peptides and Peptide Nanolipids
Biophysical characterizations of the lipid-free peptide constructs involved circular dichroism (CD), analytical ultracentrifugation (UC), pyrene fluorescence studies, surface tension measurements, and size exclusion chromatography (SEC) (Table S1; Figures S2–S5). CD analyses showed that the multivalent peptide constructs exhibited a higher degree of helicity, as judged by the magnitude of the minima at 222 nm, than the monomeric peptide in the presence or absence of lipid. The monomers and dimers exhibited increased helicity in the presence of lipid, as is typical of amphiphilic α-helical peptides. On the other hand, the trimers and tetramers were either unchanged or were marginally less helical in the presence of lipid, suggesting that these structures are folded due to intramolecular interactions even in the absence of lipid. UC studies for the 16-mer family were suggestive of reversible self-association of the multivalent species into dimeric species in the μM concentration range, whereas monomer16 did not form aggregates even at the highest concentration tested (210 μM). Surface tension values were measured for the 23-mer family of peptides at a concentration of 1 × 10−4 M; all the peptides had values within a narrow range of 44.6 and 47.2 mN/m. Surface tension values dropped with increasing monomer concentrations until reaching a point that corresponded well with the self-association saturation point determined by pyrene fluorescence (Figure S5).
Membrane interacting proteins or peptides have often been formulated with phospholipids to generate lipid nanoparticles (“lipidated”) with the aid of cholate (as detergent), followed by dialysis to remove most of the cholate from the preparation.1c Because we wished to avoid any complications resulting from any residual cholate, we employed a cholate-free approach. Lipid nanoparticles were prepared by incubating each peptide construct with an aqueous (phosphate-buffered saline, PBS) suspension of 3 mM (R)-(+)-1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) multilamellar vesicles at 22 °C for 12–16 h.1c Despite having unnatural, branched topologies, all multivalent constructs generated discoidal nanoparticles of similar size and morphology as discoidal HDL (Figure 4; Table 1; see Figure S6 for kinetics of lipid clearance). The nanoparticle sizes could be controlled within a range of ~9–15 nm by varying the ratio of peptide to lipid (Figure 4a,b). At peptide/lipid ratios that mimic the~1:100 apoA-I/lipid ratio in native HDL particles, when corrected for the number of helices per molecule (1:10 for monomer, 1:20 for dimer, 1:30 for trimer, 1:40 for tetramer), the nanodiscs generated by each construct were in the size range of 9–12 nm, as determined by SEC (Figure 4; Table 1). For a given peptide valency, the SEC nanoparticle size for the 23-mer variant was 1–2 nm larger than that for the analogous 16-mer construct (Table 1). In both series of constructs, the construct:DMPC ratio in purified nanoparticles indicated that the trimer bound considerably more lipids than expected based on the mixing ratio of 1:30 (Table 1), suggesting that a trimeric structure may be advantaged in lipid binding compared to the other valencies tested. The discoidal morphology of the nanoparticles determined by transmission electron microscopy (TEM) was similar to that of synthetic and native HDL nanoparticles formed from apoA-I (Figures 4d, S7, and S8).11
Figure 4.

Characterization of lipid nanoparticles formed by incubation of multivalent peptide constructs with DMPC multilamellar vesicles. (a) SEC profiles shown for DMPC nanoparticles generated from the monomer23 peptide at various peptide/lipid ratios. (b) Plot of nanoparticle size measured by SEC as a function of lipid/helix ratio for the 23-residue series of constructs. (c) SEC profiles for nanoparticles generated from each 16-residue constructs at a peptide/lipid ratio of 1:10, 1:20, 1:30, and 1:40 for the monomer16, dimer16, trimer16, and tetramer16, respectively. The multivalent species efficiently form nanoparticles, whereas the monomeric peptide remains largely lipid-free. (d) Negative-stain (phosphotungstic acid) TEM images of purified nanoparticles generated by the peptide constructs. All adopt a similar morphology to discoidal HDL particles. (e) Hypothetical molecular model of a lipid nanoparticle assembled from ten 16-amino-acid α-helices per leaflet (20 helices per particle), viewed from two directions, based on the double-belt model of native apoA-I in discoidal HDL particles.
Table 1.
Biophysical Properties of DMPC-Based Nanoparticles Generated with Peptides.a
| peptide | peptide:DMPC mixing ratio |
peptide:DMPC ratio in nanoparticlesb |
SEC Stokes di- ameter (nm) |
TEM diameter (nm) |
lipids per particlec |
helices per particled |
|---|---|---|---|---|---|---|
| 4F | 1:10 | 1:15 ± 2 | 11 ± 2 | ND | 300 | 20 |
| monomer23 | 1:10 | 1:14 ± 1 | 11 ± 1 | 15 ± 4 | 300 | 21 |
| dimer23 | 1:20 | 1:30 ± 2 | 12 ± 1 | 12 ± 3 | 360 | 24 |
| trimer23 | 1:30 | 1:43 ± 7 | 12 ± 1 | 12 ± 2 | 360 | 25 |
| tetramer23 | 1:40 | 1:39 ± 4 | 11 ± 2 | 12 ± 2 | 300 | 31 |
| monomer16 | 1:10 | 1:5.8 ± 0.3 | 10 ± 1 | 12 ± 3 | 250 | 43 |
| dimer16 | 1:20 | 1:28 ± 3 | 10 ± 1 | 14 ± 2 | 250 | 18 |
| trimer16 | 1:30 | 1:54 ± 7 | 11 ± 1 | 15 ± 2 | 300 | 17 |
| tetramer16 | 1:40 | 1:44 ± 3 | 9 ± 1 | 14 ± 2 | 200 | 18 |
| apoA-I | 1:100 | ND | 15, 10e | 11 ± 3e | ND | ND (20) |
Synthetic nanoparticles were prepared by mixing a given peptide construct (300 μM) with DMPC liposomes at the molar ratios shown in PBS, and incubating overnight prior to analysis by SEC or TEM. SEC Stokes diameter was determined using a standard curve of four globular proteins. TEM size was determined by averaging the longest diameter of 50 nanoparticles for each peptide species.
The peptide:DMPC ratios were determined by isolating the nanoparticles using SEC and then separately measuring the peptide concentration using UV absorbance at 280 nm and the phospholipid concentration using a commercially available assay kit (Biovision).
The average number of lipids per particle was estimated by assuming a bilayer topology (two leaflets with an equal number of lipids), calculating the area of a circle having the observed SEC Stokes diameter, and dividing this total area by the area of a single DMPC molecule (0.63 nm2).13
The number of helices per particle was estimated by dividing the number of lipids per particle by the DMPC:peptide ratio (corrected for the number of helices per construct). It is thought that two apoA-I molecules (10 helices/molecule) reside in discoidal HDL particles.
