

SUMMARY
The K650E gain-of-function mutation in the tyrosine kinase domain of FGF receptor 3 (FGFR3) causes Thanatophoric Dysplasia type II, a neonatal lethal congenital dwarfism syndrome, and when acquired somatically, it contributes to carcinogenesis. In this report, we determine the crystal structure of the FGFR3 kinase domain harboring this pathogenic mutation and show that the mutation introduces a network of intramolecular hydrogen bonds to stabilize the active-state conformation. In the crystal, the mutant FGFR3 kinases are caught in the act of trans-phosphorylation on a kinase insert autophosphorylation site, emphasizing the fact that the K650E mutation circumvents the requirement for A-loop tyrosine phosphorylation in kinase activation. Analysis of this trans-phosphorylation complex sheds light onto the determinants of tyrosine trans-phosphorylation specificity. We propose that the targeted inhibition of this pathogenic FGFR3 kinase may be achievable by small molecule kinase inhibitors that selectively bind the active-state conformation of FGFR3 kinase.
INTRODUCTION
Fibroblast growth factor receptor (FGFR) tyrosine kinases mediate the pleiotropic effects of FGFs in human biology and pathology. Gain-of-function mutations in the tyrosine kinase domains of FGFRs underlie a wide array of human developmental disorders and malignancies (Beenken and Mohammadi, 2009; Goriely et al., 2009; Webster and Donoghue, 1997b; Wilkie, 2005). FGFs act in concert with either heparin sulfate cofactors or Klotho coreceptors to bind and dimerize the ecto-domains of FGFRs, bringing the cytoplasmic tyrosine kinase domains into proper proximity and orientation, which allows trans-phosphorylation on activation loop (A-loop) tyrosines to occur (Goetz and Mohammadi, 2013). This event triggers kinase activation and causes additional trans-phosphorylation reactions on tyrosines in the kinase C-terminal tail and juxtamembrane (JM) region to create specific recruitment sites for substrates, thereby increasing the proximity of substrate to the kinase and facilitating substrate phosphorylation (Bae et al., 2009; Chen et al., 2008; Goetz and Mohammadi, 2013; Mohammadi et al., 1996a).
Crystallographic and nuclear magnetic resonance (NMR) studies of FGFR kinase domains in the unphosphorylated and A-loop phosphorylated states, as well as constitutively active FGFR kinases that harbor pathogenic gain-of-function mutations (Bae et al., 2009; Chen et al., 2007, 2013; Mohammadi et al., 1996a), show that the FGFR kinase is principally regulated at the protein dynamics level by an autoinhibitory network of hydrogen bonds in the kinase hinge/interlobe region (termed “molecular brake”), which restrains the kinase from transitioning into the active state (Chen et al., 2007, 2013). Upon A-loop tyrosine phosphorylation, the phosphate moiety of the phosphorylated A-loop tyrosine makes intramolecular hydrogen bonds with an RTK-invariant arginine at the N-terminal end of the A-loop, helping to restructure the A-loop into the active conformation. The change in the conformation of the A-loop is then allosterically communicated to the kinase hinge region, causing disengagement of the molecular brake, and hence maximal kinase activation (Chen et al., 2007, 2013).
In FGFRs, the kinase hinge and A-loop are the two hotspots for pathogenic gain-of-function mutations (Bellus et al., 1995, 2000; Kan et al., 2002; Wilkie, 2005). Previously, we explored the mechanisms for FGFR activation by the pathogenic mutations at the kinase hinge, which led to the discovery of the autoinhibitory molecular brake (Chen et al., 2007). The K650E mutation in the A-loop of FGFR3 kinase is one of the most activating, and hence pathogenic mutations (Bellus et al., 2000; Naski et al., 1996; Webster et al., 1996; Webster and Donoghue, 1997a). When occurring in the germline, the K650E mutation causes Thanatophoric Dysplasia type II, a neonatal lethal dwarfism syndrome (Tavormina et al., 1995; Wilcox et al., 1998).If acquired somatically, it contributes to the progression of multiple myeloma, bladder cancer and benign skin tumors (Cappellen et al., 1999; Chesi et al., 1997; Logié et al., 2005).
