

Abstract
RET (rearranged during transfection) is a receptor tyrosine kinase involved in the development of neural crest derived cell lineages, kidney, and male germ cells. Different human cancers, including papillary and medullary thyroid carcinomas, lung adenocarcinomas, and myeloproliferative disorders display gain-of-function mutations in RET. Accordingly, RET protein has become a promising molecular target for cancer treatment.
RET is a dependence receptor: In the absence of ligand, it induces apoptosis. Gain-of-function mutations in RET can lead to several human cancers.
The human RET (rearranged during transfection) gene maps on 10q11.2 and is composed of 21 exons spanning a region of 55,000 bp. It encodes a single-pass trans-membrane protein, RET, that belongs to the receptor tyrosine kinase (RTK) family (Pasini et al. 1995). The RET extracellular segment contains four cadherin-like domains, followed by a domain containing cysteine residues involved in the formation of intramolecular disulfide bonds (Fig. 1A) (Anders et al. 2001; Airaksinen and Saarma 2002). RET protein is highly glycosylated and N-glycosylation is necessary for its transport to the cell surface. Only the fully mature glycosylated 170 kDa RET protein isoform is exposed to the extracellular compartment, whereas the mannose-rich 150 kDa isoform is confined to the Golgi (Takahashi et al. 1993; Carlomagno et al. 1996). The transmembrane segment is composed of 22 amino acids, among which S649 and S653 mediate self-association and dimerization of RET, possibly via formation of inter-molecular hydrogen bonding (Kjaer et al. 2006). The intracellular portion of RET contains the tyrosine kinase domain split into two subdomains by the insertion of 27 amino acids. The RET COOH-terminal tail varies in length as a result of alternative splicing of the 3′ end (carboxy terminal with respect to glycine 1063), generating three different isoforms that contain 9 (RET9), 43 (RET43), or 51 (RET51) amino acids (Myers et al. 1995). RET9 and RET51 are the most abundant isoforms, and they activate similar signaling pathways through interaction with diverse protein complexes, and may exert a differential role in development (Fig. 1A) (de Graaff et al. 2001).
Figure 1.

Illustration of the mechanisms of activation of wild-type (wt) RET and RET-derived oncoproteins. (A) Wild-type RET activation is mediated by ligand (GFL)-induced dimerization; ligand binding to RET is not direct and mediated by GFR-α coreceptors (not shown); major RET autophosphorylation sites and downstream signaling pathways are indicated. RET extracellular cadherin-like domains are represented in red. The split intracellular RET tyrosine kinase domain, as well as the three alternative carboxy-terminal RET tails, are also depicted. (B) RET/PTC activation is mediated by coiled-coil-induced dimerization (left); activation of RET cysteine mutants associated with MEN2A or FMTC is mediated by disulfide bonds-mediated dimerization (right).
RET shows several autophosphorylation sites (Fig. 1A) (Liu et al. 1996; Kawamoto et al. 2004). RET tyrosine 1062 (Y1062) functions as a multidocking site for signaling molecules containing a phosphotyrosine-binding (PTB) domain (Asai et al. 1996). Phospho-Y1062 binding proteins include SHC, N-SHC (RAI), FRS2, IRS1/2, DOK1, and DOK4/5 that, in turn, contribute to the activation of RAS-MAPK (mitogen-activated protein kinases) and PI3K (phosphatidyl inositol 3 kinase)-AKT pathways. Y1096, specific to the RET51 splicing variant, couples to the PI3K-AKT and RAS-MAPK pathways, as well. These signaling cascades mediate RET-dependent cell survival, proliferation, and motility (Alberti et al. 1998; Murakami et al. 1999; Segouffin-Cariou and Billaud 2000; Melillo et al. 2001a,b; Schuetz et al. 2004). Y905 is located in the activation loop of the RET kinase and its phosphorylation is associated with RET kinase activation (Knowles et al. 2006). Finally, Y981 and Y1015 have been shown to be coupled to important signaling molecules such as SRC and PLC-γ, respectively (Borrello et al. 1996; Encinas et al. 2004).
RET is the receptor for a group of neurotrophic growth factors that belong to the glial cell line-derived neurotrophic factor (GDNF) family (GFLs, GDNF family ligands), namely, GDNF, Neurturin (NRT), Artemin (ART), and Persephin (PSF) (Airaksinen and Saarma 2002). GFLs mediate RET protein dimerization and activation (Fig. 1A). GFLs are presented to RET by GPI (glycosylphosphatidylinositol)-anchored coreceptors, called GFR-α (GDNF family receptor α 1-4). Differential tissue expression dictates the specificity of action displayed by alternative GLF-GFR-α pairs during development and adult life (Baloh et al. 2000; Airaksinen and Saarma 2002).
Together with other membrane (DCC and p75NTR) or nuclear (androgen receptor, AR) receptors, RET belongs to the family of so-called “dependence” receptors (Mehlen and Bredesen 2011). In the absence of ligand, RET exerts a proapoptotic activity, that is blocked on ligand stimulation (Bordeaux et al. 2000). Such pro-apoptotic activity is RET kinase-independent and mediated by cleavage of RET cytosolic portion by caspase-3, which, in turn, releases a carboxy-terminal RET peptide that is able to induce cell death (Bordeaux et al. 2000). It is feasible that such activity is important for RET developmental function, because it may control migration of RET-expressing cells by limiting survival of cells that move beyond ligand availability (Bordeaux et al. 2000; Cañibano et al. 2007). Whether modulation of this function is also important for RET-associated diseases is still unknown. However, it is interesting to note that a cancer-associated RET mutant (RET-C634R, see below) does not exert cleavage-dependent proapoptotic effects, whereas RET mutants associated with defective development (Hirschsprung disease, see below) exert strong proapoptotic activity that is refractory to modulation by ligand (Bordeaux et al. 2000).