Two peaks were observed in the nanoparticle region of the SEC trace for apoA-I. The 10-nm particle, purified by SEC, was used for TEM analysis.
HDL nanoparticles are highly heterogeneous and dynamic, but the generally accepted structure for discoidal HDL involves two antiparallel molecules of apoA-I wrapped like a double belt around the edge of a phospholipid bilayer. Thus, each discoidal HDL particle contains 20 individual α-helices (10 helices/apoA-I) (Figure 4e). The lipid nanoparticles assembled from most of our multivalent species contain 17–25 helices per particle (Table 1), in general agreement with the assumption that their structures involve helices aligned in an end-to-end fashion around the edge of a bilayer, like the structure adopted by apoA-I. The helical subunits of the multivalent constructs likely adopt a number of arrangements in relation to each other around the edge of the particles, made possible by the flexible linkers, involving inter- and intramolecular associations between individual helices. A constraint of the branched nature of the multivalent constructs is that a continuous antiparallel arrangement of helices around the particle (like that in native discoidal HDL) is not possible, assuming all of the helices are bound to particle surface.
We also found that the nanoparticles could be freeze-dried, stored at −20 °C, and reconstituted with water, without apparent changes in particle size or morphology (Figures S9 and S10). This process could be useful for practical formulations of these materials, as well as other peptide-based nanoparticle therapeutics.
For the 16-mer peptide series, inspection of the SEC traces for nanoparticle formation revealed that the multivalent peptides packaged lipids more efficiently than did the monomeric peptide, for which much of the lipid-free peptide remained after the incubation period (Figure 4c). Furthermore, the observed construct:DMPC ratio for monomer16 in purified nanoparticles was 1:5.8, whereas the analogous values for monomer23 and 4F (Ac-DWFKAFYDKVAEKFKEAF-NH2; a well-studied monomeric, α-helical apoA-I mimetic peptide5) were 1:14 and 1:15, respectively (Table 1), indicating that more molecules of the monomer16 peptide are necessary to form stable nanoparticles. In contrast, the observed peptide:DMPC ratios for the multivalent 16-mer peptides were similar to those for the 23-mer family, suggesting that multimerization of the 16-residue helix overcame its deficiencies of the monomer16 in lipid packaging.
Effects on Cholesterol Efflux and Plasma HDL Remodeling
HDL particles promote the efflux of cholesterol from macrophage cells in the early, rate-limiting step of RCT.12 To determine the capacity of our peptide-lipid nanoparticles to mediate efflux, cholesterol-laden mouse macrophages (J774 cells) were treated with the two families of nanoparticles for 12 h. In both series, the multivalent constructs promoted cholesterol efflux more efficiently than the monomeric peptide (Figure 5). In the 16-mer family, the observed efflux EC50 values followed the trend of tetramer16 > trimer16 > dimer16; monomer16 failed to cause any notable efflux (Figure 5). No such trend was observed for the 23-residue constructs, for which the dimer23 structure was most efficient, although all of the nanoparticles from multivalent peptides were more efficient than those from monomer23. For both families of peptides, similar trends in efflux activity were observed for the lipid-free peptide constructs (Figure S11).
Figure 5.
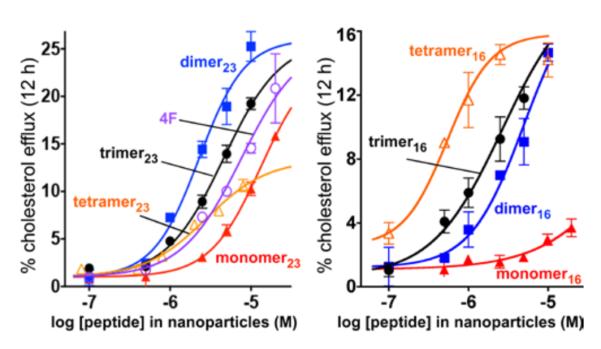
HDL-mimetic nanoparticles efficiently promote cellular cholesterol efflux. Efflux was measured with cholesterol-laden mouse macrophage J774 cells over 12 h for the 23-mer series (left) and the 16-mer series (right). Values are shown as mean ± SD of samples in triplicate. The peptide concentrations in the nanoparticles at half-maximal efflux (EC50) were: 4F, 7.6 ± 1.5 μM; monomer23, 13.3 ± 4.0 μM; dimer23, 2.3 ± 0.7 μM; trimer23, 4.6 ± 1.6 μM; tetramer23, 2.8 ± 1.9 μM; monomer16, ~150 μM; dimer16, 4.1 ± 1.2 μM; trimer16, 2.3 ± 0.6 μM; and tetramer16, 0.5 ± 0.4 μM. In both peptide series, nanoparticles prepared from multivalent constructs were superior to monomeric peptides. Similar trends in efflux activity were observed for the lipid-free peptide constructs (Figure S11).
Small, dense HDL particles are the initial acceptors of cellular cholesterol in RCT (Figure 1).14 To evaluate the capacity of our peptide-lipid nanoparticles to interact with and remodel mature plasma HDL (7.5–11 nm in size) into smaller, more dense particles (~7 nm), we treated human plasma with each nanoparticle for 1 h at 37 °C, and then analyzed the samples by immunoblotting for apoA-I.15 All peptide nanoparticles induced an increase in small, dense HDL levels, with the exception of monomer16 (Figures 6, S12, and S13). When corrected for the number of helices present in each molecule, the increases in dense HDL were similar for the entire panel of multivalent constructs. Controls involving DMPC alone did not affect the plasma HDL.
Figure 6.