There is a wealth of literature on the molecular mechanisms by which the hyperactive K650E mutant reduces proliferation of chondrocytes, causing them to prematurely enter into hypertrophic differentiation in the developing cartilage tissue. The effects of the K650E mutation on FGFR activation, maturation/trafficking, and downstream signaling pathways have been extensively studied (Foldynova-Trantirkova et al., 2012; Guo et al., 2008). Webster and colleagues showed that the A-loop tyrosines are dispensable for the constitutive activity of the K650E FGFR3 mutant, implying that K650E mutation activates the kinase in an A-loop phosphorylation-independent fashion (Webster et al., 1996). Hence, it was proposed that this pathogenic mutation mimics the action of A-loop tyrosine phosphorylation to activate the kinase (Webster et al., 1996). The molecular mechanism by which this mutation imparts constitutive activation upon FGFR3 kinase is perplexing, however, because in accordance with the crystal structures of A-loop phosphorylated FGFR1 and FGFR2 kinases, the corresponding lysine residues, namely, K656 and K659, make intramolecular hydrogen bonds with the phosphate moiety of the phosphorylated A-loop tyrosine and residues in the A-loop that actually help supporting the active A-loop conformation (Bae et al., 2010; Chen et al., 2007). Lievens and Liboi showed that the K650E mutation affects receptor maturation and that the constitutively active mutant accumulates as a mannose-rich and phosphorylated form in the endoplasmic reticulum, where it signals through activating STAT1 and Src (Lievens and Liboi, 2003). Using a neurite outgrowth differentiation assay in PC12 cells as readout, Nowroozi and colleagues suggested that activation of STAT1 and 3 may not be required for the K650E mutant to induce hypertrophic differentiation of the growth plate (Nowroozi et al., 2005). Agazie and colleagues showed that the K650E mutant requires SHP2 to induce oncogenic transformation of cells (Agazie et al., 2003).
The importance of elucidating the molecular mechanism by which the K650E mutation confers gain-of-function on FGFR3 kinase is 2-fold; first, it may provide new insight into the physiological mechanisms of FGFR kinase regulation, and second, it may facilitate the rational design of small molecule inhibitors that can specifically silence the hyperactivity of this pathogenic FGFR3 mutant. Here, we report the crystal structure of FGFR3 kinase that harbors the K650E mutation. The structure shows that the mutation functionally mimics the action of A-loop phosphorylation in activating the kinase by introducing a network of hydrogen bonds that stabilize the active-state conformation of the A-loop. Moreover, the structure provides a snapshot of two FGFR kinase molecules engaged in the act of trans-phosphorylation on the kinase insert tyrosine. Comparison of this trans-phosphorylation complex with a previously reported kinase trans-phosphorylation complex involving the C-terminal tail tyrosine (Chen et al., 2008) provides valuable insights into the molecular mechanism that controls trans-phosphorylation specificity. Based on our structural data, targeted inhibition of this pathogenic FGFR3 kinase can be achieved by small molecule kinase inhibitors that selectively bind the active-state conformation of FGFR3 kinase.
RESULTS
To elucidate the molecular basis by which the pathogenic K650E mutation imparts constitutive activation upon FGFR3 kinase in human diseases, we solved the crystal structure of the FGFR3KK650E mutant in complex with AMP-PCP, a nonhydrolyzable ATP analog, at 2.35Å resolution (Figure 1A). Data collection and structure refinement statistics are given in Table 1. The refined model consists of residues L459 to T755, one AMP-PCP molecule, two Mg2+ ions, and 128 water molecules. FGFR3KK650E exhibits the canonical two-lobe architecture of protein kinases with the smaller N-lobe comprising a twisted five-stranded β sheet and the α helix C, and the larger C-lobe consists mainly of α helices (Figure 1). The ATP analog AMP-PCP is tucked snugly in the ATP-binding cleft between the two lobes, where it makes numerous hydrogen bonds and hydrophobic contacts with residues in both lobes as well as in the kinase hinge region (Figure 1). The entire A-loop, bracketed between 635DFG and LPV658 motifs, is ordered, including the two tyrosine residues (Y647 and Y648), the phosphorylation of which is necessary for kinase activation. Most of the loop between the αD and αE helices, better known as the kinase insert region, is ordered as well (Figure 1A). The ordering of this highly flexible loop is attributable to the fact that the autophosphorylation site Y577 from this loop inserts into the catalytic pocket of a neighboring kinase in the crystal, thereby forming an enzyme-substrate trans-phosphorylation complex.
Figure 1. The Pathogenic K650E Mutation Drives the FGFR3 Kinase into the Active Conformation.

(A) Ribbon diagrams of the FGFR3KK650E structure. β strands and α helices are colored cyan and green, respectively. The A-loop, catalytic loop, nucleotide-binding loop, kinase insert, and kinase hinge are colored magenta, orange, smudge, wheat, and yellow, respectively. In all of the figures, the ATP analog (AMP-PCP) and magnesium ions are rendered as sticks and blue spheres, respectively.