RET is expressed in enteric ganglia, adrenal medulla chromaffin cells, thyroid C cells, sensory and autonomic ganglia of the peripheral nervous system, a subset of central nervous system nuclei, developing kidney and testis germ cells (Manié et al. 2001; de Graaff et al. 2001). RET null mice display impaired development of superior cervical ganglia and enteric nervous system, kidney agenesia, reduction of thyroid C cells, and impaired spermatogenesis (Manié et al. 2001). Accordingly, individuals with germline loss-of-function mutations of RET are affected by intestinal aganglionosis causing congenital megacolon (Hirschsprung disease) (Brooks et al. 2005). RET loss-of-function mutations have also been identified in congenital anomalies of kidney and urinary tract (CAKUT), either isolated or in combination with Hirschsprung disease (Jain 2009).
Several genetic alterations convert RET into a dominantly transforming oncogene. This review will describe RET-derived oncogenes that are associated with different types of human neoplasia (Fig. 1B).
RET/PTC IN PAPILLARY THYROID CARCINOMA
RET/PTC Oncogenes
Papillary thyroid carcinoma (PTC) is the most frequent thyroid cancer and endocrine malignancy overall (Nikiforov and Nikiforova 2011). PTC originates from endodermal-derived thyroid follicular cells and is etiologically associated with exposure to ionizing radiation (Williams 2008). PTC features genetic lesions targeting the RTK-RAS-MAPK pathway. Roughly half of PTC cases display activation of BRAF oncogene (most commonly secondary to V600E mutation) and a small proportion of them, mainly belonging to the follicular variant-PTC, carry mutations of RAS genes (Xing 2005; Nikiforov and Nikiforova 2011). Although quite uncommon, rearrangements of the NTRK1 RTK are found in PTC as well (Greco et al. 2010).
In PTC cases that are negative for BRAF, RAS, or NTRK1 mutations, chromosomal rearrangements targeting the long arm of chromosome 10 cause the disruption of RET gene and its fusion to various heterologous genes (Grieco et al. 1990). Such chromosomal aberrations give rise to chimeric oncogenes named RET/PTC (Fig. 1B). RET/PTC oncogenes are composed by the tyrosine kinase and COOH-tail encoding sequence of RET (from exon 12 to the 3′-end) and fused at the 5′ end to the promoter sequence and 5′-terminal exons of heterologous genes (Fig. 2) (Nikiforov and Nikiforova 2011). The fusion partner genes encode proteins that share the presence of protein–protein interaction domains, such as coiled-coil motifs able to mediate RET TK dimerization (Figs. 1B and 2). Most common RET/PTC rearrangements (90% of the cases) are RET/PTC1 and RET/PTC3, the fusions between RET and CCDC6 or NCOA4 (RFG, ARA70) genes, respectively. RET/PTC1 and RET/PTC3 (and RET/PTC4, another NCOA4-RET fusion variant) are generated through a paracentric inversion of the long arm of chromosome 10, where RET, CCDC6, and NCOA4 map (Grieco et al. 1990; Santoro et al. 1994). Instead, the other RET/PTC variants are generated by translocations between different chromosomes and are either rare (RET/PTC2) or identified only in single cases of radiation-induced PTC (Fig. 2).
Figure 2.

Schematic representation of RET/PTC oncoproteins. On the top, wild-type RET protein is illustrated. For each RET/PTC rearrangement, the name of the fusion partner is indicated on the left and the corresponding chromosomal alteration is indicated on the right. The fusion points are indicated by arrowheads. The length in amino acids of the partner protein portion is also indicated. Boxes in red indicate dimerization (coiled-coil) domains. SP, Signal peptide; TM, transmembrane domain; TK, tyrosine kinase domain; CCDC6, coiled-coil domain-containing protein 6; PRKAR1A, protein kinase, cAMP-dependent, regulatory, type I, alpha; NCOA4 (RFG, ARA70), nuclear coactivator 4 (RET-fused gene, androgen receptor-associated protein 70); RFG5 (GOLGA5), RET-fused gene 5 (Golgin A5); TRIM24, tripartite motif-containing 24; RFG7 (TRIM33), RET-fused gene 7 (tripartite motif-containing 33); KTN1, kinectin 1; RFG9, RET-fused gene 9; ELKS (RAB6IP2), glutamate, leucine, lysine, serine-rich sequence (RAB6-interacting protein 2); PCM1, pericentriolar material 1; RFP (TRIM27), RET finger protein (tripartite motif-containing 27); HOOK3, hook homolog 3.
RET/PTC Prevalence
The frequency of RET/PTC rearrangements (average 25% of the cases) varies considerably in different patient series (Nikiforov and Nikiforova 2011). This may depend on patients’ exposure to different etiologic factors. As an example, in pediatric patients and in cases from areas contaminated by radioiodine isotopes, RET/PTC frequency can reach 50%–70% (Zhu et al. 2006). It is also possible that variable prevalence of RET/PTC may depend on the methodology used for the detection. Accordingly, the rearrangement can be present only in a subset of cancer or even benign cells (nonclonal RET/PTC); and in these cases, it is detected only when highly sensitive techniques (such as nested reverse transcriptase-PCR or real-time reverse transcriptase PCR) or single-cell assays (such as FISH: fluorescent in situ hybridization) are used (Unger et al. 2004; Rhoden et al. 2006; Zhu et al. 2006).