Double-labeled fluorescent imaging and Western-blot analyses to track interactions between synthetic lipid nanoparticle components and native HDLs. Nanoparticles were formed from DMPC/1% rhodamine-labeled lipid (red signal) and fluorescein labeled peptide constructs (green signal) denoted by (F*). Particles in the rightmost two lanes contained labeled lipid, but unlabeled peptide. (a, b) The particles were incubated with human plasma for various times, separated by NDGGE, and fluorescently imaged. Lipid-poor HDLs co-migrate at 7.1 nm with human albumin, which autofluoresces and binds labeled lipid and peptide to some extent (see Figure S13). (c) Overlay of fluorescence images. (d) The gel was then Western blotted for human apoA-I to get a reading on the native HDL particles. (e) An overlay of the fluorescence images and Western blot revealed that the multivalent peptides have rapidly incorporated into the HDLs, and coexist with apoA-I. Over time, the lipid-poor HDL bands disappear and mature HDL bands increased in size, likely due to HDL metabolism.17
Interaction with Native HDL
To investigate how our synthetic nanolipids would interact with biological lipoproteins in vitro, we prepared peptide constructs alkylated on the Cys side chain with a labeling agent instead of iodoacetamide.16 Fluorescein was used for the 23-mer series, whereas biotin was used for the 16-mer series because fluorescein labeling interfered with nanoparticle formation with these small constructs. The labeled constructs were assembled into nanoparticles with DMPC containing 1% of a rhodamine-labeled DMPC analogue, yielding particles that were similar in size to those obtained from nonlabeled components (albeit with a larger size distribution) (Figure S14). We then independently tracked the peptide and lipid components of synthetic nanoparticles as they interacted with lipoproteins in a plasma sample (Figure 6; see Figure S13 for an analogous study with the 16-mer peptides). Human plasma was incubated with the labeled nanoparticles and subjected to nondenaturing gradient gel electrophoresis (NDGGE). The gel was fluorescently imaged to visualize the labeled peptide and lipid components (Figure 6a-c), and then Western blotted for apoA-I to visualize the HDL species (Figure 6d; Figure S15 shows there was no cross-reactivity between the apoA-I antibody and the peptide constructs). The synthetic nanoparticles were rapidly incorporated into biological lipoproteins; within 15 min, the synthetic nanoparticles (11–14 nm) were not apparent in the gel. The dimer23 and trimer23 fluorescence was overlaid with the apoA-I-containing HDL bands at 7.5–11 nm. In the case of the monomer, little fluorescence was observed in the HDL region; instead, the monomer was largely associated with human albumin (7.1-nm band). The labeled lipid was present in the HDL regions, in larger particles that did not enter the gel (probably low-density and very low-density lipoproteins), and bound to albumin. Interestingly, over time, the small, dense HDL bands disappeared and the mature HDL bands increased in size, possibly due to HDL metabolism.17 Thus, in human plasma in vitro the multivalent peptides were readily transferred from the synthetic nanoparticles to native HDL particles, caused HDL remodeling, and at that point coexisted in the particles with apoA-I (Figure 6e). A similar overall behavior was observed for the dimer16, trimer16, and tetramer16 constructs (Figure S13).
Proteolytic Stability and Pharmacokinetics
Peptides administered systemically are subject to renal clearance and enzymatic degradation by abundant proteases.18 A prolonged circulatory half-life would be beneficial for clinical use of apoA-I mimetic peptides. To investigate the stability of our peptides, we first followed the disappearance of the intact peptides incubated in mouse serum at 37 °C by LC-MS with single-ion monitoring (SIM) (Figure S16). For both series of peptides, we found that the monomeric peptides were degraded quickly, with ~50% lost in the first 2 h, whereas the multivalent constructs were much more stable, with minimal degradation during the 12–24 h incubations (Figures S17 and S18). We also tested the stability of the peptides against isolated proteases in vitro: chymotrypsin, thermolysin, pronase, and pepsin. The monomers were readily digested by all proteases, but the multivalent apoA-I mimetic constructs were remarkably more stable (Figure 7; see also Figures S17 and S18 for time courses of degradation and data for the dimeric and tetrameric constructs). All of the peptides were more resistant to degradation when formulated as DMPC/peptide nanoparticles, but the observed stability trends remained the same (Figures 7, S17, and S18).
Figure 7.
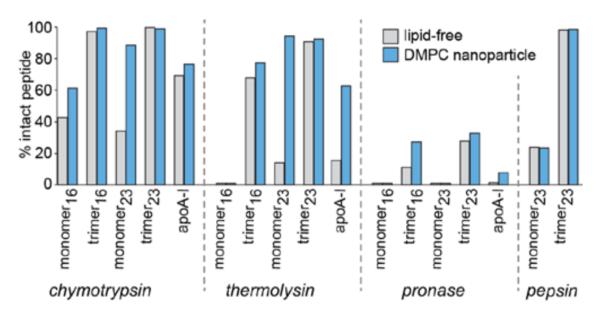
Proteolytic stabilities of peptide constructs and DMPC nanoparticles compared to apoA-I and recombinant HDL (rHDL) particles. Samples (0.5 mg/mL peptide or protein) were incubated at 37 °C with each protease (20 mU/mL, chymotrypsin, thermolysin, and pronase; 0.5 U/mL, pepsin) for 2 h in PBS. The multivalent constructs were more stable than monomeric peptides, for both the 23-mer and 16-mer families, and peptide nanolipids were more stable than the corresponding lipid-free peptides. The trimeric constructs and their lipid nanoparticles were more stable than native apoA-I and rHDL.
When administered intraperitoneally (i.p.) to wild-type mice (BALB/cByJ), the multivalent nanoparticles exhibited superior blood concentrations and much longer plasma residence times compared with the nanolipid particles from monomers (Figure 8). Peptide doses were 60 mg/kg (equal doses on a helix basis; 0.63, 0.32, 0.21, and 0.16 μmol of monomer16, dimer16, trimer16 and tetramer16 per mouse, respectively) for the 16-mer series, and 75 mg/kg (0.55, 0.36, and 0.18 μmol of monomer23, dimer23, and trimer23 per mouse, respectively) for the 23-mer nanoparticles. Plasma concentrations of the monomer16 peptide were only above the limit of detection (1.7 μM) at 0.5 h (Cmax of 12 μM), whereas plasma concentrations of the 16-residue multivalent peptides peaked at ~4 h and had much longer residency (Cmax of 48, 44, and 20 μM for dimer16, trimer16, and tetramer16, respectively); tetramer16 remained at detectable levels even after 24 h. The plasma levels of the dimer23 and trimer23 constructs were also high (Cmax of 26, 88, and 44 μM for monomer23, dimer23, and trimer23, respectively) and were above the concentrations required for in vitro HDL remodeling (10 and 7 μM for dimer23 and trimer23, respectively) from 1 h post-injection up to 31 and 18 h, respectively (Figure 8). These long plasma half-lives are remarkable for peptides comprised of all natural L-amino acids. A similar pharmacokinetics (PK) profile was observed for the corresponding trimer23 nanoparticles assembled with a trimeric construct synthesized from all D-amino acids (Figure 9a), further suggesting that proteolysis is not a primary in vivo elimination mode for trimer23. Subcutaneous injection of the trimer23 nanoparticles into mice (n = 3; 75 mg/kg dose) gave PK and in vivo plasma HDL remodeling profiles (Figure S19) similar to the profiles from i.p. injection. Likewise, i.p. injection of the trimer nanoparticles (n = 3; 30 mg/kg) to rats gave a long elimination half-life of ~10 h and a high Cmax of 27 μM (Figure 9b). For comparison, the plasma half-life of apoA-I in mice is 11 hours.19
Figure 8.