(B) Close-up view of the catalytically critical salt bridge between K508 and E525 in the kinase N-lobe important for ATP coordination and hydrolysis.
(C) The carboxylate group of E650 introduces a network of intramolecular hydrogen bonds that tether the flexible A-loop to the rigid core of the C-lobe, thereby stabilizing the active-state conformation of the A-loop. Side chains of selected residues are shown as sticks. Atom colorings in this figure and the following figures are as follows: oxygens in red, nitrogens in blue, and coloring for carbons follow the coloring scheme of the specific region of the kinase to which they belong. The mutant residue E650 and kinase insert tyrosine Y577 are labeled in red; other residues are labeled in black. The hydrogen bonds are shown as black dashed lines.
See also Figures S1–S3.
Table 1.
X-Ray Data Collection and Refinement Statistics
| Construct | FGFR3KK650E |
|---|---|
| Data Collection | |
| Resolution (Å) | 50–2.35 (2.39–2.35)a |
| Space group | P212121 |
| Unit cell parameters (Å, °) | a = 54.226 α = 90.00 |
| b = 61.427 β = 90.00 | |
| c = 100.666 γ = 90.00 | |
| Content of the asymmetric unit | 1 |
| No. of measured reflections | 67,586 |
| No. of unique reflections | 14,385 |
| Data redundancy | 4.7 |
| Data completeness (%) | 97.0 (61.1) |
| Rsym (%)b | 7.1 (24.3) |
| I/sig | 32.4 |
|
| |
| Refinement | |
| R factor/R free | 17.92/22.05 |
| No. of protein atoms | 2,322 |
| No. of solvent atoms | 128 |
| No. of nonprotein/solvent atoms | 33 |
| Rmsd bond length (Å) | 0.003 |
| Rmsd bond angle (°) | 0.776 |
| PDB ID | 4K33 |
The numbers in parentheses refer to the highest resolution shell.
Rsym = ∑∣I – ⟨I⟩∣/∑I, where I is the observed intensity of a reflection, and ⟨I⟩ is the average intensity of all of the symmetry-related reflections.
The K650E Mutation Confers Gain-of-Function by Stabilizing the Active Conformation of the Kinase A-Loop
Superimposition of the FGFR3KK650E structure onto the structures of unphosphorylated (low activity state) and A-loop phosphorylated (active state) FGFR1 (Bae et al., 2009; Mohammadi et al., 1996a) and FGFR2 (Chen et al., 2007, 2008) kinases unambiguously shows that FGFR3KK650E is in the active-state conformation (Figure 1B; Figure S1 available online). The A-loop of FGFR3KK650E superimposes onto that of phosphorylated FGFR1 and FGFR2 kinases with root-mean-square deviations (rmsd) of 0.81 and 1.2Å, respectively, and features the β strands 10 and 12 that are characteristics of the activated kinases (Figure 1). A network of intramolecular hydrogen bonds introduced by the K650E mutation facilitates and stabilizes the active-state conformation of the A-loop in the FGFR3KK650E structure (Figure 1B). Specifically, the carboxylate group of E650 makes five hydrogen bonds: two with R616 in the catalytic loop, one each with the backbone amides of T651 and T652, and one with Y648 within the A-loop (Figures 1B and S2). The latter hydrogen bond helps position the side chain of Y648 into a nearly identical location as that of the corresponding phosphorylated tyrosine residue in the A-loop phosphorylated wild-type (WT) FGFR1 and FGFR2 kinases (Figure S1). The hydroxyl group of Y648 in FGFR3KK650E makes an intramolecular hydrogen bond with R640 at the N-terminal end of the A-loop, which is reminiscent of the hydrogen bonds that the phosphate moiety of the phosphorylated A-loop tyrosine makes with the invariant A-loop arginine (Figures 1B and S2). This hydrogen bond likely also contributes to the active-state conformation of the A-loop.