RET/PTC Oncogenic Activity
Adoptive expression of RET/PTC oncogenes induces thyroid cell transformation in vitro (Santoro et al. 1993; Wang et al. 2003; Melillo et al. 2005). Moreover, targeted expression of RET/PTC in thyroid follicular cells induce thyroid hyperplasia or neoplasia in transgenic mice (Santoro et al. 1996; Powell et al. 1998). This evidence supports the causal contribution of RET/PTC formation to PTC development. Nevertheless, the low penetrance of the disease in transgenic animals, as well as the presence of RET/PTC rearrangements in papillary microcarcinoma, that may not progress to invasive cancer (Viglietto et al. 1995), suggests that additional oncogenic events should occur and cooperate with RET/PTC to generate an overt disease. Notably, the acute expression of RET/PTC in immortalized rat thyroid follicular cells in vitro activates a proapoptotic response because of unscheduled activation of the RAS-MAPK pathway (Castellone et al. 2003; Wang et al. 2003). Moreover, expression of RET/PTC in primary human thyrocytes induces oncogene-induced senescence (OIS) (Vizioli et al. 2011). It is not uncommon that normal cells oppose a barrier to neoplastic transformation by switching on suicidal or senescent programs in response to oncogene activation (Hanahan and Weinberg 2011). Thus, it is conceivable that RET/PTC rearrangements may be fairly frequent events in thyroid cells but insufficient alone to induce a full-blown cancer, unless further mutational events or epigenetic modifications occur to enable cells to escape cell death or growth arrest defenses.
Mechanisms of RET/PTC Formation
Breakage of RET and partner genes and their fusion are believed to result from unfaithful repair of DNA double-strand breaks (Ameziane-El-Hassani et al. 2010; Gandhi et al. 2010a). RET and its most common fusion partners (CCDC6 and NCOA4) seem to be particularly susceptible to breakage because they map in DNA fragile sites (Gandhi et al. 2010b). Fragile sites are nonrandom DNA loci that are stable under normal conditions but become hot spots of chromosome breakage under exposure to different agents, such as ethanol, caffeine, and hypoxia (Durkin and Glover 2007).
In addition, RET gene disruption may be caused by genotoxic agents such as ionizing radiation and reactive oxygen species. RET/PTCs are enriched in patients with a known history of exposure to internal (because of thyrocyte ability to concentrate radioiodine) (Williams 2008) or external beam (Collins et al. 2002) radiation, as well as in atomic bomb survivors (Hamatani et al. 2008). Accordingly, RET/PTC formation can be experimentally induced by irradiation of cultured thyrocytes and thyroid tissue xenografts in SCID mice (Ito et al. 1993; Mizuno et al. 1997, 2000; Caudill et al. 2005). However, it is important to note that most patients with RET/PTC-positive cancer do not have a documented exposure to radiation. In these cases, genotoxic agents other than radiation may cause DNA breakage (Gandhi et al. 2010b). Of note, H2O2, a potent DNA-damaging agent, is produced in large amounts by thyrocytes during the process of thyroid hormone biosynthesis and may represent one such agent (Ameziane-El-Hassani et al. 2010). Therefore, RET/PTC-specific occurrence in thyroid cancer might be explained by the fact that thyroid gland is commonly exposed to agents such as ionizing radiation or H2O2 that can disrupt RET and its fusion partners. In addition, in thyroid tissue, simultaneous breakage of RET and fusion partners, as well as their recombination, may be facilitated by thyroid cell-specific architecture of nuclear chromatin. It has been shown, indeed, that CCDC6, NCOA4, and RET loci display close proximity specifically in thyroid follicular cell chromatin (Nikiforova et al. 2000; Gandhi et al. 2006).
Mechanisms of RET/PTC Oncogenic Activation
Two major mechanisms underlie RET oncogenic conversion on RET/PTC formation. First, secondary to gene fusion, RET tyrosine kinase encoding domain is placed under the transcriptional control of the promoter and regulatory elements of RET fusion partners. Differently from RET, whose expression is restricted to neuroectoderm-derived cells, partner genes are ubiquitously expressed and able to drive RET expression in thyroid follicular cells. In addition, fusion to heterologous proteins containing protein homodimerization motifs results in constitutive RET kinase dimerization, ligand-independent activation, and autophosphorylation followed by continuous activation of downstream signaling pathways (Fig. 1B) (Bongarzone et al. 1993; Monaco et al. 2001).
The expression of a constitutively active RET kinase leads to chronic exposure of thyroid follicular cells to the activation of intracellular signaling pathways, such as RAS-MAPK, which is initiated at the level of RET tyrosine 1062 (Y1062) (Fig. 1A). Intriguingly, this pathway includes BRAF, the other oncogenic protein commonly activated in PTC (Fig. 3). Constitutive signaling is, in turn, responsible for the acquisition of several hallmarks of cancer cells including cell autonomy (independence from growth factors like TSH—thyroid-stimulating hormone), cell motility, and invasion (Melillo et al. 2005). RET/PTC signaling is also able to induce remodeling of the tumor stroma that may facilitate tumor growth. Accordingly, several reports have shown that RET/PTC via the RAS-MAPK cascade endorses an inflammatory-like response characterized by the production of several cytokines and chemokines that, in turn, recruit macrophages, lymphocytes, and mast cells within the tumor thereby promoting cell survival, invasion, and angiogenesis (Russell et al. 2003; Borrello et al. 2005; Melillo et al. 2005, 2010; Puxeddu et al. 2005).