HDL-mimetic nanoparticles prepared from multivalent peptide constructs have superior in vivo PK and plasma HDL remodeling properties compared to the monomeric peptides in mice. (a) Mice (BALB/c, n = 3) were injected i.p. with a 75 mg/kg dose (equal doses on a helix basis; 0.55 μmol monomer23, 0.36 μmol dimer23, 0.18 μmol trimer23) of peptide-lipid nanoparticles. Measured area under the curve values for the monomer23, dimer23, and trimer23 were 110 ± 10, 1570 ± 130, and 500 ± 40 μM•h, respectively. Plasma samples from the PK study were Western blotted for mouse apoA-I. The multivalent peptides, in contrast to the monomer, increased the level of lipid-poor HDLs in the mice, which are beneficial for combating atherosclerosis. (b) Mice (BALB/c, n = 3) were injected i.p. with a 60-mg/kg dose (equal doses on a helix basis; 0.63 μmol monomer16, 0.32 μmol dimer16, 0.21 μmol trimer16, 0.16 μmol tetramer16) of peptide nanoparticles. Measured AUC values for the monomer16, dimer16, trimer16, and tetramer16 were <10, 340 ± 30, 180 ± 30, and 260 ± 30 μM•h, respectively. Western blotting indicated that the tetramer16 nanoparticles most effectively increased the in vivo levels of small, dense HDLs in this series of compounds.
Figure 9.
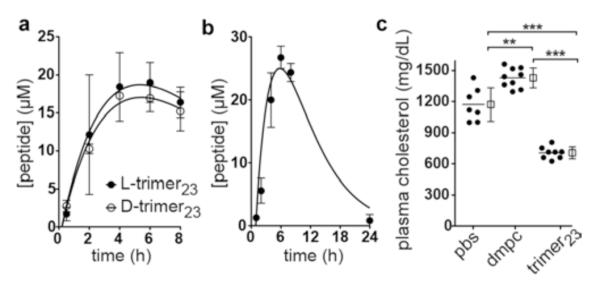
Pharmacokinetics and plasma cholesterol reduction studies of trimer23 nanolipid particles. (a) Mice (BALB/cByJ, n = 3) were injected i.p. (50-mg/kg dose) with DMPC nanoparticles synthesized from l-trimer23 or d-trimer23 (synthesized from all d-amino acids). Rates of clearance were similar for the two peptides, indicating that enzymatic degradation is not a major route of clearance in vivo. (b) Rats (Sprague–Dawley (Crl:CD(SD)), n = 3) were injected i.p. with a 30-mg/kg dose of trimer23/DMPC nano-particles. (c) Two-week, 50-mg/kg i.p. administration of the trimer23/DMPC nanoparticles (n = 8) reduced the plasma total cholesterol levels of 10-week old female LDLr-null mice fed a HFD compared to PBS (n = 7) or DMPC liposome/PBS (n = 9) controls. Data points are shown as the mean ± SD; **, p ≤ 0.01; ***, p ≤ 0.001 by one-way ANOVA, post hoc Tukey–Kramer test.
In vivo plasma HDL remodeling blots from these PK studies showed a marked enhancement of lipid-poor HDL bands from 2–8 h post-injection for the dimer23- and trimer23-based nanoparticles (but not for monomer23), indicating successful remodeling of mouse HDL in vivo (Figures 8 and S19). Likewise, the small, dense HDL bands were increased by tetramer16 nanoparticles from 0.5–8 h post-injection, indicating successful remodeling of mouse HDL in vivo.
Exploratory in vivo efficacy/toxicity studies were carried out using the trimer23 nanoparticles in low-density lipoprotein receptor (LDLr)-null (LDLr)−/−) mice, which are a widely used animal model for atherosclerosis research.20 At 10 weeks of age, the mice were switched to a high-fat diet (HFD; 1.25% cholesterol, 15.8% fat, and no cholate), and daily 50-mg/kg i.p. injections of the trimer23/DMPC nanoparticles were commenced. After two weeks, treatment with the trimer23 nanoparticles had reduced plasma total cholesterol levels by 40% and 51%, respectively, compared to controls involving i.p. injection of PBS or DMPC liposomes (Figure 9c). As determined by SEC fractionation of pooled plasma samples, the reductions in plasma total cholesterol stemmed mainly from reduced levels of VLDL and LDL, but, encouragingly, the plasma HDL was also remodeled in favor of the smaller HDL particle sizes (Figure S20). No differences were observed between the treated and control mice in food intake, water intake, body weight, or plasma liver enzyme levels (ALT and AST) over the course of the two-week study.
DISCUSSION
We carried out the first systematic study of branched, multivalent peptide constructs as a new approach for generating synthetic HDL-like nanoparticles, by using two distinct families of 16-mer and 23-mer peptide constructs. In the 16-mer series, cholesterol efflux capacity and proteolytic stability directly depended on the number of helices present in the peptide construct, lipid nanoparticle formation was more efficient for the multivalent species compared to monomer16, and only the tetramer16 showed notable plasma HDL remodeling in vivo. The functional properties observed for the 23-mer family, in contrast, did not directly depend on the number of helices, although the lipid nanoparticles from the multivalent constructs were superior to those from the monomer23 peptide in cholesterol efflux, proteolytic stability, PK, and HDL remodeling in vivo. Thus, although the sizes of the nanoparticles could be controlled by adjusting the peptide/lipid ratio during their preparation, the resultant functional properties were more complex and not directly tunable according to the peptide construct used.
Because the shorter peptides possess less inherent capacity for lipid association, multimerization had a more dramatic effect on the function of these constructs. Consistent with this observation is that more molecules of the monomer16 peptide were necessary per phospholipid to form a stable nanoparticle compared to the multivalent variants (Table 1), which likely stems from the monomer being less folded and/or having lower lipid affinity. It is notable that the multivalent approach permitted the use of monomer16, a short peptide that was functionally deficient as the monomer in generating lipid nanoparticles, effluxing cholesterol, and remodeling plasma HDLs. Consistent with our findings, other peptides composed of tandem α-helical segments exhibit improved cholesterol efflux and HDL remodeling properties compared to monomeric helices.7e
An important issue is whether the observed functional improvements can be explained by a simple increase in the effective concentration of peptides present or, put another way, would a four-fold higher concentration of the monomeric peptide be functionally equivalent to a tetrameric construct? In this case, the answer is clearly no. In the context of cholesterol efflux, tetramer16 is nearly 10-fold more potent than dimer16, and dimer16 is at least 10-fold more potent than monomer16; in the 23-mer series, the dimer construct most efficiently promoted efflux, despite there being a higher effective concentration of peptides in the trimer and tetramer species at a given concentration. Likewise, improvements in PK area under the curve (AUC) values were greater than would be expected on the basis of effective concentration– dimer23 exhibited a 14-fold greater AUC value than monomer23, and all of the multivalent 16-mer constructs had >10-fold improvements compared to monomer16.