As in the crystal structures of A-loop phosphorylated, activated FGFR1 and FGFR2 kinases (Bae et al., 2009; Chen et al., 2007), the autoinhibitory molecular brake at the kinase hinge/interlobe region of FGFR3KK650E is disengaged, providing further evidence that the mutation drives the kinase into the active state (Figure S3). The active-state conformation of FGFR3KK650E is also apparent by the formation of a catalytically important salt bridge between the invariant E525 in the αC helix and K508 in the β3 strand of the kinase N-lobe (Figure 1C). This salt bridge primes K508 for hydrogen bonding with the α and β phosphate groups of AMP-PCP (Figure 1C). Analysis of crystal packing contacts provides additional strong evidence that the mutated FGFR3 kinase has adopted the active-state conformation. In the crystal, FGFR3KK650E molecules are trapped in the act of trans-phosphorylation, whereby one kinase molecule acts as the enzyme, while the other serves as the substrate, offering a phosphorylatable tyrosine residue from the kinase insert region (Y577) (Figure 2). The hydroxyl moiety Y577 of the substrate-acting kinase engages in two short-range hydrogen bonds with the catalytic base D617 of the enzyme-acting kinase and is about 5Å away from the γ-phosphate of the ATP analog AMP-PCP, indicating that Y577 from the substrate-acting kinase is poised for deprotonation and phosphorylation by the enzyme-acting kinase (Figure 2). Given the fact that substrate binding is strictly dependent on kinase activation (Hubbard and Till, 2000), this observation lends further support for the fact that the K650E mutation has forced the FGFR3 kinase into the active state. Taken together, these structural findings unambiguously demonstrate that the pathogenic K650E mutation confers gain-of-function by functionally mimicking the action of A-loop tyrosine phosphorylation in stabilizing the active-state conformation of the A-loop.
Figure 2. Crystallographic Snapshot of the Kinase Insert trans-Phosphorylation Reaction.
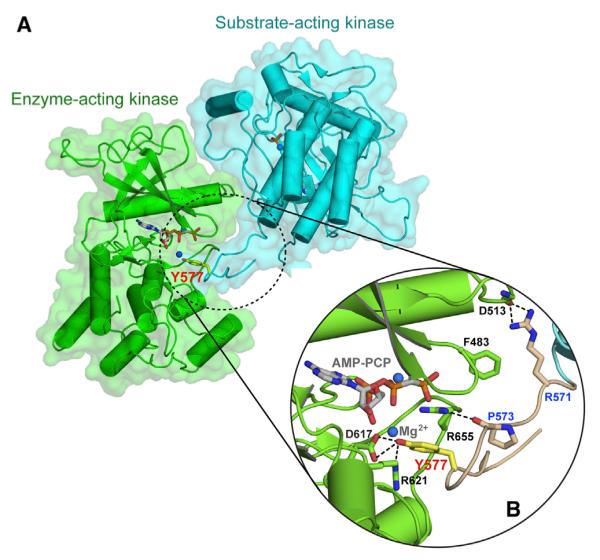
(A) The substrate-acting kinase (in cyan) interacts with both N- and C-lobe of the enzyme-acting kinase (in green) during the trans-phosphorylation on Y577.
(B) A detailed view of the interface formed between the enzyme-acting and substrate-acting kinases. The interacting residues are shown as sticks and labeled in black for the enzyme-acting kinase and in blue for the substrate-acting kinase. The kinase insert region is colored wheat, and the side chain of the Y577 residue is shown as yellow sticks and labeled in red. The hydrogen bonds are shown as black dashed lines. Atom coloring is as in Figure 1.
See also Figure S1.
The K650E Mutation Activates FGFR3 Kinase Independent of A-Loop Tyrosine Phosphorylation
To functionally validate our structural finding that the K650E mutation circumvents the requirement for A-loop tyrosine phosphorylation for FGFR3 kinase activation, we next compared the kinase activity of FGFR3KK650E with that of FGFR3KK650E harboring Y647F or Y648F single mutations or the Y647F/Y648F double mutation in vitro (Figures 3A–3E). As expected, FGFR3KK650E exhibited greater activity than wild-type FGFR3K (FGFR3KWT) at every time point examined, with the greatest difference (over 20-fold) seen at 20 s, the earliest measured time point (Figure 3F). The difference in activity between FGFR3KK650E and FGFR3KWT tapered off at later time points, however. This is expected, as A-loop tyrosine phosphorylation takes place over time, amplifying the catalytic activity of FGFR3KWT. The activity of the FGFR3KK650E mutants in which one or both A-loop tyrosine residues has been replaced with phenylalanine remains comparable to that of the parent FGFR3KK650E molecule (Figure 3F), corroborating that kinase activation by the K650E mutation occurs independent of A-loop tyrosine phosphorylation. Our data are consistent with the previously published data by Webster and colleagues showed that the A-loop tyrosines are dispensable for the constitutive activity of the K650E FGFR3 mutant, implying that K650E mutation activates the kinase in an A-loop phosphorylation-independent fashion (Webster et al., 1996). Notably, unlike FGFR3KWT, the activity of FGFR3KK650E did not increase significantly over time (Figure 3F), further substantiating that the K650E mutation activates the kinase independent of A-loop tyrosine phosphorylation. Taken together, these biochemical data support the structural finding that the K650E mutation confers constitutive activation by essentially mimicking the action of A-loop tyrosine phosphorylation in stabilizing the active-state conformation of the kinase.