Figure 3.

RAS-MAPK oncogenic signaling cascade activated by RET oncoproteins in MTC (left) and PTC (right). In red are indicated other mutational targets (RAS and BRAF) associated with MTC or PTC, respectively. In most of the cases, these mutations are alternative and not present simultaneously in a single tumor.
RET Fusion Partners
It is possible that, besides causing activation of RET kinase, RET/PTC rearrangements affect also the function of RET fusion partners, this in turn contributing to thyroid tumorigenesis. According to this possibility, the rearrangement might cause a genetic double hit, inducing simultaneously the gain of RET oncogenic activity and the knockdown of the tumor suppressor function of RET partner gene. The RET/PTC2 rearrangement nicely illustrates this possibility. In this case, the RET fusion partner is represented by the PRKARIA gene, which encodes the regulatory subunit RIα of protein kinase A. PRKARIA is a bona fide tumor suppressor gene that is targeted by germline inactivating mutations in patients affected by the Carney complex. This is a rare autosomal dominant cancer syndrome characterized by lentiginosis, atrial, and cutaneous myxoma, pituitary adenoma, testicular tumors, ovarian cysts, schwannoma, and thyroid neoplasia (Kirschner et al. 2000; Boikos and Stratakis 2006). Another example may be represented by TRIM24 (also known as HTIF1), the RET fusion partner in RET/PTC6 (Klugbauer and Rabes 1999). TRIM24 null mice develop hepatic cell carcinoma, and TRIM24 gene, on chromosome 7q32, shows frequent genetic aberrations in human hepatic cell carcinoma, strongly suggesting its role as a tumor suppressor gene (Wong et al. 1999; Khetchoumian et al. 2007). Finally, several studies also suggest that CCDC6 and NCOA4, the genes involved in most common RET/PTC variants (RET/PTC1 and RET/PTC3, respectively), might display tumor suppressor function. CCDC6 gene product is a ubiquitously expressed 65 kDa protein that displays proapoptotic activity and is involved in ATM-mediated cellular response to DNA damage (Celetti et al. 2004; Merolla et al. 2007). A role of CCDC6 in the repression of CREB1, a transcriptional factor essential for thyroid cell growth and differentiation, has also been described (Leone et al. 2010). NCOA4 gene encodes a 70 kDa protein that functions as a coactivator of PPARγ (peroxisome-proliferator-activated receptor γ) and AR (androgen receptor) (Yeh and Chang 1996; Heinlein et al. 1999). The ectopic overexpression of NCOA4 in prostate cancer cells reduces cell proliferation and NCOA4 expression is reduced in aggressive prostate and breast cancers, thus suggesting that this gene may function as a suppressor of tumorigenesis (Kollara et al. 2001; Li et al. 2002; Ligr et al. 2010).
RET MUTATIONS IN MEDULLARY THYROID CARCINOMA AND MEN2 SYNDROMES
Multiple Endocrine Neoplasia Type 2
Medullary thyroid carcinoma (MTC) arises from neural crest-derived calcitonin-producing thyroid parafollicular C cells and represents 5%–10% of all thyroid cancers. Although most MTCs are sporadic and affect adults, around 25% of cases are familial occurring in the frame of inherited cancer syndromes named multiple endocrine neoplasia type 2 (MEN2) syndromes (online Mendelian inheritance in men, OMIM: #171400) (de Groot et al. 2006). MEN2 comprises three related disorders: MEN2A, MEN2B, and familial medullary thyroid carcinoma (FMTC). MEN2A, first described in 1961 by J.H. Sipple, is characterized by MTC associated with pheochromocytoma (a benign tumor of adrenal medulla) in 50% of cases and parathyroid hyperplasia or adenoma in 10%–30% of cases; more rarely, MEN2A patients show other disease features such as cutaneous lichen amyloidosis and congenital megacolon (see below). In MEN2B syndrome, MTC is associated with pheochromocytoma, ganglioneuromatosis of the intestine, thickening of corneal nerves, and marfanoid habitus (de Groot et al. 2006). Finally, MTC is the only disease phenotype of patients displaying FMTC. Recently, some investigators have suggested FMTC as a phenotypic variant of MEN2A with decreased expression.
RET and MEN2
Specific germline missense mutations of RET gene cause the MEN2 syndromes (Fig. 4). Most MEN2A and FMTC mutations affect cysteines in the extracellular cysteine-rich domain of RET. MEN2A is associated most frequently with mutations of cysteine 634 (85%), particularly C634R, whereas FMTC mutations are evenly distributed among the various cysteines (C609, C611, C618, C620, C630) (de Groot et al. 2006). Rare MEN2A or FMTC mutations in RET ectodomain do not target cysteine-rich domain (de Groot et al. 2006; Fazioli et al. 2008; Castellone et al. 2010). FMTC can also be associated with changes in the RET kinase domain (including E768D, L790F, V804L, and V804M).
Figure 4.

Representation of the most common germline missense mutations in RET gene found in MEN2 syndromes. The location of the mutation with respect to RET protein domains and RET exons is indicated. MEN2 phenotypes associated with the various mutations are indicated on the right.
Most MEN2B patients carry the M918T mutation in RET kinase domain, whereas only a small fraction of them harbor the A883F substitution (Fig. 4). Very rarely, the MEN2B phenotype is sustained by double mutations targeting either the same or two different RET alleles (de Groot et al. 2006).
Finally, several additional rare germline RET variants have been identified through the systematic screening of MTC patients. However, their pathological significance is not always obvious, especially if data on cosegregation of the mutation with the disease and functional studies are not available. A comprehensive database annotating all RET variants and their pathogenetic relevance has been generated (Margraf et al. 2009).