As an initial step toward understanding the mechanisms that lead to the functional improvements for multivalent agents, we labeled the peptide and phospholipid components of the synthetic nanoparticles and tracked their interactions with native HDL in human plasma in vitro. Interestingly, monomer23 was largely bound to human albumin, whereas the dimer23 and trimer23 constructs associated almost exclusively with HDL particles. These findings are consistent with data from Wool et al. showing that monomeric 4F only weakly associates with HDL and that it mainly bound to plasma proteins.7f By contrast, a tandem peptide with two 4F segments linked by proline associated with all lipoprotein classes, including HDL.7f Likewise, a “peptibody” derived from two copies of 4F attached to a murine IgG Fc fragment coimmunoprecipitated with apoA-I, indicating that the pepti-body coexisted with apoA-I in the HDL particles.7h Thus, it appears that multimerization of amphiphilic α-helical peptides results in more selective, higher affinity binding to lipoproteins.
A common limitation of peptide-based therapeutics is a short half-life in vivo. Stability studies in vitro and in vivo indicate that the multivalent species described here are considerably more robust than the analogous monomers. We suggest that intramolecular self-assembly of the multivalent constructs might be responsible for the high proteolytic stabilities observed. Also, the observed higher affinity association with native lipoproteins for the multivalent constructs may contribute to their greater plasma residency times. Previous studies have shown that branched, multiple antigen peptides (MAPs) exhibit resistance to proteases, which has been ascribed to steric hindrance limiting access to the protease active site.21 In addition to excellent proteolytic stability, the multivalent constructs may be more resistant to renal clearance than the monomers because of their larger sizes.22
The observed plasma cholesterol reduction promoted by trimer23 is exceptional within the realm of apoA-I mimetic peptides. Other apoA-I mimetic peptides have typically been reported to lack any significant effect, or have only modest effects, on plasma cholesterol levels, even when they effectively reduce the development of atherosclerotic lesions.23 For example, mouse plasma total cholesterol levels were unchanged by daily, 8-week administration of D4F (4F peptide with all D-amino acids) at 45-mg/kg, p.o.,23c or daily, 6-week administration of ATI-5261 at 30-mg/kg, i.p.,23a and just modestly reduced (9–14%) by daily, 3-week or 6-week administration of mR18L at 50-mg/kg, p.o.23b Likewise, neither apoA-I nor rHDLs cause significant reduction in plasma cholesterol levels.24 These observations raise a question about whether our peptides function by the same mechanisms as other apoA-I mimetic peptides. Our data indicate that the peptides can promote HDL remodeling to produce lipid-poor particles and induce cellular cholesterol efflux, two important aspects of the RCT pathway. Therefore, it is reasonable to propose that the mechanism of plasma cholesterol reduction involves the promotion of RCT. We are currently working to clarify these mechanistic issues.
Synthetic peptides with a monomeric, amphiphilic α-helical structure can functionally mimic apoA-I in generating lipid nanoparticles and modulating the biological properties of HDL.5,25 Assorted studies with different peptide derivatives in mouse models of atherosclerosis have demonstrated antiatherogenic effects, which led to human clinical studies with certain agents, such as the 18-mer apoA-I mimetic peptides 4F26 and D4F,27 and the 22-mer peptide ETC-642.28 Considering the process of multimerization described herein, it remains to be seen whether other monomeric apoA-I mimetic peptides, such as those mentioned above, can be functionally improved in a similar manner. More broadly, it is encouraging that every one of the constructs that we tested, despite their abiologic branched topologies, generated robust nanolipid particles. These results suggest that more diverse structural topologies, as well as other types of amphiphilic molecules (besides amphiphilic peptides), could be useful in forming lipid nanoparticles with HDL-like functions.
CONCLUSIONS
Our findings with branched, multivalent peptide constructs support their utility as scaffolding agents for nanolipid particles with HDL-like properties. Our modular synthetic approach of attaching multiple copies of a purified α-helical peptide to different scaffolds is synthetically efficient and affords a level of molecular design beyond the peptide primary sequence. The resulting multivalent apoA-I mimetics can be functionally superior to the monomeric peptides in several respects. Furthermore, enhanced proteolytic stability in vitro and improved in vivo PK profiles confer a real practical advantage to such multivalent peptide constructs. This key benefit from our approach circumvents the high cost and potential complications of other strategies to stabilize apoA-I mimetics, involving peptides synthesized from all D-amino acids29 or chemical additives to improve their proteolytic stability.30 Indeed, we have obtained encouraging preliminary results for the antiatherogenic potential of our multivalent nanolipid particles on chronic daily dosing in LDLr-null mice (to be reported separately in due course). From a basic science standpoint, our approach lays the groundwork for further studies involving constructs with more than four helical subunits, aimed bridging the gap between monomeric apoA-I mimetics and full-length apoA-I to understand Nature’s reasons for using a protein with 10 repetitive helical segments to stabilize HDL particles. Thus, our results provide a new path for advancing the chemical biology of HDL and for developing therapeutic agents to manage atherosclerosis.
EXPERIMENTAL SECTION
General
For more detailed experimental methods, and additional procedures, including the synthesis of peptides and scaffolds, biophysical characterizations, and LC-MS quantitation of peptide concentrations, see the Supporting Information. All procedures involving human samples and live animals were approved by TSRI Institutional Review Board and TSRI Institutional Animal Care and Use Committee, respectively.
Synthesis of Multivalent Constructs
All peptide ligation reactions involved a 1.5-fold excess of purified peptide relative to the number of thioesters in the scaffold. Ligations were performed in 200 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer containing 7 M guanidine hydrochloride (Gdn·HCl), 100 mM tris(2-carboxyethyl)phosphine (TCEP), pH 7.5, at room temperature for 6–12 h. At the completion of the ligation reaction, iodoacetamide (~50-fold excess relative to Cys) was added to cap the free thiol moieties on Cys residues. After 5 min, CF3CO2H was added to quench the reaction, and the product was purified by RP-HPLC.
Peptide–Lipid Nanoparticle Preparation and Characterization
DMPC multilamellar vesicles (MLVs) were prepared as described in the supporting information. A stock solution of peptide in PBS was added to 0.5 or 3 mM MLVs at the appropriate initial molar ratio desired, and the solutions were vortexed for 1 h and incubated with shaking for 12 h at 22 °C. To obtain fluorescently labeled nanolipid particles, a solution of 5-IAF-labeled peptide (23-residue series) or biotin-labeled peptide (16-residue series) in PBS was added at a 1:10 (helix:lipid) molar ratio to 0.5 or 3 mM DMPC MLVs containing 1% 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl (RhBDMPE, Avanti). The mixture was vortexed for 12 h at 22 °C. The lipid nanoparticles were purified by SEC and characterized by using SEC and TEM. Cholesterol and phospholipid amounts were determined in purified nanoparticles using cholesterol and phospholipid assay kits purchased from BioVision; peptide concentrations were determined using UV absorption.
