

Abstract
Gammagard IVIg is a therapeutic approach to treat Alzheimer's disease currently in phase 3 clinical trials. Despite the reported efficacy of the approach the mechanism of action is poorly understood. We have previously shown that intracranial injection of anti-Aβ antibodies into the frontal cortex and hippocampus reveals important information regarding the time course of events once the agent is in the brain. In the current study we compared IVIg, mouse-pooled IgG, and the anti-Aβ antibody 6E10 injected intracranially into the frontal cortex and hippocampus of 7-month-old APP/PS1 mice. We established a time course of events ranging from 1 to 21 d postinjection.
IVIg and pooled mouse IgG both significantly reduced Aβ deposition to the same degree as the 6E10 anti-Aβ antibody; however, the clearance was much slower to occur, happening between the 3 and 7 d time points. In contrast, as we have previously shown, Aβ reductions were apparent with the 6E10 anti-Aβ group at the 1 d time point. Also, neuroinflammatory profiles were significantly altered by the antibody treatments. APP/PS1 transgenic mice at 7 months of age typically exhibit an M2a inflammatory phenotype. All antibody treatments stimulated an M2b response, yet anti-Aβ antibody was a more rapid change. Because the neuroinflammatory switch occurs before the detectable reductions in amyloid deposition, we hypothesize that the IVIg and pooled mouse IgG act as immune modulators and this immune modulation is responsible for the reductions in amyloid pathology.
Introduction
Alzheimer's disease (AD) is characterized by deposition of amyloid plaques, composed of aggregated amyloid-β (Aβ) peptide, and neurofibrillary tangles, composed of aggregated tau protein (Hyman et al., 2012). Anti-Aβ immunotherapy has been shown to improve cognition while lowering brain Aβ in numerous mouse models (Morgan et al., 2000; Wilcock et al., 2004c; Maier et al., 2006). It has been shown that anti-Aβ immunotherapy lowers brain Aβ through both central and peripheral mechanisms (Wilcock and Colton, 2009). We previously showed that anti-Aβ antibodies activate microglia, and the microglial activation is important for the clearance of compact amyloid deposits (Wilcock et al., 2003; Wilcock et al., 2004a). Antibodies can also catalytically disaggregate amyloid deposits in vitro (Solomon et al., 1997). When administered systemically, anti-Aβ antibodies can enter the CNS and activate microglia (Wilcock et al., 2004c), but they also result in the efflux of Aβ from the brain via a mechanism called the peripheral sink (DeMattos et al., 2001).
Adverse cerebrovascular events continue to plague the anti-Aβ immunotherapy field. These adverse events include microhemorrhages and vasogenic edema (termed ARIA-H and ARIA-E, respectively) (Wilcock et al., 2004b; Sperling et al., 2011). We have previously published data that suggests activation of matrix metalloproteinases (MMPs) MMP2 and MMP9 are associated with the increase in microhemorrhage by anti-Aβ immunotherapy (Wilcock et al., 2011a).
Intravenous Ig (IVIg) is a term applied to the pharmaceutical preparation of pooled human IgG derived from plasma. IVIg is used to treat immune deficiency disorders including idiopathic thrombocytopenic purpura and hypogammaglobulinemia (Hartung et al., 2009). The described immunomodulatory effects of IVIg include downregulation of antibody production, inhibition of B-cell-mediated IL-6 production and inhibition of nitric oxide secretion, among many other immunological effects (for review, see Dodel et al., 2010). After early reports of IVIg being tested in AD patients (Dodel et al., 2004), an open label dosing of Gammagard, a commercially available IVIg, reported good tolerance and increased plasma Aβ following infusion. MMSE scores also increased in the small number of patients receiving the IVIg (Relkin et al., 2009). Phase 3 trials are ongoing after passing the futility analysis in January 2012. Despite the early indicators of a positive clinical benefit of IVIg in AD patients, the underlying mechanism(s) of action are unknown. Some have suggested the presence of anti-Aβ antibodies accounts for the effects of IVIg (Magga et al., 2010), while others suggest immune modulation effects (Dodel et al., 2010).
We have previously shown that anti-Aβ antibody administration systemically results in significant changes in inflammatory markers in the brain (Wilcock et al., 2011b). We are using the macrophage phenotypes of M1, M2a, M2b, and M2c to characterize the neuroinflammatory phenotype of the brain. The use of the multiple markers of neuroinflammation allows us to better characterize the inflammatory state of the brain beyond examining typical microglial cell surface markers of “activation.” In the current study we use a time course to examine the effects of IVIg and mouse IgG on neuroinflammation and Aβ load, and contrast these effects with anti-Aβ antibody.
Materials and Methods
Animals.
APP/PS1 transgenic mice (C57BL6 mice carrying human APPSwe and PS1-dE9 mutations; Jankowsky et al., 2004) were bred in-house and aged to 7 months. Then 120 7-month-old APP/PS1 mice were assigned to one of four injection groups: saline, anti-Aβ antibody (6E10, Aβ3–8, IgG1; Covance), IVIg (trade name Gammagard from Baxter and composed primarily of IgG1 and IgG2; Schwab and Nimmerjahn, 2013), or mouse IgG (Sigma-Aldrich; composed primarily of IgG1 and IgG2a; Sant'Anna et al., 1985). The anti-Aβ antibody, IVIg, and mouse IgG were diluted to a final concentration of 1 mg/ml in sterile saline. Six mice in each group (three female and three male) were killed at each time point (1, 3, 7, 14, and 21 d). The study was approved by the University of Kentucky Institutional Animal Care and Use Committee and conformed to the National Institutes of Health Guide for the Care and Use of Animals in Research.
Bilateral intracranial injection.
On the day of surgery mice were weighed, anesthetized with isoflurane, and placed in a stereotaxic apparatus (51733D digital dual manipulator mouse stereotaxic frame; Stoelting). A mid-sagittal incision was made to expose the cranium and four burr holes were drilled with a dental drill mounted in the stereotaxic frame over the frontal cortex and hippocampus to the following coordinates: frontal cortex, anteroposterior, +1.7 mm, lateral ± 2.0 mm; hippocampus, anteroposterior −2.7 mm; lateral, ± 2.5 mm, all taken from bregma. A 26 gauge needle attached to a 10 ml Hamilton syringe (Hamilton) containing the solution to be injected was lowered 3.0 mm ventral to bregma, and a 2 μl injection was made over a 2 min period. The incision was cleaned and closed with surgical staples for the 1–7 d time points, and sutures for the 14 and 21 d time points. Sutures were removed at 10 d.