Genetic testing in MTC patients is important not only to distinguish sporadic from familial cases (and thus to identify mutation carriers in the family at risk of developing the disease) but also to assess risk of developing aggressive MTC and MTC-associated neoplastic lesions, such as pheochromocytoma and parathyroid adenoma. Accordingly, most common RET mutations have been classified to different disease risk levels, ranging from A (less severe) to D (most severe). Risk level classification guides decisions regarding timing of prophylactic thyroidectomy and intraoperative management of the parathyroid glands (Kloos et al. 2009).
RET and Sporadic MTC
MTC arises sporadically in about 75% of cases and RET somatic mutations, mainly M918T, occur in about 50% of sporadic MTC, but very rarely in sporadic pheochromocytoma (Beldjord et al. 1995; Lindor et al. 1995). The presence of somatic RET mutation correlates with an aggressive MTC disease phenotype (Romei et al. 1996; Schilling et al. 2001). Recently, sporadic MTC negative for RET mutations have been shown to frequently display mutation of RAS genes (Moura et al. 2011). Thus, similarly to follicular cell-derived thyroid tumors, thyroid C-cell-derived carcinomas commonly feature the activation of RET-RAS-MAPK pathway (Fig. 3).
MECHANISMS OF RET ONCOGENIC CONVERSION SECONDARY TO POINT MUTATIONS
The mechanisms leading to RET oncogenic conversion in MEN2 and MTC depend on the site of the amino-acid change. In the case of cysteine mutants, cysteine removal is believed to prevent the formation of intramolecular disulfide bonds, thus allowing free cysteine residues to form intermolecular bonds and mediate the formation of covalent RET dimers with constitutive kinase and signaling activity (Fig. 3) (Santoro et al. 1995). Moreover, differently from wild-type (wt) RET, C634 RET mutant is resistant to intracellular domain cleavage and does not show cytotoxic activity in the absence of ligand, thereby losing the “dependence” receptor feature. This implies that a single mutation (e.g., C634) may induce at the same time increased mitogenic signaling and reduced proapoptotic activity (Bordeaux et al. 2000). Mutations associated with FMTC, which generally target cysteines other than C634 (Fig. 4), are less potently transforming than MEN2A-associated C634 mutations because of their weaker ability to induce formation of RET dimers (Carlomagno et al. 1997; Ito et al. 1997; Chappuis-Flament et al. 1998).
A change in substrate specificity together with a ligand-independent activation of the enzymatic function has been implicated in the mechanism of activation induced by M918T mutation (Santoro et al. 1995; Songyang et al. 1995). In line with this model, M918T mutants differ from wild-type RET in the stoichiometry of phosphorylation of RET tyrosines and of various intracellular proteins (Santoro et al. 1995; Salvatore et al. 2001). RET/M918T-expressing tumors have different gene expression profiles compared with RET/C634-expressing tumors (Jain et al. 2004). Moreover, X-ray crystallographic analysis of RET tyrosine kinase domain has shown that wild-type RET kinase adopts a head-to-tail autoinhibited dimeric state and that this inactive conformation is destabilized by M918T mutation (Knowles et al. 2006). The mechanism through which RET intracellular mutations (other than M918T) activate constitutively RET enzymatic function has not been clearly elucidated.
RET Gain- and Loss-of-Function in Disease
As mentioned above, germline mutations in RET have been implicated in both sporadic and familial cases of Hirschsprung disease (HSCR). HSCR is characterized by the congenital absence of enteric innervation, causing block of peristalsis and bowel obstruction (congenital megacolon) (Brooks et al. 2005). RET mutations found in HSCR patients are heterogeneous, ranging from gene deletions to nonsense and missense point mutations, and in most cases they cause a loss of RET signaling. Moreover, some HSCR mutations were found to cause a constitutive caspase-3-mediated RET cleavage and proapoptotic activity probably attributable to the “dependence receptor” features of RET (Bordeaux et al. 2000).
In a few cases, HSCR cosegregates with MEN2A/FMTC that instead, as described above, are associated with RET gain-of-function. These promiscuous HSCR-MEN2A/FMTC cases typically display mutations in RET cysteines other than C634. The most reasonable explanation of this paradox relies on the possibility that these particular mutations have a Janus-faced effect, as they feature both a decreased cell surface expression and a constitutive, although low-level, kinase activity. This may on the one hand be sufficient to cause thyroid C-cells transformation, but on the other hand not be sufficient to sustain correct enteric neurons development for the reduced ability of such mutants of interacting with the ligand on the cell surface (Carlomagno et al. 1997; Ito et al. 1997; Chappuis-Flament et al. 1998; Arighi et al. 2004).