Size-Exclusion Chromatography (SEC)
Samples were passed through a 0.45-μm polyvinylidene fluoride (PVDF) filter and injected to a Superdex 200 HR 10/30 column (GE Healthcare) at room temperature at a flow rate of 0.5 mL/min using an AKTA purifier chromatography system. Tris-buffered saline (10 mM Tris, pH 7.4, 100 mM NaCl, 0.25 mM EDTA, 0.0005% NaN3) or phosphate-buffered saline (10 mM phosphate, 136 mM NaCl, pH 7.4) was used as a running buffer. The void volume was at 7.1 mL. The standards were: thyroglobulin (669 kDa, 17 nm), ferritin (440 kDa, 12.2 nm), bovine serum albumin (67 kDa, 7.1 nm), and ribonuclease A (13.7 kDa), which gave the following retention volumes (mL): 8.3, 11.0, 14.0, and 17.2, respectively.
Transmission Electron Microscopy (TEM)
The nanoparticle fractions collected by SEC were imaged without further purification. Glow-discharged copper grids (400 mesh) coated with carbon and formvar (G400-Cu, Electron Microscopy Sciences, Hatfield, PA) were then immediately inverted, carbon surface down, onto 7 μL droplets of sample solutions placed on Para-film. After 3 min, excess liquid was wicked off and the grids immediately placed onto individual droplets of aqueous 2% phosphotungstic acid at pH 7.0. After 2 min, excess stain was removed and the grids were allowed to dry thoroughly. Images were taken on a Philips CM100 electron microscope (FEI, Hillsbrough, OR) at 80 kV and collected using a Megaview III CCD camera (Olympus Soft Imaging Solutions, Lakewood, CO). Size measurements were made on at least 50 nanoparticles, quantified by using SimpleDigitizer 3.1.8 or Photoshop CS4 software.
Cholesterol Efflux
Cholesterol efflux from macrophage cells was determined by using a modification of a reported procedure.7e J774A.1 cells (TIB-67; American Type Culture Collection, Manassas, VA) were cultured to 80–90% confluence in DMEM (Dulbecco’s Modified Eagle’s Medium) with 10% fetal bovine serum at 5% CO2. Cells were then incubated with 5% lipoprotein-deficient serum (LPDS), 2 μg/mL Sandoz acyl-CoA:cholesterol acyltransferase inhibitor (Sigma) and 25 μg/mL acetylated low-density lipoprotein in DMEM for 48 h, followed by 12 h in DMEM with 5% LPDS, 2 μg/mL Sandoz, with 0.3 mM 8-(4-chlorophenylthio)adenosine-3′,5′-cyclic monophosphate (Sigma). Peptides were then applied in DMEM with 5% LPDS. Aliquots of medium were taken at 12 h and centrifuged (1000 rpm, 10 min) to remove floating cells. The cell monolayers were rinsed three times with ice-cold DMEM and lysed by the addition of 200 μL Cytobuster lysis buffer. After incubation at room temperature for 10 min, cell lysates were homogenized by using a homogenizer (PowerGen 125, Fisher Scientific). Cell-free medium and cell lysates were analyzed for total cholesterol amounts by using a cholesterol Amplex assay kit (Invitrogen). Cholesterol efflux at each time point was calculated as the amount of cholesterol in the medium as a percentage of the total cholesterol (medium + lysate).
Remodeling of HDL in Human Plasma
These procedures were modified from a reported method.15 Human plasma was incubated with a peptide at 37 °C for various times, and then quenched by adding 50% sucrose (10-fold dilution). Samples were Western blotted for human apoA-I as described in the Supporting Information.
Protease/Serum Stability Assays
Mouse serum samples were obtained from the Department of Animal Resources at TSRI. Peptides or apoA-I were incubated in serum at a final peptide concentration of 0.1 mg/mL at 37 °C, or at a final concentration of 0.5 mg/mL with 20 mU/mL proteases (pronase, chymotrypsin, or thermolysin) in PBS (pH 7.4). After various incubation times, aliquots were removed. Serum samples were extracted with 1.5 volumes of acetonitrile containing 0.1% CF3CO2H and 0.2% triton X-100, centrifuged at 13,000 rpm for 10 min, and the supernatants were acidified with 0.5 volumes of 1% acetic acid. Aliquots of protease samples were mixed with 1.5 volumes of PBS containing protease inhibitors (Complete™) and acidified with 0.5 volumes of 1% acetic acid. Peptide susceptibility to pepsin digestion was assessed by incubating peptide/DMPC nanoparticles (0.5 mg/mL peptide) with pepsin (0.5 U/mL) at 37 °C in 10% acetic acid (pH 2.2). Aliquots of samples were taken at indicated times, and the digestion was stopped by addition of pepstatin (10 μg/mL) and dilution with acetonitrile. The concentrations of peptide or apoA-I in the samples were determined by LC-MS SIM or HPLC, respectively, as described in the Supporting Information.
Mouse Pharmacokinetics
Male mice (20 g) on a BALB/cByJ background were obtained from the Rodent Breeding Colony of the Department of Animal Resources at TSRI, and were maintained on a chow diet. Peptides-DMPC nanoparticles (1:10, helix:lipid molar ratio) were spin concentrated with a 3000 molecular weight cutoff centrifugal filter (Millipore). Mice were fasted beginning 8 h before dosing, and continued the fast until after the 8-h time point blood draw was completed. Groups of three mice received a dose of peptide-DMPC nanoparticles via intraperitoneal injection (0.3 mL). Blood (30 or 60 μL) was drawn from the retro-orbital sinus into a heparinized capillary tube before dosing (0 min) and at different intervals after dosing. Plasma was isolated immediately from the whole blood by centrifugation. Immediately after plasma separation, 20 μL of plasma were extracted with 30 μL of acetonitrile containing 0.1% CF3CO2H and 0.2% triton X-100. After vortexing for 30 s, the mixture was centrifuged at 13,000 rpm for 10 min at 4°C. 40 μL of supernatant were removed and acidified with 20 μL of 1% acetic acid. This solution was used for quantifying peptide levels in the plasma by using LC-MS SIM as described in the Supporting Information, with a lower limit of quantification of 1.7, 0.8, 0.6, and 0.8 μM in plasma for monomer16, dimer16, trimer16, and tetramer16, respectively; 0.5, 0.2, and 0.2 μM, for monomer23, dimer23, and trimer23, respectively. To determine the level of in vivo HDL remodeling, aliquots of the plasma samples from the PK studies were Western blotted for mouse apoA-I, as described in the Supporting Information.
Plasma Cholesterol Studies in LDLr-Null Mice
LDLr-null (LDLr−/−) mice were fed a chow diet until they were 10-weeks old, when they were switched to a high-fat Western diet (HFD) (Harlan Teklad 94059). At the time that the HFD was started, trimer23-DMPC nanoparticles were administered by daily i.p. injection for two weeks in the continued presence of HFD. Mice receiving i.p. injections of PBS or DMPC ULVs served as controls. The mice were bled after an overnight fast (~15 h) after two weeks of treatment; the plasma was used to determine cholesterol levels using a commercially available assay kit (Amplex red cholesterol assay kit, No. A12216, Life Technologies) according to the manufacturer’s instructions).