Tissue processing and histology.
After injection with a lethal dose of pentobarbital, mice were perfused intracardially with 25 ml of normal saline. Brains were rapidly removed and bisected in the mid-sagittal plane. The left half was immersion fixed in freshly prepared 4% paraformaldehyde. The right half was dissected with the frontal cortex and hippocampus being isolated, flash frozen in liquid nitrogen, and stored at −80°C. The left hemibrain was passed through a series of 10, 20, and 30% sucrose solutions as cryoprotection and 25 μm frozen horizontal sections were collected using a sliding microtome and stored floating in PBS containing sodium azide at 4°C.
Sections spaced 300 μm spanning the estimated injection site were initially mounted and stained by cresyl violet to identify the injection site. For all subsequent histology and immunohistochemistry six sections spanning the injection site, spaced ∼100 μm apart were selected and analyzed. Free-floating immunohistochemistry for CD45 (rat monoclonal; Thermo Scientific) and Aβ (rabbit polyclonal Aβ1–16; Invitrogen) were performed as described previously (Wilcock et al., 2003).
ELISA measurement.
Protein was extracted for Aβ analysis from the right frontal cortex using a two-step extraction method. First, the brain was homogenized in PBS containing a complete protease and phosphatase inhibitor (Pierce Biotechnology). These samples were centrifuged at 16,000 × g at 4°C for 1 h. The supernatant was removed and became the “soluble” extract. The resulting pellet was homogenized in 100 μl of 70% formic acid and centrifuged at 16,000 × g at 4°C for 1 h. The supernatant was removed and neutralized 1:20 with 1 m Tris-HCl and became the “insoluble” extract. Protein concentration for both the soluble and insoluble extracts was determined using the bicinchoninic acid protein assay according to manufacturer's instructions (Thermo Scientific). We used the Meso-Scale Discovery multiplex ELISA system to measure Aβ38, Aβ40, and Aβ42 (MSD). ELISA kits were run according to the manufacturer's instructions. We took the soluble extract remaining after Aβ analysis and measured levels of MMP9, tissue inhibitor of metalloproteinases 1 (TIMP1), MMP2, and TIMP2 by ELISA according to manufacturer instructions (R&D Systems). We also measured the levels of mouse and human IgG to detect the injected antibodies. We used the Easy-Titer IgG assay kits (Thermo Scientific) for mouse IgG and human IgG.
Quantitative real-time reverse transcription (RT)-PCR.
RNA was extracted from left hippocampus using the Trizol Plus RNA Purification System (Ambion, Invitrogen) according to the manufacturer's instructions. RNA was quantified using the BioSpec Nano spectrophotometer (Shimadzu) and cDNA was reverse transcribed using the cDNA High Capacity kit (Applied Biosystems) according to the manufacturer's instructions. Real-time PCR was performed using the 384-well microfluidic card custom TaqMan assays containing TaqMan Gene Expression probes for our genes of interest (Applied Biosystems, Invitrogen). All gene expression data were normalized to 18S rRNA expression. Fold change compared was determined using the δ δCt method.
Analysis.
Data are presented as mean ± SEM. Statistical analysis was performed using the JMP statistical analysis program (SAS). Statistical significance was assigned where the p value was lower than 0.05. One-way ANOVA and two-way ANOVA were used, where appropriate, to detect treatment differences and differences within treatment groups along the time course.
Results
Measurement of mouse IgG in the protein extracted from the right frontal cortex showed that mice injected with mouse IgG and anti-Aβ IgG had significantly increased levels of IgG in the brain 1 d after injection (Fig. 1A). This was reduced dramatically by the 3 d time point and was down to the levels of mice receiving saline or human IVIg by the 7 d time point. At no time point did we observe a difference between the mouse IgG and anti-Aβ antibody-injected mice for the amount of IgG present suggesting the clearance kinetics of the IgG was identical (Fig. 1A). Similarly, quantification of human IgG showed significant increases only in the mice receiving the human IVIg at the 1 d time point. This was reduced but still significantly elevated at 3 d and was undetectable at the 7 d time point (Fig. 1B).
Figure 1.
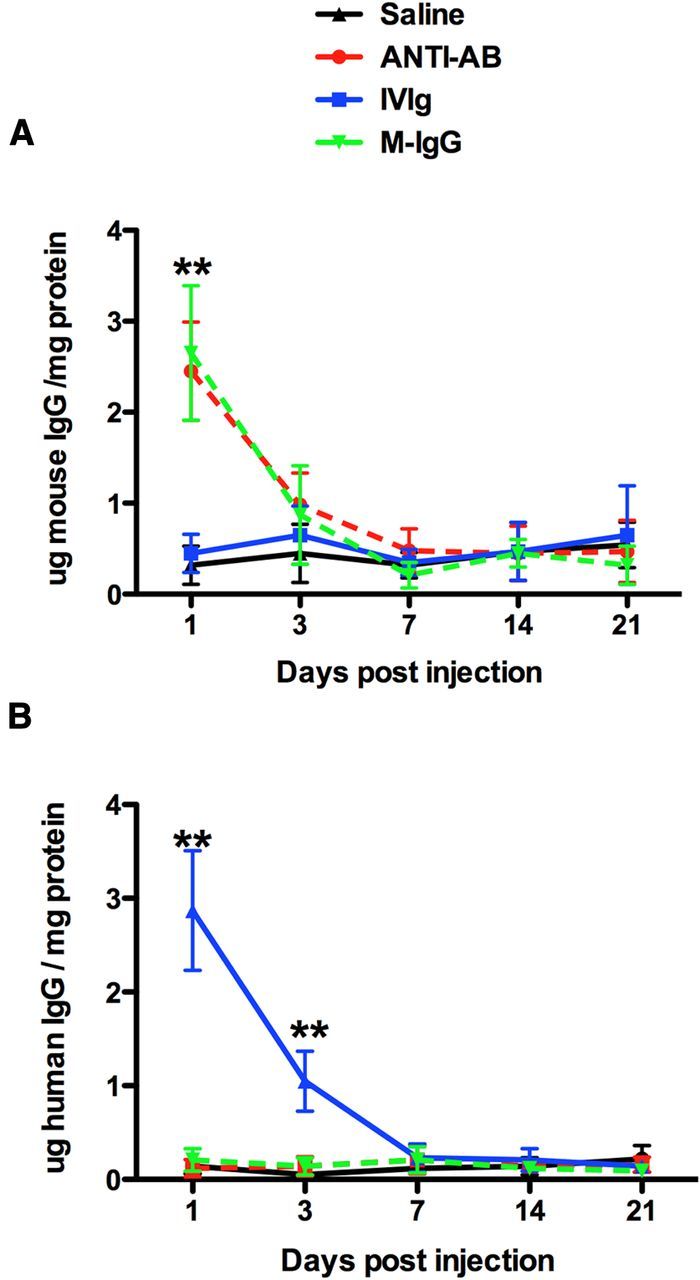
IgG levels in the brain are rapidly decreased between 1 and 3 d following injection. A, Mouse IgG levels in the right frontal cortex over the time course from 1 to 21 d. B, Human IgG levels in the right frontal cortex over the time course form 1 to 21 d. **p < 0.01 compared with saline.