RET IN MALIGNANCIES OTHER THAN THYROID CARCINOMA
RET in Lung Cancer
For many years, RET oncogenic conversion has been thought to be confined to thyroid cancer. More recently, structural RET alterations or changes of its expression have been described in neoplasms affecting organs other than thyroid. Systematic high-throughput sequencing screening identified the M918T RET mutation in one single case of non-small-cell lung cancer (NSCLC). In this case, RET mutation co-occurred with K-RAS mutation, whereas K-RAS is generally alternative to RTK mutation (Thomas et al. 2007). Very recently, RET has been shown to play an important role in a subset of NSCLC patients. In about 1% NSCLC, particularly in adenocarcinoma, chromosomal inversions cause the fusion of the RET-encoded TK domain (from exon 12 to the 3′-end of RET, as in RET/PTC rearrangements) to different 5′-terminal exons (15, 16, 22, 23, or 24 exons in different rearrangement variants) of the KIF5B (kinesin family member 5B) gene (Ju et al. 2012; Kohno et al. 2012; Li et al. 2012; Lipson et al. 2012; Takeuchi et al. 2012). Less commonly, the RET-encoded TK domain was found to be fused to CCDC6, NCOA4, or TRIM33 genes (as in RET/PTC1, RET/PTC3 and RET/PTC7, respectively) (Li et al. 2012; Wang et al. 2012; Drilon et al. 2013). Similarly to RET/PTC rearrangements, KIF5B-RET fusion proteins are likely to form homodimers through the coiled-coil domain present in the NH2-terminal portion of KIF5B (Fig. 5). The coiled-coil domain is retained in all variants of KIF5B-RET rearranged proteins. Consistently, KIF5B-RET fusion proteins display ligand-independent activation of RET kinase and are able to transform fibroblasts in vitro (Kohno et al. 2012).
Figure 5.
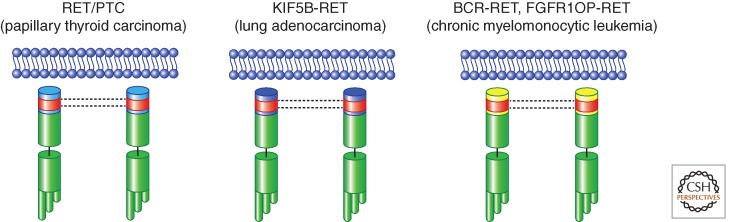
Schematic representation of RET gene rearrangements in human cancer. RET/PTC rearrangements in PTC are shown, together with KIF5B-RET (found in lung adenocarcinoma) and BCR-RET and FGFR1OP-RET (found in chronic myelomonocytic leukemia). All the RET fusion partners invariably code for protein–protein dimerization motifs (depicted as red boxes).
RET in Leukemia
RET gene was found up-regulated in a subtype of acute myeloid leukemia with myelomonocytic stage of differentiation (Camos et al. 2006). More recently, gene rearrangements causing the fusion of the RET-encoding TK domain (from exon 12 to the 3′-end of RET, as in RET/PTC and KIF5B-RET) in one case to the first 5′-terminal 4 exons of BCR (breakpoint cluster region) and in another case to the first 5′-terminal 12 exons of FGFR1OP (fibroblast growth factor receptor 1 oncogenic partner) genes have been cloned from two cases of chronic myelomonocytic leukemia (CMML) (Ballerini et al. 2012). BCR-RET and FGFR1OP-RET fusion proteins act as bona fide oncoproteins, display aberrant activation of RET kinase, and transform hematopoietic cells in vitro. Thus, although the prevalence of these rearrangements is still unknown, CMML appears to be another neoplasia that, together with PTC and lung adenocarcinoma, is associated with RET activation through gene rearrangement (Fig. 5).
RET Role in Additional Cancer Types
Systematic DNA sequencing in a set of colon cancers has revealed the rare occurrence of somatic RET sequence variants (Wood et al. 2007). Similarly, sequencing of around 1500 cancer-related genes uncovered the presence of rare RET variants of unknown functional significance in hormone receptor positive breast cancer (Kan et al. 2010).
Apart from mutations, many reports have indicated a positive correlation between RET overexpression and ER (estrogen receptor)-positive breast carcinoma (Tozlu et al. 2006; Esseghir et al. 2007). RET transcriptional promoter was found to display three EREs (estrogen receptor elements) that may mediate RET up-regulation by estrogens (Boulay et al. 2008; Kang et al. 2010). A functional link between RET and ER is indicated by the observation that RET inhibition restored a hormone-sensitive phenotype of antiestrogen-resistant breast cancer cells (Plaza-Menacho et al. 2010).
Some frequent RET sequence variants (polymorphisms) have been correlated to specific tumor types. RET G691S polymorphism cosegregates with MTC, pancreatic cancer, and desmoplastic subtype of cutaneous malignant melanoma (Robledo et al. 2003; Cebrian et al. 2005; Sawai et al. 2005; Narita et al. 2009). G691S caused increased receptor-signaling responses to GDNF stimulation, with augmented cell proliferation, migration, and invasion (Narita et al. 2009).
RET protein was found overexpressed in pancreatic carcinoma and GDNF polarized migration of pancreatic cancer cells toward nerves mediating neural invasion (Veit et al. 2004; Gil et al. 2010). Finally, although the molecular basis was not elucidated, overactivation of RET protein has been observed in glioblastoma multiforme and involved in maintaining a robust downstream signaling able to limit the efficacy of therapies with kinase inhibitors (Stommel et al. 2007).
RET PROTEIN AS AN ANTINEOPLASTIC THERAPEUTIC TARGET
RET Kinase Inhibitors
Small molecule protein kinase inhibitors (PKI) are an important class of anticancer agents. In most of the cases, they compete with ATP, thereby obstructing autophosphorylation and signal transduction downstream from the targeted kinase (Zhang et al. 2009). Prominent examples of successful PKI are represented by imatinib (Gleevec) against BCR-ABL in chronic myeloid leukemia (CML), gefitinib (iressa), and erlotinib against EGFR in non-small-cell lung carcinoma (NSCLC), and vemurafenib (PLX4032) against BRAF in melanoma (Zhang et al. 2009).