Figure 3. The Pathogenic K650E Mutation Circumvents the Requirement for A-Loop Tyrosine Phosphorylation for FGFR3 Kinase Activation.

(A–E) The substrate phosphorylation activities of FGFR3KWT, FGFR3KK650E, and its A-loop tyrosine mutant derivatives (FGFR3KK650E/Y647F, FGFR3KK650E/Y648F, and FGFR3KK650E/Y647F/Y648F) were compared using native-PAGE (upper panel) coupled with time-resolved MALDI Q-TOF MS (lower panel).
(F) The percentage of at least one site phosphorylation on the substrate was quantitated using the peak intensity data generated by mass spectrometry. For the sake of accuracy, only the MS data of the early time points (20, 40, and 60 s), which are in the linear phase of the kinase assay, were processed and presented.
Structural Basis for Specificity of Tyrosine trans-Phosphorylation in FGFR3 Kinase
The crystal structure of FGFR3KK650E depicts, as already mentioned, a snapshot of a trans-phosphorylation reaction on the kinase insert tyrosine residue Y577. We previously reported the crystal structure of FGFR2 kinases caught in the act of trans-phosphorylation on the FGFR-invariant C-terminal tail tyrosine (Chen et al., 2008). Comparison of the two kinase trans-phosphorylation complexes provides important insights into the molecular determinants of specificity of kinase tyrosine trans-phosphorylation. Similar to the C-terminal tail tyrosine trans-phosphorylation complex, the kinase insert tyrosine trans-phosphorylation complex is also asymmetric, wherein the C-lobe of the substrate-acting kinase engages both the N- and C-lobe of the enzyme-acting kinase (Figure 2). The enzyme-substrate interface can be roughly divided into a proximal and a distal site, with the former in the vicinity of the catalytic cleft of the enzyme-acting kinase and the latter remote from the catalytic cleft of the enzyme-acting kinase.
At the proximal interface, several contacts in the kinase insert tyrosine trans-phosphorylation complex (Figure 4A) diverge from those observed in the C-terminal tail tyrosine trans-phosphorylation complex (Figure 4B). Notably, in stark contrast to the C-terminal tail tyrosine trans-phosphorylation complex, where the P+1 pocket of the enzyme plays an important role in providing specificity at the proximal enzyme-substrate interface (Figure 4B), this pocket is not utilized in the kinase insert tyrosine trans-phosphorylation complex. Instead, proximal specificity in this complex is primarily achieved by R655 from the A-loop of the enzyme-acting kinase. The guanidinium moiety of R655 stacks against the phenyl ring of Y577 via a π-cation interaction and also forms a hydrogen bond with the backbone carbonyl oxygen of P573 in the kinase insert of the substrate-acting kinase (Figure 4A).
Figure 4. Structural Determinants of Tyrosine trans-Phosphorylation Specificity in FGFR Kinases.

(A) Contacts between R571 of substrate and F483 of enzyme at the distal site mediate FGFR3 tyrosine trans-phosphorylation specificity on the kinase insert tyrosine Y577. Van der Waals surfaces of R571 and F483 are shown in mesh to emphasize their contacts. Likewise, meshed surfaces are used to indicate the p-cation interaction between the guanidinium moiety of R655 and the phenyl ring of Y577.
(B) Based on the crystal structure of FGFR2 kinases trapped in the act of trans-phosphorylation on C-terminal tail tyrosine, the hydrophobic contacts between L761 of substrate and the P+1 pocket of the enzyme are critical for tyrosine trans-phosphorylation specificity on the C-terminal tail tyrosine Y760 in FGFR3. To highlight the hydrophobic contacts between the substrate and the enzyme at the P+1 pocket, the molecular surfaces of P+1 and P+3 residues (L761 and L763) of the substrate and residues in the P+1 pocket of the enzyme (L656, V700, and F704) are shown. Enzyme-acting kinases in both trans-phosphorylation complexes are colored green. Substrate-acting kinases are colored cyan and salmon in kinase insert and C-terminal tail tyrosine trans-phosphorylation complexes, respectively.