Total Aβ load measured by immunohistochemistry in the frontal cortex and hippocampus of the saline-injected mice was as previously shown for this mouse model (Fig. 2A–C; Jankowsky et al., 2004). We found that at the earliest time point, 1 d postinjection, the anti-Aβ antibody had significantly reduced the brain Aβ load in both frontal cortex and hippocampus (Fig. 2D; hippocampus shown; Fig. 2M; quantification). There was a slight further reduction in Aβ with anti-Aβ antibody between 1 and 3 d (Fig. 1D,E,M) and then no further reductions were observed. In fact, Aβ levels began to steadily increase from the 7–21 d time points (Fig. 2M). In contrast, both reductions in Aβ load were not observed with IVIg or mouse IgG until the 7 d time point, indicating that this reduction occurred between the 3 and 7 d time points (Fig. 2H,I,K–M). At no time point did we observe a significant difference between the IVIg and mouse IgG treatment groups.
Figure 2.
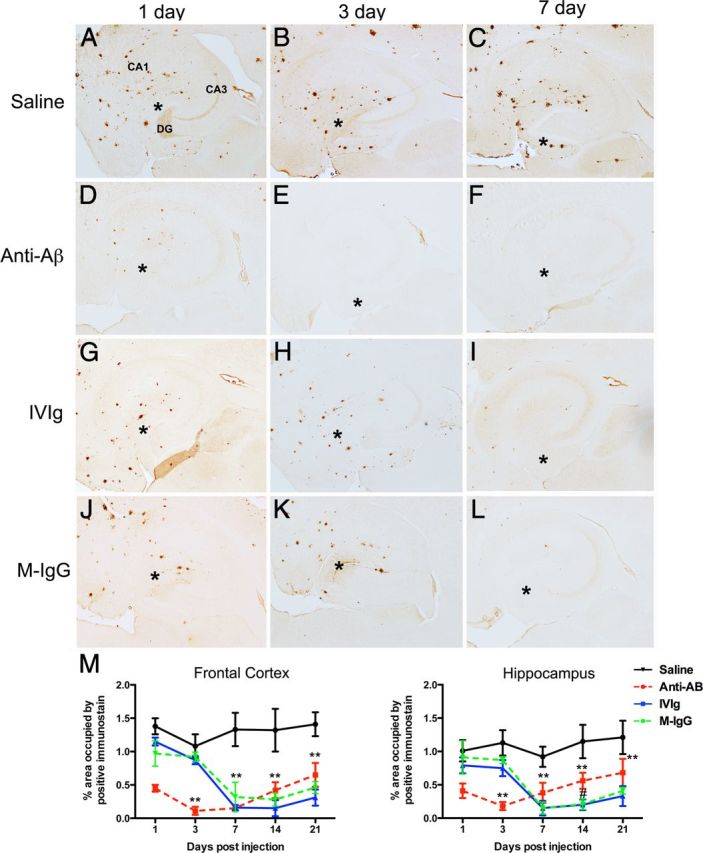
Brain Aβ is reduced by anti-Aβ antibodies, IVIg, and mouse IgG. A–C, Show the hippocampus of mice receiving saline 1 (A), 3 (B), and 7 (C) d following injection. D–F, Show the hippocampus of mice receiving anti-Aβ antibody 1 (D), 3 (E), and 7 (F) d following injection. G–I, Show the hippocampus of mice receiving IVIg 1 (G), 3 (H), and 7 (I) d following injection. J–L, Show the hippocampus of mice receiving mouse IgG 1 (J), 3 (K), and 7 (L) d following injection. In A, the dentate gyrus (DG), cornu ammonis 3 (CA3), and cornu ammonis 1 (CA1) regions are indicated to provide orientation. Asterisks in A–L indicate the injection site within the hippocampus. M, Shows quantification of Aβ across the full time course, from 1 to 21 d, in the frontal cortex and hippocampus. **p < 0.01 compared with saline for all points under the asterisk; #p < 0.05 for IVIg and mouse IgG compared with anti-Aβ antibody.
Measurement of Aβ levels by ELISA showed similar patterns as the immunohistochemistry. The soluble pools of Aβ1–40 and Aβ1–42 were significantly reduced by anti-Aβ antibody at 1 d compared with saline, and a further slight reduction was observed at the 3 d time point. Interestingly, soluble Aβ1–40 increased dramatically between the 3 and 21 d time point, reaching the levels observed in mice receiving saline injection (Fig. 3A). While a similar pattern was observed with soluble Aβ1–42, the increase between 3 and 21 d was less dramatic (Fig. 3B). Both mouse IgG and IVIg injection reduced soluble Aβ1–40 and Aβ1–42 between the 3 and 7 d time points and showed a gradual increase between the 7 and 21 d time points, although this increase was less dramatic for the Aβ1–40 than the anti-Aβ antibody group (Fig. 3A,B). In contrast to the soluble pools of Aβ, insoluble Aβ1–40 was reduced between the 1 and 3 d time points by the anti-Aβ antibody, while insoluble Aβ1–42 was already reduced at the 1 d time point. Mouse IgG and IVIg both showed significant reductions in insoluble Aβ1–40 and Aβ1–42 between the 3 and 7 d time points (Fig. 3C,D). Of note, is the apparent increased efficacy of IVIg to lower insoluble Aβ1–40 compared with the reduction seen by anti-Aβ antibody or mouse IgG (Fig. 3C).
Figure 3.