Based on its involvement in cancer and particularly in MTC, a neoplasm that does not respond to conventional chemotherapy, RET has raised interest as a molecular target of kinase inhibitors (Schlumberger et al. 2008; Gild et al. 2011). Preclinical studies have shown that MTC cell lines are addicted to RET oncogenic signaling and that RET PKIs are able to block MTC cell proliferation (Schlumberger et al. 2008). Some compounds were isolated that exerted potent RET inhibition. They include vandetanib (ZD6474), sorafenib (BAY 43-9006), sunitinib (SU11248), cabozantinib (XL184), lenvatinib (E7080), and ponatinib (AP24534) (Carlomagno et al. 2002, 2006; Kim et al. 2006; Verbeek et al. 2011; De Falco et al. 2013). These compounds are multitargeted, being able to inhibit RET and also additional kinases.
Therapeutic Targeting of RET in Human Cancer
Most of RET PKIs have been or are being evaluated in clinical trials for MTC treatment (Schlumberger et al. 2008). In particular, vandetanib (ZD6474), an inhibitor of RET, EGFR (epidermal growth factor receptor) and VEGFR (vascular endothelial growth factor receptor), and cabozantinib, an inhibitor of RET, MET, and VEGFR, have been approved by the FDA for MTC treatment, based on significant progression-free survival prolongation in clinical trials (Kurzrock et al. 2011; Wells et al. 2012). However, it should be noted that some specific RET mutations (codons 804 and 806) confer resistance to vandetanib and would require alternative inhibitors (Carlomagno et al. 2004, 2009).
Medullary thyroid cancer (MTC) may not remain the only cancer that benefits from RET kinase inhibition. Papillary thyroid carcinoma (PTC) in general responds well to adjuvant radioiodine treatment and therefore RET kinase inhibitors may not prove particularly useful for this tumor (Gild et al. 2011). Given the recent discovery of RET gain-of-function in lung adenocarcinoma and CMML, it is feasible that RET kinase inhibitors may also find applications in these cancers. Consistently, KIF5B-RET transformed fibroblasts growth was inhibited by vandetanib (ZD6474) (Kohno et al. 2012), treatment with cabozantinib induced objective response in some patients with RET mutant lung adenocarcinoma (Drilon et al. 2013), and treatment with sorafenib (BAY 43-9006), another RET kinase inhibitor, induced cytological, and clinical remission in a patient carrying the BCR-RET fusion (Ballerini et al. 2012).
CONCLUSIONS
After its initial isolation from patients affected by papillary thyroid carcinoma, RET receptor tyrosine kinase also proved to be a key player in the pathogenesis of medullary thyroid carcinoma (MTC). More recently, rare forms of leukemia and non-small-cell lung carcinoma have been associated with oncogenic conversion of RET, thus expanding the spectrum of cancer diseases associated with mutations of this gene. This knowledge will foster efforts to develop agents able to target RET oncoproteins for cancer treatment.
Footnotes
Editors: Joseph Schlessinger and Mark A. Lemmon
Additional Perspectives on Signaling by Receptor Tyrosine Kinases available at www.cshperspectives.org
REFERENCES
- Airaksinen MS, Saarma M 2002. The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: 383–394 [DOI] [PubMed] [Google Scholar]
- Alberti L, Borrello MG, Ghizzoni S, Torriti F, Rizzetti MG, Pierotti MA 1998. Grb2 binding to the different isoforms of Ret tyrosine kinase. Oncogene 17: 1079–1087 [DOI] [PubMed] [Google Scholar]
- Ameziane-El-Hassani R, Boufraqech M, Lagente-Chevallier O, Weyemi U, Talbot M, Métivier D, Courtin F, Bidart JM, El Mzibri M, Schlumberger M, et al. 2010. Role of H2O2 in RET/PTC1 chromosomal rearrangement produced by ionizing radiation in human thyroid cells. Cancer Res 70: 4123–4132 [DOI] [PubMed] [Google Scholar]
- Anders J, Kjar S, Ibáñez CF 2001. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. J Biol Chem 276: 35808–35817 [DOI] [PubMed] [Google Scholar]
- Arighi E, Popsueva A, Degl’Innocenti D, Borrello MG, Carniti C, Perälä NM, Pierotti MA, Sariola H 2004. Biological effects of the dual phenotypic Janus mutation of ret cosegregating with both multiple endocrine neoplasia type 2 and Hirschsprung’s disease. Mol Endocrinol 18: 1004–1017 [DOI] [PubMed] [Google Scholar]
- Asai N, Murakami H, Iwashita T, Takahashi M 1996. A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with shc adaptor proteins. J Biol Chem 271: 17644–17649 [DOI] [PubMed] [Google Scholar]
- Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, Dobbelstein S, Petit A, Lapillonne H, Gautier EF, et al. 2012. RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 26: 2384–2399 [DOI] [PubMed] [Google Scholar]
- Baloh RH, Enomoto H, Johnson EM Jr, Milbrandt J 2000. The GDNF family ligands and receptors—Implications for neural development. Curr Opin Neurobiol 10: 103–110 [DOI] [PubMed] [Google Scholar]
- Beldjord C, Desclaux-Arramond F, Raffin-Sanson M, Corvol JC, De Keyzer Y, Luton JP, Plouin PF, Bertagna X 1995. The RET protooncogene in sporadic pheochromocytomas: Frequent MEN 2-like mutations and new molecular defects. J Clin Endocrinol Metab 80: 2063–2068 [DOI] [PubMed] [Google Scholar]