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Malcolm Wood for assistance with TEM, Drs. Yeeting Chong and Paul Schimmel for assistance with AUC, Dr. Margery Connelly for helpful discussions, Ms. Kathleen Jo Hightshoe, Ms. Audrey S. Black, and Mr. David J. Bonnet for assistance with animal handling, and the NIH for financial support (NHLBI HL104462). We also thank American Heart Association Western States Affiliate for a postdoctoral fellowship (12POST12040298) to Dr. Y. Zhao, and National Institute of Advanced Science and Technology (AIST) for a visiting research fellowship to Dr. T. Imura.
Footnotes
Supporting Information. Detailed experimental procedures, Table S1, and Figures S1–S20. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interests.
REFERENCES
- (1) (a).Thaxton CS, Daniel WL, Giljohann DA, Thomas AD, Mirkin CA. J. Am. Chem. Soc. 2009;131:1384–1385. doi: 10.1021/ja808856z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Luthi AJ, Patel PC, Ko CH, Mutharasan RK, Mirkin CA, Thaxton CS. Trends Mol. Med. 2010;16:553–560. doi: 10.1016/j.molmed.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bricarello DA, Smilowitz JT, Zivkovic AM, German JB, Parikh AN. ACS Nano. 2011;5:42–57. doi: 10.1021/nn103098m. [DOI] [PubMed] [Google Scholar]; (d) Luthi AJ, Zhang H, Kim D, Giljohann DA, Mirkin CA, Thaxton CS. ACS Nano. 2012;6:276–285. doi: 10.1021/nn2035457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2) (a).Rothblat GH, Phillips MC. Curr. Opin. Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Asztalos BF, Tani M, Schaefer EJ. Curr. Opin. Lipidol. 2011;22:176–185. doi: 10.1097/MOL.0b013e3283468061. [DOI] [PubMed] [Google Scholar]; (c) Rosenson RS, Brewer HB, Jr., Chapman MJ, Fazio S, Hussain MM, Kontush A, Krauss RM, Otvos JD, Remaley AT, Schaefer EJ. Clin. Chem. 2011;57:392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- (3) (a).High density lipoproteins, dyslipidemia, and coronary heart disease; Schaefer EJ, editor. Springer; New York, N.Y.: 2010. Kontush A, Chapman MJ. High-density lipoproteins. Structure, metabolism, function and therapeutics. Wiley; Hoboken, N. J.; 2012.
- (4) (a).Murphy AJ, Remaley AT, Sviridov D. Curr. Pharm. Des. 2010;16:4134–4147. doi: 10.2174/138161210794519228. [DOI] [PubMed] [Google Scholar]; (b) Spillmann F, Schultheiss H-P, Tschoepe C, Van Linthout S. Curr. Pharm. Des. 2010;16:1517–1530. doi: 10.2174/138161210791050988. [DOI] [PubMed] [Google Scholar]; (c) Tardif J-C. J. Clin. Lipidol. 2010;4:399–404. doi: 10.1016/j.jacl.2010.08.018. [DOI] [PubMed] [Google Scholar]
- (5) (a).Van Lenten BJ, Wagner AC, Anantharamaiah GM, Navab M, Reddy ST, Buga GM, Fogelman AM. Curr. Atheroscler. Rep. 2009;11:52–57. doi: 10.1007/s11883-009-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Getz GS, Wool GD, Reardon CA. Curr. Atheroscler. Rep. 2010;12:96–104. doi: 10.1007/s11883-010-0097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hovingh G, Bochem AE, Kastelein JJ. Curr. Opin. Lipidol. 2010;21:481–486. doi: 10.1097/MOL.0b013e3283404507. [DOI] [PubMed] [Google Scholar]; (d) Osei-Hwedieh DO, Amar M, Sviridov D, Remaley AT. Pharmacol. Ther. 2011;130:83–91. doi: 10.1016/j.pharmthera.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gordon SM, Davidson WS. Curr. Opin. Endocrinol. Diabetes Obes. 2012;19:109–114. doi: 10.1097/MED.0b013e32835056d4. [DOI] [PubMed] [Google Scholar]
- (6).Frank PG, Marcel YL. J. Lipid Res. 2000;41:853–872. [PubMed] [Google Scholar]
- (7) (a).Mishra VK, Palgunachari MN, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. J. Biol. Chem. 1995;270:1602–11. doi: 10.1074/jbc.270.4.1602. [DOI] [PubMed] [Google Scholar]; (b) Demoor L, Boutillon C, Fievet C, Vanloo B, Baert J, Rosseneu M, Fruchart J-C, Tartar A. Eur. J. Biochem. 1996;239:74–84. doi: 10.1111/j.1432-1033.1996.0074u.x. [DOI] [PubMed] [Google Scholar]; (c) Nion S, Demoor L, Boutillon C, Luchoomun J, Vanloo B, Fievet C, Castro G, Rosseneu M, Fruchart J-C, Tartar A, Clavey V. Atherosclerosis. 1998;141:227–235. doi: 10.1016/s0021-9150(98)00176-2. [DOI] [PubMed] [Google Scholar]; (d) Sethi AA, Stonik JA, Thomas F, Demosky SJ, Amar M, Neufeld E, Brewer HB, Davidson WS, D’Souza W, Sviridov D, Remaley AT. J. Biol. Chem. 2008;283:32273–32282. doi: 10.1074/jbc.M804461200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wool GD, Reardon CA, Getz GS. J. Lipid Res. 2008;49:1268–1283. doi: 10.1194/jlr.M700552-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wool GD, Vaisar T, Reardon CA, Getz GS. J. Lipid Res. 2009;50:1889–1900. doi: 10.1194/jlr.M900151-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) D’Souza W, Stonik JA, Murphy A, Demosky SJ, Sethi AA, Moore XL, Chin-Dusting J, Remaley AT, Sviridov D. Circ. Res. 2010;107:217–227. doi: 10.1161/CIRCRESAHA.110.216507. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Lu SC, Atangan L, Won Kim K, Chen MM, Komorowski R, Chu C, Han J, Hu S, Gu W, Veniant M, Wang M. J. Lipid Res. 2012;53:643–52. doi: 10.1194/jlr.M020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776–9. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- (9) (a).Sadler K, Tam JP. Rev. Mol. Biotechnol. 2002;90:195–229. doi: 10.1016/s1389-0352(01)00061-7. [DOI] [PubMed] [Google Scholar]; (b) Zhong W, Skwarczynski M, Fujita Y, Simerska P, Good MF, Toth I. Aust. J. Chem. 2009;62:993–999. [Google Scholar]; (c) Angeles-Boza AM, Erazo-Oliveras A, Lee Y-J, Pellois J-P. Bioconj. Chem. 2010;21:2164–2167. doi: 10.1021/bc100130r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Breurken M, Lempens EHM, Temming RP, Helms BA, Meijer EW, Merkx M. Bioorg. Med. Chem. 2011;19:1062–1071. doi: 10.1016/j.bmc.2010.07.058. [DOI] [PubMed] [Google Scholar]