Both soluble and insoluble Aβ1–40 and Aβ1–42 are reduced by anti-Ab antibodies, IVIg, and mouse IgG. A, B, Show ELISA measurement of Aβ1–40 (A) and Aβ1–42 (B) in our soluble protein extract. C, D, Show ELISA measurement of Aβ-140 (C) and Aβ1–42 (D) in our insoluble, formic acid protein extract. *p < 0.05; **p < 0.01 compared with saline for all points under the asterisk; #p < 0.05 for IVIg and mouse IgG compared with anti-Aβ antibody. ∧p < 0.05 for IVIg compared with both mouse IgG and anti-Aβ antibody.
To determine the role of neuroinflammation we first examined CD45 immunohistochemistry. Saline-injected mice showed low levels of microglial activation around plaques in the frontal cortex (images not shown) and hippocampus (Fig. 4A–C). One day following injection a small number of CD45-positive cells could be seen associated with the injection site (Fig. 4A), but these were not detectable at the 3 d time point and beyond (Fig. 4B,C). In contrast, diffuse microglial activation could be seen throughout the dentate gyrus, CA3, and CA2 regions of the hippocampus 1 d following injection (Fig. 4D). This activation then localized to the granule cell layer of the dentate gyrus and the pyramidal cell layer of the CA3 and CA2 regions (Fig. 4E). We have previously shown this similar pattern of microglial activation following anti-Aβ antibody injection along this same time course (Wilcock et al., 2003). Some microglial activation in these cell layers remained at 7 d and the activation resolved close to saline-injected levels by 21 d (Fig. 4F,M). While IVIg and mouse IgG showed some increased CD45 expression at 1 and 3 d, it was not until 7 d after injection that we observed significant microglial activation that resembled the anti-Aβ antibody-injected mice (Fig. 4G–L). Quantification of the CD45 immunohistochemistry showed that microglial activation peaked 3 d following injection with anti-Aβ antibody, but 7 d after injection with IVIg and mouse IgG in both the frontal cortex and hippocampus (Fig. 4M).
Figure 4.
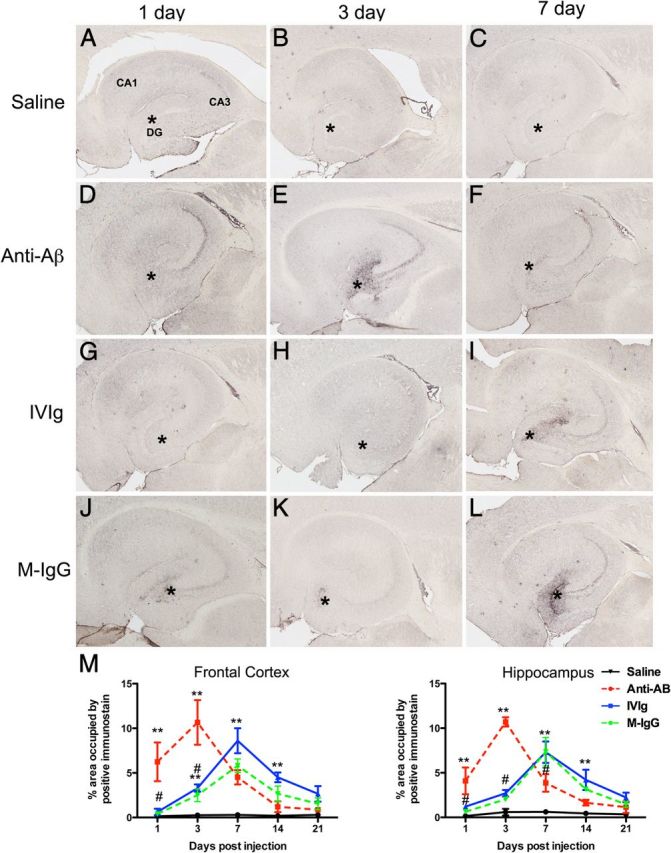
Microglial cells are activated by anti-Aβ antibodies, IVIg, and mouse IgG. A–C, Show the hippocampus of mice receiving saline 1 (A), 3 (B), and 7 (C) d following injection. D–F, Show the hippocampus of mice receiving anti-Aβ antibody 1 (D), 3 (E), and 7 (F) d following injection. G–I, Show the hippocampus of mice receiving IVIg 1 (G), 3 (H), and 7 (I) d following injection. J–L, Show the hippocampus of mice receiving mouse IgG 1 (J), 3 (K), and 7 (L) d following injection. In A, the dentate gyrus (DG), cornu ammonis 3 (CA3), and cornu ammonis 1 (CA1) regions are indicated to provide orientation. Asterisks in A–L indicate the injection site within the hippocampus. M, Shows quantification of CD45 immunoreactivity across the full time course, from 1 to 21 d, in the frontal cortex and hippocampus. **p < 0.01 compared with saline for the data point(s) directly under the asterisk; #p < 0.05 the difference between IVIg and mouse IgG compared with anti-Aβ antibody.
To better characterize the neuroinflammatory response to the injected antibodies, we isolated RNA from the right hippocampus and performed real-time RT-PCR for genes relatively specific for the four phenotypes of macrophages: M1, M2a, M2b, and M2c. We found that all M1 associated genes (IL-1β, MARCO, and TNF-a) were significantly increased at 3 d following anti-Aβ injection and quickly resolved by the 7 d time point (Fig. 5A). Mouse IgG and IVIg showed significant increases at the 7 d time point but this increase was slower to resolve (Fig. 5A). In contrast to the M1 markers, we found that M2a-associated genes (Arg1, IL-1Ra, and YM1) were significantly decreased at the 3 d time point with anti-Aβ antibody and at the 7 d time point with mouse IgG and IVIg (Fig. 5B). Again, the M2a markers were slower to reach control levels with mouse IgG and IVIg than the anti-Aβ antibody group, much like the M1 markers (Fig. 5B). Of particular interest is the M2b phenotype, which is known to be initiated by immune complexes. Markers of the M2b phenotype were significantly increased at the 1 d time point following anti-Aβ injection, and for two of the three markers (CD86 and FcγR1) peaked at 3 d before resolving rapidly at the 7 d time point (Fig. 5C). In contrast, Significant increases in M2b markers were observed at 3 d for the IVIg and mouse IgG-injected mice and, for the CD86 and FcγR1, peaked at 7 d before resolving at the 14 d time point (Fig. 5C). Of note for the M2c phenotype, TGF-β was significantly elevated at the 7 d time point following anti-Aβ injection but only a small increase was seen in the IVIg and mouse IgG-injected mice at the 14 d time point (Fig. 5D). The individual time point values with statistical significances are shown in Table 1.