(C–E) Analysis of autophosphorylation by mass spectrometry corroborates the importance of R571 and L761 in substrate recognition in the kinase insert and C-terminal tail tyrosine trans-phosphorylation. The R571A mutation diminishes trans-phosphorylation on the kinase insert tyrosine Y577 without a major impact on the C-terminal tail tyrosine phosphorylation (D). The L761A mutation (equivalent to L770A in FGFR2) impairs the trans-phosphorylation of the C-terminal tail tyrosine Y760 (E).
At the distal interface, the contacts in the kinase insert tyrosine trans-phosphorylation complex are completely different from those observed in the C-terminal tail tyrosine trans-phosphorylation complex. The main specific contacts in the distal site of the kinase insert tyrosine trans-phosphorylation complex are two hydrogen bonds between R571 (from the N-terminal end of the kinase insert of the substrate-acting kinase) and the backbone carbonyl oxygen atom of D513 in the loop between β3 strand and αC helix of the enzyme-acting kinase (Figure 4A). All of the remaining interactions at this distal contact site are of van der Waals nature with the most notable ones being the contacts between F483 from the nucleotide-binding loop of the enzyme-acting kinase and R571 of the substrate-acting kinase.
To functionally probe the observed mode of trans-phosphorylation specificity on the kinase insert tyrosine in the FGFR3KK650E crystal, we studied the impact of mutating R571 to alanine on phosphorylation of the kinase insert tyrosine Y577. Consistent with the structural data, the R571A mutation drastically impaired phosphorylation of the kinase insert tyrosine (Y577) but had only a minor effect on the phosphorylation of the C-terminal tail tyrosine (Y760) (Figure 4D). As expected based on our previous crystal structure of the C-terminal tail tyrosine trans-phosphorylation complex (Chen et al., 2008), substitution of L761, which engages the P+1 pocket of the enzyme, with alanine caused a major decrease in the kinase C-terminal tail tyrosine phosphorylation but had little impact on phosphorylation of the kinase insert tyrosine (Figure 4E).
DISCUSSION
In this study we elucidated the molecular mechanism by which the pathogenic K650E mutation in FGFR3, which underlies a lethal skeletal dysplasia in humans, confers gain-of-function on FGFR3. Our data unambiguously demonstrate that the K650E mutation mimics the action of A-loop tyrosine phosphorylation to cause ligand-independent activation of FGFR3. It should be noted that enzymes, in general, and tyrosine kinases, in particular, are intrinsically dynamic protein moieties, and in fact the catalytic turnover rate of enzymes is strongly correlated with magnitude and timescales of internal motions that enzymes undergo (Eisenmesser et al., 2005; Villali and Kern, 2010). Using X-ray crystallography and NMR spectroscopy, we have recently shown that FGFR kinases swing between an inhibited state, which is conformationally rigid and stable, and an active state, which is conformationally dynamic and hence inhomogeneous (Chen et al., 2013). The unphosphorylated wild-type FGFR kinase can sample the active state albeit with low frequency and hence predominately populates the inhibited state. Pathogenic mutations, such as the K650E mutation, introduce intramolecular contacts that facilitate the ability of the kinase to adopt and remain longer in the active state, thereby increasing the population of kinase molecules in the active state (Chen et al., 2013). In other words, the crystallographically fixed intramolecular hydrogen bonding contacts introduced by the mutation are dynamic in solution, and the FGFR3KK650E pathogenic kinase also oscillates between inhibited and active states with the equilibrium being skewed toward the active state.
Comparison of the kinase insert tyrosine trans-phosphorylation complex in the FGFR3KK650E crystal with the FGFR2 kinase C-terminal tail tyrosine trans-phosphorylation complex (Chen et al., 2008) shows that kinase trans-phosphorylation entails a great degree of sequence specificity and structural complementarities. It remains to be determined whether Y577 of FGFR3 is phosphorylated in living cells. However, the equivalent tyrosine in FGFR1, Y583, is a bona fide in vivo phosphorylation site (Mohammadi et al., 1996b) and is also a second major autophosphorylation site in vitro (Furdui et al., 2006). Y577 is readily phosphorylated in vitro, both in the wild-type and in the K650E mutant FGFR3 kinases. Based on these observations, we surmise that Y577 undergoes phosphorylation in vivo as well. Future efforts should be directed toward elucidating the structural basis for trans-phosphorylation on the A-loop tyrosines, the earliest trans-phosphorylation event necessary for kinase activation. A full comprehension of the molecular details of FGFR trans-phosphorylation reactions will be essential to understanding FGFR signaling and devising novel strategies for targeted modulation of FGF signaling for therapeutic purposes.