- (10) (a).Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Arterioscler. Thromb. Vasc. Biol. 1996;16:328–38. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]; (b) Panagotopulos SE, Witting SR, Horace EM, Hui DY, Maiorano JN, Davidson WS. J. Biol. Chem. 2002;277:39477–39484. doi: 10.1074/jbc.M207005200. [DOI] [PubMed] [Google Scholar]
- (11) (a).Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. J. Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]; (b) Chromy BA, Arroyo E, Blanchette CD, Bench G, Benner H, Cappuccio JA, Coleman MA, Henderson PT, Hinz AK, Kuhn EA, Pesavento JB, Segelke BW, Sulchek TA, Tarasow T, Walsworth VL, Hoeprich PD. J. Am. Chem. Soc. 2007;129:14348–14354. doi: 10.1021/ja074753y. [DOI] [PubMed] [Google Scholar]
- (12) (a).Rothblat GH, de la Llera-Moya M, Favari E, Yancey PG, Kellner-Weibel G. Atherosclerosis. 2002;163:1–8. doi: 10.1016/s0021-9150(01)00713-4. [DOI] [PubMed] [Google Scholar]; (b) de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. Arterioscler. Thromb. Vasc. Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kane JP, Malloy MJ. Curr. Opin. Lipidol. 2012;23:367–371. doi: 10.1097/MOL.0b013e328353eef1. [DOI] [PubMed] [Google Scholar]
- (13).Khelashvili G, Pabst G, Harries D. J. Phys. Chem. B. 2010;114:7524–7534. doi: 10.1021/jp101889k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14) (a).von Eckardstein A, Nofer J-R, Assmann G. Arterioscler. Thromb. Vasc. Biol. 2001;21:13–27. doi: 10.1161/01.atv.21.1.13. [DOI] [PubMed] [Google Scholar]; (b) Oram JF, Heinecke JW. Physiol. Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- (15).Troutt JS, Alborn WE, Mosior MK, Dai J, Murphy AT, Beyer TP, Zhang Y, Cao G, Konrad RJ. J. Lipid Res. 2008;49:581–587. doi: 10.1194/jlr.M700385-JLR200. [DOI] [PubMed] [Google Scholar]
- (16).Li H-H, Thomas MJ, Pan W, Alexander E, Samuel M, Sorci-Thomas MG. J. Lipid Res. 2001;42:2084–2091. [PubMed] [Google Scholar]
- (17).Ishida BY, Albee D, Paigen B. J. Lipid Res. 1990;31:227–236. [PubMed] [Google Scholar]
- (18).Werle M, Bernkop-Schnuerch A. Amino Acids. 2006;30:351–367. doi: 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- (19).Graversen Jonas H, Laurberg Jacob M, Andersen Mikkel H, Falk E, Nieland J, Christensen J, Etzerodt M, Thogersen Hans C, Moestrup Soren K. J. Cardiovasc. Pharmacol. 2008;51:170–7. doi: 10.1097/FJC.0b013e31815ed0b9. [DOI] [PubMed] [Google Scholar]
- (20) (a).Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Arterioscler. Thromb. Vasc. Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]; (b) Van Craeyveld E, Gordts SC, Singh N, Jacobs F, De Geest B. Acta Cardiol. 2012;67:11–21. doi: 10.1080/ac.67.1.2146561. [DOI] [PubMed] [Google Scholar]
- (21).Pini A, Falciani C, Bracci L. Curr. Protein Pept. Sci. 2008;9:468–477. doi: 10.2174/138920308785915227. [DOI] [PubMed] [Google Scholar]
- (22).Graversen JH, Laurberg JM, Andersen MH, Falk E, Nieland J, Christensen J, Etzerodt M, Thogersen HC, Moestrup SK. J. Cardiovasc. Pharmacol. 2007;51:170–177. doi: 10.1097/FJC.0b013e31815ed0b9. [DOI] [PubMed] [Google Scholar]
- (23) (a).Bielicki JK, Zhang H, Cortez Y, Zheng Y, Narayanaswami V, Patel A, Johansson J, Azhar S. J. Lipid Res. 2010;51:1496–503. doi: 10.1194/jlr.M003665. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Handattu SP, Datta G, Epand RM, Epand RF, Palgunachari MN, Mishra VK, Monroe CE, Keenum TD, Chaddha M, Anantharamaiah GM, Garber DW. J. Lipid Res. 2010;51:3491–9. doi: 10.1194/jlr.M006916. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Navab M, Reddy ST, Anantharamaiah GM, Imaizumi S, Hough G, Hama S, Fogelman AM. J. Lipid Res. 2011;52:1200–10. doi: 10.1194/jlr.M013144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24) (a).Benoit P, Emmanuel F, Caillaud JM, Bassinet L, Castro G, Gallix P, Fruchart J-C, Branellec D, Denefle P, Duverger N. Circulation. 1999;99:105–110. doi: 10.1161/01.cir.99.1.105. [DOI] [PubMed] [Google Scholar]; (b) Shah PK, Yano J, Reyes O, Chyu K-Y, Kaul S, Bisgaier CL, Drake S, Cercek B. Circulation. 2001;103:3047–3050. doi: 10.1161/hc2501.092494. [DOI] [PubMed] [Google Scholar]; (c) Busseuil D, Shi Y, Mecteau M, Brand G, Kernaleguen AE, Thorin E, Latour JG, Rheaume E, Tardif JC. Br. J. Pharmacol. 2008;154:765–773. doi: 10.1038/bjp.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Segrest JP, Garber D, Brouillette CG, Harvey SC, Anantharamaiah GM. Adv. Protein Chem. 1994;45:303–69. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- (26).Watson CE, Weissbach N, Kjems L, Ayalasomayajula S, Zhang Y, Chang I, Navab M, Hama S, Hough G, Reddy ST, Soffer D, Rader DJ, Fogelman AM, Schecter A. J. Lipid Res. 2011;52:361–373. doi: 10.1194/jlr.M011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bloedon LT, Dunbar R, Duffy D, Pinell-Salles P, Norris R, DeGroot BJ, Movva R, Navab M, Fogelman AM, Rader DJ. J. Lipid Res. 2008;49:1344–52. doi: 10.1194/jlr.P800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28). [accessed in February 2013]; http://www.prnewswire.co.uk/news-releases/esperion-reports-positive-results-for-second-phase-1-study-of-etc-642-155462665.html.
- (29).Navab M. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- (30).Navab M, Ruchala P, Waring AJ, Lehrer RI, Hama S, Hough G, Palgunachari MN, Anantharamaiah GM, Fogelman AM. J. Lipid Res. 2009;50:1538–47. doi: 10.1194/jlr.M800539-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.