Figure 5.
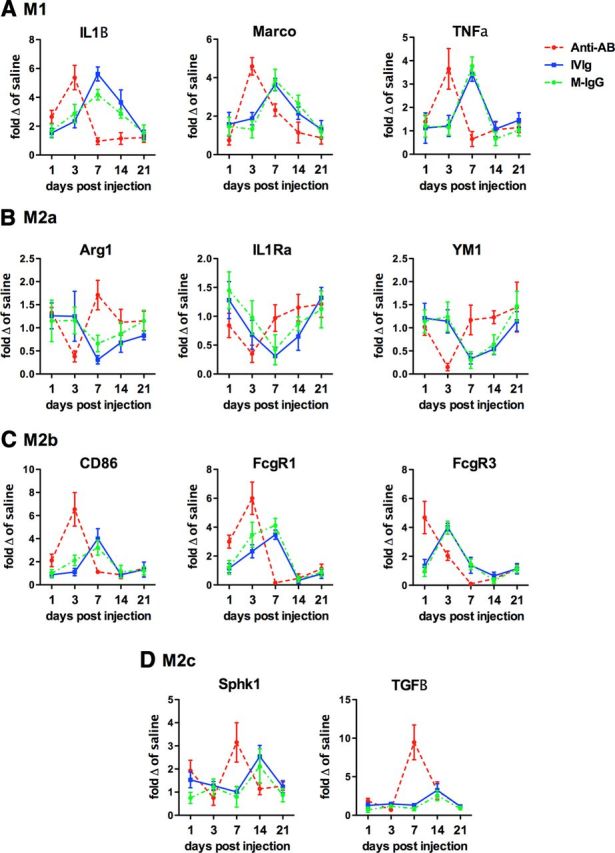
Neuroinflammatory phenotypes are altered by anti-Aβ antibodies, IVIg, and mouse IgG. The figure shows relative gene expression for genes representative of the M1 (A), M2a (B), M2b (C), and M2c (D) phenotypes. Data are shown as fold change relative to APP/PS1 mice receiving saline injection at the given time point. Due to the multiple statistical differences the statistical significance of the differences is highlighted in Table 1.
Table 1.
Individual data points for the neuroinflammatory marker gene expression
| T'ment | Survival | M1 |
M2a |
M2b |
M2c |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL1-β | MARCO | TNF-α | AG1 | IL1Ra | YM1 | CD86 | FcγR1 | FcγR3 | Sphk1 | TGF-β | ||
| Anti-Aβ | 1 d | 2.6 ± 0.5 | 0.8 ± 0.3 | 1.4 ± 0.3 | 1.3 ± 0.1 | 0.8 ± 0.2 | 1.0 ± 0.2 | 2.1 ± 0.5 | 3.0 ± 0.5 | 4.7 ± 1.1 | 1.9 ± 0.5 | 1.9 ± 0.3 |
| 3 d | 5.4 ± 0.8* | 4.6 ± 0.5* | 3.7 ± 0.9* | 0.4 ± 0.1* | 0.4 ± 0.2* | 0.2 ± 0.1* | 6.5 ± 1.5* | 6.0 ± 1.1* | 2.0 ± 0.3* | 0.8 ± 0.3 | 0.7 ± 0.1 | |
| 7 d | 0.9 ± 0.2 | 2.3 ± 0.3 | 0.65 ± 0.3 | 1.7 ± 0.3 | 1.0 ± 0.3 | 1.2 ± 0.3 | 1.1 ± 0.1 | 0.2 ± 0.1 | 0.1 ± 0.05 | 3.2 ± 0.9* | 9.5 ± 2.3* | |
| 14 d | 1.2 ± 0.4 | 1.2 ± 0.5 | 1.0 ± 0.2 | 1.1 ± 0.3 | 1.1 ± 0.2 | 1.2 ± 0.1 | 0.9 ± 0.3 | 0.5 ± 0.3 | 0.45 ± 0.3 | 1.1 ± 0.3 | 3.2 ± 1.1 | |
| 21 d | 1.2 ± 0.3 | 0.9 ± 0.3 | 1.1 ± 0.2 | 1.2 ± 0.2 | 1.2 ± 0.2 | 1.5 ± 0.5 | 1.5 ± 0.2 | 1.1 ± 0.3 | 1.1 ± 0.3 | 1.3 ± 0.2 | 1.1 ± 0.3 | |
| IVIg | 1 d | 1.5 ± 0.6 | 1.6 ± 0.6 | 1.1 ± 0.6 | 1.3 ± 0.3 | 1.3 ± 0.3 | 1.2 ± 0.3 | 0.9 ± 0.2 | 1.2 ± 0.3 | 1.3 ± 0.5 | 1.5 ± 0.6 | 1.3 ± 0.3 |
| 3 d | 2.3 ± 0.5 | 1.9 ± 0.3 | 1.2 ± 0.5 | 1.3 ± 0.5 | 0.7 ± 0.3 | 1.1 ± 0.3 | 1.1 ± 0.3 | 2.3 ± 0.5 | 4.0 ± 0.6* | 1.3 ± 0.2 | 1.5 ± 0.3 | |
| 7 d | 5.6 ± 0.5* | 3.7 ± 0.3* | 3.4 ± 0.3* | 0.3 ± 0.1* | 0.3 ± 0.2* | 0.3 ± 0.1* | 3.9 ± 0.9* | 3.5 ± 0.3* | 1.4 ± 0.6 | 1.0 ± 0.2 | 1.3 ± 0.2 | |
| 14 d | 3.6 ± 0.9 | 2.2 ± 0.3 | 1.1 ± 0.4 | 0.7 ± 0.2 | 0.7 ± 0.2 | 0.5 ± 0.1 | 0.9 ± 0.2 | 0.3 ± 0.2 | 0.7 ± 0.3 | 2.5 ± 0.5* | 3.3 ± 0.9* | |
| 21 d | 1.4 ± 0.3 | 1.3 ± 0.5 | 1.5 ± 0.6 | 0.8 ± 0.1 | 1.3 ± 0.2 | 1.1 ± 0.2 | 1.3 ± 0.7 | 0.8 ± 0.3 | 1.1 ± 0.4 | 1.2 ± 0.3 | 1.1 ± 0.3 | |
| M-IgG | 1 d | 1.4 ± 0.5 | 1.5 ± 0.5 | 1.2 ± 0.5 | 1.2 ± 0.5 | 1.5 ± 0.3 | 1.1 ± 0.3 | 1.0 ± 0.3 | 1.2 ± 0.5 | 0.9 ± 0.4 | 0.8 ± 0.2 | 0.7 ± 0.3 |
| 3 d | 2.9 ± 0.6 | 1.3 ± 0.5 | 1.1 ± 0.3 | 1.2 ± 0.3 | 1.0 ± 0.3 | 1.2 ± 0.3 | 2.1 ± 0.4 | 3.5 ± 0.9 | 3.9 ± 0.5* | 1.2 ± 0.4 | 1.2 ± 0.3 | |
| 7 d | 4.2 ± 0.4* | 3.9 ± 0.6* | 3.8 ± 0.4* | 0.7 ± 0.2* | 0.4 ± 0.2* | 0.3 ± 0.2* | 3.2 ± 0.6* | 4.1 ± 0.5* | 1.5 ± 0.4 | 0.8 ± 0.5 | 0.9 ± 0.3 | |
| 14 d | 2.9 ± 0.3 | 2.7 ± 0.4 | 0.7 ± 0.3 | 0.9 ± 0.3 | 0.9 ± 0.1 | 0.6 ± 0.2 | 1.2 ± 0.5 | 0.4 ± 0.2 | 0.3 ± 0.2 | 2.1 ± 0.8* | 2.6 ± 0.7* | |
| 21 d | 1.5 ± 0.5 | 1.2 ± 0.3 | 1.0 ± 0.2 | 1.2 ± 0.2 | 1.1 ± 0.3 | 1.5 ± 0.3 | 1.2 ± 0.3 | 0.9 ± 0.3 | 1.1 ± 0.3 | 0.9 ± 0.3 | 0.9 ± 0.3 | |