EXPERIMENTAL PROCEDURES
Please refer to the Supplemental Experimental Procedures for full details.
Protein Expression and Purification
The human FGFR3 kinase domains FGFR3K450-758 and FGFR3K429-768, including their mutated forms, and the C-terminal tail peptide of FGFR2 kinase (FGFR2K761-821) were all expressed using pET bacterial expression vectors with an N-terminal 6XHis-tag to aid in protein purification.
Crystallization and Structure Determination
Crystals of the FGFR3KK650E protein in complex with the ATP-analog (AMP-PCP) were grown by vapor diffusion at 20°C using crystallization buffer composed of 0.1 M HEPES (pH 7.5), 20% (w/v) PEG 4000, and 4% (v/v) (±)-1,3 Butanediol. Diffraction data were processed using the HKL2000 suite (Otwinowski and Minor, 1997), and the structure was solved by the molecular replacement program AMoRe (Navaza, 1994), using the crystal structure of FGFR2 kinase that harbors the K659N mutation (Protein Data Bank [PDB] ID code 2PVY) (Chen et al., 2007) as the search model. Model building was carried out using O (Jones et al., 1991), and at later stages Coot (Emsley and Cowtan, 2004) was used, and refinement was completed using PHENIX (Adams et al., 2002).
Dissection of the Role of A-Loop Tyrosine Phosphorylation in Gain-of-Function by the K650E Mutation Using Peptide Substrate Phosphorylation
Peptide substrate phosphorylation activities of wild-type and mutated FGFR3 kinases (FGFR3KWT, FGFR3KK650E, FGFR3KK650E/Y647F, FGFR3KK650E/Y648F, and FGFR3KK650E/Y647F/Y648F) were analyzed by MALDI-TOF MS (Bruker Auto-flex MALDI-TOF, Bruker Daltonics) in positive ion linear mode.
Analysis of the Specificity of Tyrosine trans-Phosphorylation
The trans-phosphorylation on kinase insert and C-terminal tail tyrosines in wild-type and mutated FGFR3 kinases (FGFR3K440-778, FGFR3K440-778/R571A, FGFR3K440-778/R655A, and FGFR3K440-778/L761A) were analyzed by LTQ Orbitrap (Thermo Electron) liquid chromatography-tandem mass spectrometry.
Supplementary Material
ACKNOWLEDGMENTS
The authors are thankful to Dr. Regina Goetz, Artur Belov, and Yang Liu for their critical reading of the manuscript and thoughtful suggestions and Drs. R. Abramowitz and J. Schwanof for synchrotron beamline assistance. This work was supported by a National Institute of Dental and Craniofacial Research grant (DE13686 to M.M.), a National Institute of Neurological Disorders and Stroke grant (P30 NS050276 to T.A.N.), grants from the Natural Science Foundation of China (31270789 and 81102486 to H.C. and Z.H.), and the Zhejiang Key Group Project in Scientific Innovation (2010R10042-01 to Z.H. and X.L.). Beamlines X-4A and X-4C at the National Synchrotron Light Source, Brookhaven National Laboratory, a DOE facility, are supported by New York Structural Biology Consortium.
Footnotes
ACCESSION NUMBERS The coordinates and structure factors for the FGFR3KK650E structure have been deposited in the Protein Data Bank under the accession number 4K33.
SUPPLEMENTAL INFORMATION Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at http://dx.doi.org/10.1016/j.str.2013.07.017.