Bolded font shows points that are statistically significantly greater than saline-injected mice at the same time point. Time points with an asterisk illustrate the peak change for that gene. Italicized font shows points that are statistically significantly lower than saline-injected mice at the same time point.
We have previously shown that MMP2 and MMP9 activation is increased in mice that develop microhemorrhages due to anti-Aβ immunotherapy. Although we did not observe microhemorrhages in the current study, as expected due to the young age of the mice and the intracranial administration paradigm used, we did examine gene expression for the MMP2 and MMP9 systems to determine whether IVIg and mouse IgG have different effects on the systems than anti-Aβ antibody. We found that MMP3 (the activator of MMP9) and MMP9 gene expression were significantly elevated 1 and 3 d following injection of anti-Aβ antibody (Fig. 6A). The increase in mice injected with IVIg or mouse IgG were peaked at the 7 d time point; however, the increase in expression of MMP3 and MMP9 was significantly less than that observed with anti-Aβ antibody (Fig. 6A). The expression of TIMP1, the endogenous inhibitor of MMP9, was increased at 3 d in all groups with no significant difference between any of the antibody-injected groups (Fig. 6A). Protein levels of MMP9 and TIMP1, measured by ELISA, matched the time course of the RNA (Fig. 6B). Expression of MMP14 (the activator of MMP2) and MMP2 was significantly increased at the 1 d time point in mice receiving anti-Aβ antibody injection (Fig. 6C). We saw modest increases in MMP14 with IVIg and mouse IgG at the 3 d time point (Fig. 6C). MMP2 gene expression was also increased with IVIg and mouse IgG at the 3 d time point (Fig. 6C). TIMP2, the endogenous inhibitor of MMP2, was significantly increased at the 1 d time point following anti-Aβ antibody injection, and the 3 d time point following IVIg and mouse IgG injection (Fig. 6C). Interestingly, expression of all genes in the MMP2 system was significantly reduced to almost 0 at the 7 d time point with anti-Aβ antibody injection before rebounding back to control levels at 14 and 21 d (Fig. 6C). This was a phenomenon we did not observe with the IVIg or mouse IgG injection mice. Protein for MMP2 and TIMP2 matched the temporal patterns observed in the gene expression studies, with the exception of the lack of a reduction at the 7 d time point with anti-Aβ antibody.
Figure 6.
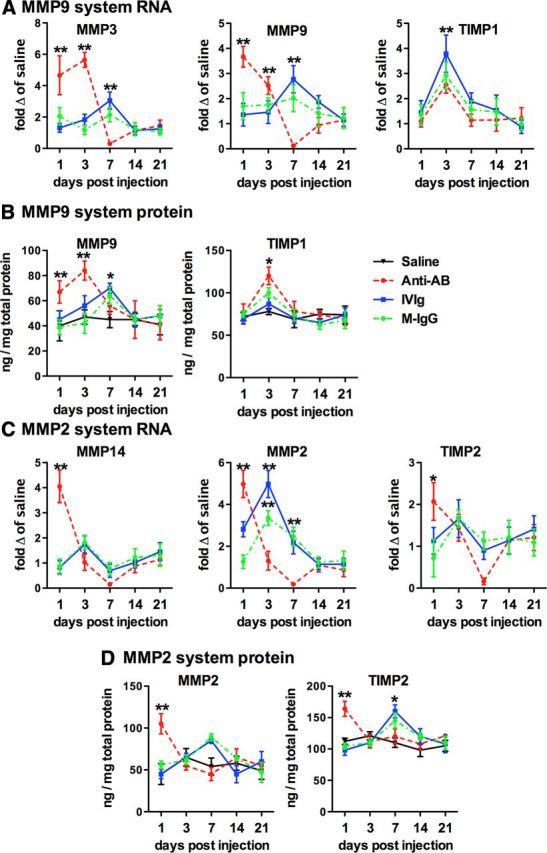
Both the MMP9 and MMP2 systems are activated by anti-Aβ antibodies, IVIg, and mouse IgG. A, C, Show relative gene expression of components of the MMP9 system (A) and the MMP2 system (C). Gene expression is shown as fold change compared with mice receiving saline injection at the given time point. B, D, Show protein expression of MMP9 and TIMP1 (B), and MMP2 and TIMP1 (D) measured by ELISA. *p < 0.05; **p < 0.01 for the data point shown compared with all other datasets at that time point. Where multiple groups are together, the statistical significance is compared with saline.
Discussion
IVIg is a therapeutic approach currently in clinical trials for the treatment of AD. Despite early indicators of some clinical benefit (Relkin et al., 2009), the mechanism(s) of action are unknown. There has been much speculation on the mechanisms, ranging from the presence of circulating anti-Aβ antibodies in IVIg (Dodel et al., 2002), and therefore acting in much the same way as anti-Aβ immunotherapy (Magga et al., 2010), to immune modulation, a known mechanism of action of IVIg used in the treatment of autoimmune disorders (Dodel et al., 2010). In the current study we applied a study design previously used to establish mechanism(s) of action of anti-Aβ antibody once it is in the brain (Wilcock et al., 2003). We intracranially administered IVIg, mouse IgG, or anti-Aβ antibody, all at the same concentration and volume, bilaterally into the frontal cortex and hippocampus of APP/PS1 transgenic mice and killed the mice 1, 3, 7, 14, and 21 d following injection. Mouse IgG was tested in addition to the human IVIg to determine whether there were any differences between these two groups. Mouse IgG should not contain antibodies to human Aβ protein and, therefore, if differences were observed between the mouse IgG and IVIg then we could conclude that there were unique antibodies in the IVIg contributing to its efficacy. We attempted to detect anti-Aβ antibodies in the mouse IgG preparation by either ELISA or decoration of plaques on tissue sections and were unable to achieve signal.
Overall, we found that IVIg and mouse IgG lower Aβ levels in brain by comparable degrees as that seen with anti-Aβ antibody. However, this reduction occurs over a slower time course. We also found that the modulation of the neuroinflammatory response occurs over a slower time course than anti-Aβ antibody and appears, in some instances, to be longer lasting. These differences in time course are not accounted for by the presence of the IgG in brain, since all the injected antibodies showed the same clearance kinetics as one another. Our data suggest that IVIg can lower Aβ as effectively as anti-Aβ immunotherapy, but the two therapies likely have distinct mechanisms of action due to the different time courses. However, it is also feasible that the same mechanisms are engaged with both anti-Aβ antibodies and IVIg/mouse IgG but simply delayed. Immune modulation is one likely mechanism of action suggested by our data. In addition, isotype differences may account for some of the differences between the IVIg/mouse IgG and anti-Aβ antibody groups. Mouse IgG and IVIg compose primarily IgG1 and IgG2 in IVIg and mouse IgG compared with IgG1 only in the anti-Aβ antibody.
Anti-Aβ antibody injection rapidly reduced Aβ burden in the brains of APP/PS1 transgenic mice, with the greatest reduction seen at the 1 d time point, as previously shown in Tg2576 mice (Wilcock et al., 2003). In contrast, we did not see significant reductions in Aβ with either the IVIg or mouse IgG until the 7 d time point, indicating the clearance occurs between the 3 and 7 d period. The difference between the anti-Aβ antibody and the IVIg/mouse IgG groups suggests that there may be distinct mechanisms of action. We focused on inflammation as an explanation for the differences in Aβ removal because we have previously found a relationship between microglial activation and Aβ clearance by anti-Aβ antibodies injected intracranially (Wilcock et al., 2003; Wilcock et al., 2004a). More recently, we reported that systemically administered anti-Aβ antibody results in altered neuroinflammatory phenotype in the brains of Tg2576 mice (Wilcock et al., 2011b). Most amyloid-depositing mice show an M2a phenotype primarily (Colton et al., 2006) and this is what we observed in our saline-injected mice. When we injected anti-Aβ antibodies we found increases in M1 genes and M2b genes at the 1 d time point, peaking at 3 d. There was also a concomitant reduction in M2a gene expression at the 3 d time point. Both IVIg and mouse IgG showed similar increases in M1 and M2a gene expression, but along a slower time course, peaking at 7 d.
While macrophages were once broadly grouped into M1 and M2 based on their expression of IL-12 and IL-10 (M1; high IL-12 and low IL-10, M2; low IL-12 and high IL-10), studies found other phenotypes that did not fit this description (Gerber and Mosser, 2001). The M2b macrophage was described in the periphery by Mosser et al. several years ago (Edwards et al., 2006). Called the type-II activated macrophage, the group elegantly showed that activation of the Fcγ receptors by immune complexes switches the macrophage phenotype to this unique state characterized by elevations in some M1 markers, particularly IL-1β, TNF-α, and IL-6, while there was increased IL-10 and no induction of IL-12 (Mantovani et al., 2004; Mosser and Edwards, 2008). There were also some markers that were identified to be more specific for an M2b phenotype, including CD86 (Edwards et al., 2006). CD86 is expressed on antigen-presenting cells and is a ligand for CD28 and CD152 on T-cells, leading to T-cell activation (Sansom et al., 2003). We have recently shown that in the AD brain samples, both early stage and late stage, the phenotype that is not observed is M2b (Sudduth et al., 2013a). In the current study, we show that the increased presence of IgG in the brain by intracranial administration, be it IVIg, mouse IgG, or specific anti-Aβ IgG, leads to the modulation of the inflammatory response away from M2a and into an M2b phenotype. We cannot be sure whether the increase in IL-1β, TNF-α, and MARCO are due to an M2b or M1 phenotype since the primary distinguishing factor between these is the balance of IL-10 and IL-12, and neither are consistently and reliably detectable in the mouse brain tissue. The concomitant increase in M2b-specific genes suggests that at least some of the increase in these M1 genes is a part of the M2b response.
Data from mouse models of disease, including neurodegenerative diseases, suggest that altering neuroinflammatory phenotype results in changes in progression of pathology. For instance, Gómez-Nicola et al. (2013) showed recently that changing the microglial phenotype from M1 to M2a in a mouse model of prion disease slowed neurodegeneration and disease progression. We have previously shown that lowering of the M2a response while increasing M1 markers is associated with reductions in Aβ with immunotherapy (Wilcock et al., 2011b); however, it is now possible that this was an M2b phenotype as opposed to an M1 based on our current data. More recently, we showed that lithium treatment increased M2a markers and decreased M1 markers in APP transgenic mice and was associated with increased amyloid deposition (Sudduth et al., 2012). Contrasting results were found with the PPARγ agonist, pioglitazone, where a switch from M1 to M2 was observed along with decreased plaque load (Mandrekar-Colucci et al., 2012). The contrasting data found in animal studies may be accounted for by nonimmunological effects of the drugs being used and suggests a clear need for better understanding of the various neuroinflammatory phenotypes in the brain. An elegant review recently discussed macrophage diversity in atherosclerosis, and there it was suggested that it is not necessarily the polarization to one phenotype or another, but rather a balance of phenotypes, that is necessary for the progression, or lack thereof, of the pathological process (Mantovani et al., 2009). It is possible that this is the case, too, for the diseased brain.