REFERENCES
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Agazie YM, Movilla N, Ischenko I, Hayman MJ. The phosphotyrosine phosphatase SHP2 is a critical mediator of transformation induced by the oncogenic fibroblast growth factor receptor 3. Oncogene. 2003;22:6909–6918. doi: 10.1038/sj.onc.1206798. [DOI] [PubMed] [Google Scholar]
- Bae JH, Lew ED, Yuzawa S, Tomé F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138:514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae JH, Boggon TJ, Tomé F, Mandiyan V, Lax I, Schlessinger J. Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc. Natl. Acad. Sci. USA. 2010;107:2866–2871. doi: 10.1073/pnas.0914157107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, Greenhaw GA, Hecht JT, Francomano CA. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat. Genet. 1995;10:357–359. doi: 10.1038/ng0795-357. [DOI] [PubMed] [Google Scholar]
- Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, Francomano CA. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. Am. J. Hum. Genet. 2000;67:1411–1421. doi: 10.1086/316892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, Chopin D, Thiery JP, Radvanyi F. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]
- Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, Mohammadi M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol. Cell. 2007;27:717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Xu CF, Ma J, Eliseenkova AV, Li W, Pollock PM, Pitteloud N, Miller WT, Neubert TA, Mohammadi M. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc. Natl. Acad. Sci. USA. 2008;105:19660–19665. doi: 10.1073/pnas.0807752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Huang Z, Dutta K, Blais S, Neubert TA, Li X, Cowburn D, Traaseth NJ, Mohammadi M. Cracking the molecular origin of intrinsic tyrosine kinase activity through analysis of pathogenic gain-of-function mutations. Cell Rep. 2013;4:376–384. doi: 10.1016/j.celrep.2013.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M, Nardini E, Brents LA, Schröck E, Ried T, Kuehl WM, Bergsagel PL. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997;16:260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum. Mutat. 2012;33:29–41. doi: 10.1002/humu.21636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furdui CM, Lew ED, Schlessinger J, Anderson KS. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell. 2006;21:711–717. doi: 10.1016/j.molcel.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, Pfeifer SP, McVean GA, Rajpert-De Meyts E, Wilkie AO. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat. Genet. 2009;41:1247–1252. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Degnin CR, Laederich MB, Lunstrum GP, Holden P, Bihlmaier J, Krakow D, Cho YJ, Horton WA. Sprouty 2 disturbs FGFR3 degradation in thanatophoric dysplasia type II: a severe form of human achondroplasia. Cell. Signal. 2008;20:1471–1477. doi: 10.1016/j.cellsig.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kan SH, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR, Rannan-Eliya S, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am. J. Hum. Genet. 2002;70:472–486. doi: 10.1086/338758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens PM, Liboi E. The thanatophoric dysplasia type II mutation hampers complete maturation of fibroblast growth factor receptor 3 (FGFR3), which activates signal transducer and activator of transcription 1 (STAT1) from the endoplasmic reticulum. J. Biol. Chem. 2003;278:17344–17349. doi: 10.1074/jbc.M212710200. [DOI] [PubMed] [Google Scholar]
- Logié A, Dunois-Lardé C, Rosty C, Levrel O, Blanche M, Ribeiro A, Gasc JM, Jorcano J, Werner S, Sastre-Garau X, et al. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum. Mol. Genet. 2005;14:1153–1160. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Schlessinger J, Hubbard SR. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996a;86:577–587. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Dikic I, Sorokin A, Burgess WH, Jaye M, Schlessinger J. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol. Cell. Biol. 1996b;16:977–989. doi: 10.1128/mcb.16.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naski MC, Wang Q, Xu J, Ornitz DM. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat. Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- Navaza J. Amore - an automated package for molecular replacement. Acta Crystallogr. A. 1994;50:157–163. [Google Scholar]
- Nowroozi N, Raffioni S, Wang T, Apostol BL, Bradshaw RA, Thompson LM. Sustained ERK1/2 but not STAT1 or 3 activation is required for thanatophoric dysplasia phenotypes in PC12 cells. Hum. Mol. Genet. 2005;14:1529–1538. doi: 10.1093/hmg/ddi161. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweets RM, editors. Methods in Enzymology. University of Virginia; Charlottesville: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, Wilcox WR, Rimoin DL, Cohn DH, Wasmuth JJ. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- Villali J, Kern D. Choreographing an enzyme’s dance. Curr. Opin. Chem. Biol. 2010;14:636–643. doi: 10.1016/j.cbpa.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. Enhanced signaling and morphological transformation by a membrane-localized derivative of the fibroblast growth factor receptor 3 kinase domain. Mol. Cell. Biol. 1997a;17:5739–5747. doi: 10.1128/mcb.17.10.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 1997b;13:178–182. doi: 10.1016/s0168-9525(97)01131-1. [DOI] [PubMed] [Google Scholar]
- Webster MK, D’Avis PY, Robertson SC, Donoghue DJ. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol. Cell. Biol. 1996;16:4081–4087. doi: 10.1128/mcb.16.8.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox WR, Tavormina PL, Krakow D, Kitoh H, Lachman RS, Wasmuth JJ, Thompson LM, Rimoin DL. Molecular, radiologic, and histopathologic correlations in thanatophoric dysplasia. Am. J. Med. Genet. 1998;78:274–281. doi: 10.1002/(sici)1096-8628(19980707)78:3<274::aid-ajmg14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev. 2005;16:187–203. doi: 10.1016/j.cytogfr.2005.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.