While microhemorrhages were not observed in the current study due to the age of the mice, and the intracranial administration route selected, they do continue to be a major concern in the development of immunotherapeutics for AD. We have previously shown that MMP2 and MMP9 activation is significantly increased in mice that develop microhemorrhages due to anti-Aβ immunotherapy (Wilcock et al., 2011a). MMP2 and MMP9 are heavily implicated in the pathogenesis of hemorrhagic transformation following ischemic stroke (Klein and Bischoff, 2011) and blood–brain barrier disruption in vascular dementia (Candelario-Jalil et al., 2011). We have also recently shown that MMP2 and MMP9 systems are activated in a mouse model of vascular dementia based on hyperhomocysteinemia (Sudduth et al., 2013b). To begin to assess the potential capacity for IVIg to induce microhemorrhages we assessed the MMP2 and MMP9 systems in the current study by quantitative RT-PCR and ELISA measurement of components of these systems. We found that IVIg, mouse IgG, and anti-Aβ antibodies activated both systems; however, a greater increase in the MMP9 system was observed with anti-Aβ antibodies.
In summary, we have shown that IVIg and mouse IgG, when present in the brain, lower Aβ deposition similarly to anti-Aβ antibodies, but with a different time course. We believe that the immunomodulatory functions of IVIg are active in the brain, turning on an M2b phenotype that may be involved in the reduction of the Aβ deposition. Overall, our data suggest that immunomodulation by IVIg may be beneficial in the treatment of AD.
Footnotes
This work was funded by a Baxter Biosciences research grant, and National Institutes of Health Grants NS079637 (D.M.W.) and P20GM103486 (D.M.W.).
Baxter Biosciences is the manufacturer of Gammagard.
References
- Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair JC, Edmonds E, Prestopnik J, Wills J, Rosenberg GA. Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke. 2011;42:1345–1350. doi: 10.1161/STROKEAHA.110.600825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodel RC, Du Y, Depboylu C, Hampel H, Frölich L, Haag A, Hemmeter U, Paulsen S, Teipel SJ, Brettschneider S, Spottke A, Nölker C, Möller HJ, Wei X, Farlow M, Sommer N, Oertel WH. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2004;75:1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodel R, Hampel H, Depboylu C, Lin S, Gao F, Schock S, Jäckel S, Wei X, Buerger K, Höft C, Hemmer B, Möller HJ, Farlow M, Oertel WH, Sommer N, Du Y. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer's disease. Ann Neurol. 2002;52:253–256. doi: 10.1002/ana.10253. [DOI] [PubMed] [Google Scholar]
- Dodel R, Neff F, Noelker C, Pul R, Du Y, Bacher M, Oertel W. Intravenous immunoglobulins as a treatment for Alzheimer's disease: rationale and current evidence. Drugs. 2010;70:513–528. doi: 10.2165/11533070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber JS, Mosser DM. Reversing lipopolysaccharide toxicity by ligating the macrophage Fc gamma receptors. J Immunol. 2001;166:6861–6868. doi: 10.4049/jimmunol.166.11.6861. [DOI] [PubMed] [Google Scholar]
- Gómez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of Microglial Proliferation during Chronic Neurodegeneration. J Neurosci. 2013;33:2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung HP, Mouthon L, Ahmed R, Jordan S, Laupland KB, Jolles S. Clinical applications of intravenous immunoglobulins (IVIg)–beyond immunodeficiencies and neurology. Clin Exp Immunol. 2009;158(Suppl 1):23–33. doi: 10.1111/j.1365-2249.2009.04024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41:271–290. doi: 10.1007/s00726-010-0689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magga J, Puli L, Pihlaja R, Kanninen K, Neulamaa S, Malm T, Härtig W, Grosche J, Goldsteins G, Tanila H, Koistinaho J, Koistinaho M. Human intravenous immunoglobulin provides protection against Abeta toxicity by multiple mechanisms in a mouse model of Alzheimer's disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier M, Seabrook TJ, Lazo ND, Jiang L, Das P, Janus C, Lemere CA. Short amyloid-beta (Abeta) immunogens reduce cerebral Abeta load and learning deficits in an Alzheimer's disease mouse model in the absence of an Abeta-specific cellular immune response. J Neurosci. 2006;26:4717–4728. doi: 10.1523/JNEUROSCI.0381-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. doi: 10.1161/ATVBAHA.108.180497. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2009;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Sansom DM, Manzotti CN, Zheng Y. What's the difference between CD80 and CD86? Trends Immunol. 2003;24:314–319. doi: 10.1016/s1471-4906(03)00111-x. [DOI] [PubMed] [Google Scholar]
- Sant'Anna OA, Mouton D, Ibanez OM, Bouthillier Y, Mevel JC, Reis MH, Biozzi G. Basal immunoglobulin serum concentration and isotype distribution in relation to the polygenic control of antibody responsiveness in mice. Immunogenetics. 1985;22:131–139. doi: 10.1007/BF00563510. [DOI] [PubMed] [Google Scholar]
- Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci U S A. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR, Jr, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367–385. doi: 10.1016/j.jalz.2011.05.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Wilson JG, Everhart A, Colton CA, Wilcock DM. Lithium treatment of APPSwDI/NOS2−/− mice leads to reduced hyperphosphorylated tau, increased amyloid deposition and altered inflammatory phenotype. PLoS One. 2012;7:e31993. doi: 10.1371/journal.pone.0031993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer's disease. Neurobiol Aging. 2013a;34:1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Powell DK, Smith CD, Greenstein A, Wilcock DM. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J Cereb Blood Flow Metab. 2013b;33:708–715. doi: 10.1038/jcbfm.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Colton CA. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets. 2009;8:50–64. doi: 10.2174/187152709787601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis. 2004a;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004b;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004c;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Morgan D, Gordon MN, Taylor TL, Ridnour LA, Wink DA, Colton CA. Activation of matrix metalloproteinases following anti-Abeta immunotherapy; implications for microhemorrhage occurrence. J Neuroinflammation. 2011a;8:115. doi: 10.1186/1742-2094-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Zhao Q, Morgan D, Gordon MN, Everhart A, Wilson JG, Lee JE, Colton CA. Diverse inflammatory responses in transgenic mouse models of AD and the effect of immunotherapy on these responses. ASN Neuro. 2011b;3:249–258. doi: 10.1042/AN20110018. [DOI] [PMC free article] [PubMed] [Google Scholar]