- Boikos SA, Stratakis CA 2006. Carney complex: Pathology and molecular genetics. Neuroendocrinology 83: 189–199 [DOI] [PubMed] [Google Scholar]
- Bongarzone I, Monzini N, Borrello MG, Carcano C, Ferraresi G, Arighi E, Mondellini P, Della Porta G, Pierotti MA 1993. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RI alpha of cyclic AMP-dependent protein kinase A. Mol Cell Biol 13: 358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, Bredesen DE, Edery P, Mehlen P 2000. The RET proto-oncogene induces apoptosis: A novel mechanism for Hirschsprung disease. EMBO J 19: 4056–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello MG, Alberti L, Arighi E, Bongarzone I, Battistini C, Bardelli A, Pasini B, Piutti C, Rizzetti MG, Mondellini P, et al. 1996. The full oncogenic activity of Ret/ptc2 depends on tyrosine 539, a docking site for phospholipase Cγ. Mol Cell Biol 16: 2151–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello MG, Alberti L, Fischer A, Degl’innocenti D, Ferrario C, Gariboldi M, Marchesi F, Allavena P, Greco A, Collini P, et al. 2005. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci 102: 14825–14830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, Wartmann M, Stumm M, Lane HA, Hynes NE 2008. The Ret receptor tyrosine kinase pathway functionally interacts with the ERα pathway in breast cancer. Cancer Res 68: 3743–3751 [DOI] [PubMed] [Google Scholar]
- Brooks AS, Oostra BA, Hofstra RM 2005. Studying the genetics of Hirschsprung’s disease: Unraveling an oligogenic disorder. Clin Genet 67: 6–14 [DOI] [PubMed] [Google Scholar]
- Camós M, Esteve J, Jares P, Colomer D, Rozman M, Villamor N, Costa D, Carrió A, Nomdedéu J, Montserrat E, et al. 2006. Gene expression profiling of acute myeloid leukemia with translocation t(8;16)(p11;p13) and MYST3-CREBBP rearrangement reveals a distinctive signature with a specific pattern of HOX gene expression. Cancer Res 66: 6947–6954 [DOI] [PubMed] [Google Scholar]
- Cañibano C, Rodriguez NL, Saez C, Tovar S, Garcia-Lavandeira M, Borrello MG, Vidal A, Costantini F, Japon M, Dieguez C, et al. 2007. The dependence receptor Ret induces apoptosis in somatotrophs through a Pit-1/p53 pathway, preventing tumor growth. EMBO J 26: 2015–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno F, De Vita G, Berlingieri MT, de Franciscis V, Melillo RM, Colantuoni V, Kraus MH, Di Fiore PP, Fusco A, Santoro M 1996. Molecular heterogeneity of RET loss of function in Hirschsprung’s disease. EMBO J 15: 2717–2725 [PMC free article] [PubMed] [Google Scholar]
- Carlomagno F, Salvatore G, Cirafici AM, De Vita G, Melillo RM, de Franciscis V, Billaud M, Fusco A, Santoro M 1997. The different RET-activating capability of mutations of cysteine 620 or cysteine 634 correlates with the multiple endocrine neoplasia type 2 disease phenotype. Cancer Res 57: 391–395 [PubMed] [Google Scholar]
- Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, Ryan AJ, Fontanini G, Fusco A, Santoro M 2002. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res 62: 7284–7290 [PubMed] [Google Scholar]
- Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, Billaud M, Santoro M 2004. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23: 6056–6063 [DOI] [PubMed] [Google Scholar]
- Carlomagno F, Anaganti S, Guida T, Salvatore G, Troncone G, Wilhelm SM, Santoro M 2006. BAY 43–9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst 98: 326–334 [DOI] [PubMed] [Google Scholar]
- Carlomagno F, Guida T, Anaganti S, Provitera L, Kjaer S, McDonald NQ, Ryan AJ, Santoro M 2009. Identification of tyrosine 806 as a molecular determinant of RET kinase sensitivity to ZD6474. Endocr Relat Cancer 16: 233–241 [DOI] [PubMed] [Google Scholar]
- Castellone MD, Cirafici AM, De Vita G, De Falco V, Malorni L, Tallini G, Fagin JA, Fusco A, Melillo RM, Santoro M 2003. Ras-mediated apoptosis of PC CL 3 rat thyroid cells induced by RET/PTC oncogenes. Oncogene 22: 246–255 [DOI] [PubMed] [Google Scholar]
- Castellone MD, Verrienti A, Magendra Rao D, Sponziello M, Fabbro D, Muthu M, Durante C, Maranghi M, Damante G, Pizzolitto S, et al. 2010. A novel de novo germ-line V292M mutation in the extracellular region of RET in a patient with phaeochromocytoma and medullary thyroid carcinoma: functional characterization. Clin Endocrinol (Oxf) 73: 529–534 [DOI] [PubMed] [Google Scholar]
- Caudill CM, Zhu Z, Ciampi R, Stringer JR, Nikiforov YE 2005. Dose-dependent generation of RET/PTC in human thyroid cells after in vitro exposure to γ-radiation: A model of carcinogenic chromosomal rearrangement induced by ionizing radiation. J Clin Endocrinol Metab 90: 2364–2369 [DOI] [PubMed] [Google Scholar]
- Cebrian A, Lesueur F, Martin S, Leyland J, Ahmed S, Luccarini C, Smith PL, Luben R, Whittaker J, Pharoah PD, et al. 2005. Polymorphisms in the initiators of RET (rearranged during transfection) signaling pathway and susceptibility to sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 90: 6268–6274 [DOI] [PubMed] [Google Scholar]
- Celetti A, Cerrato A, Merolla F, Vitagliano D, Vecchio G, Grieco M 2004. H4(D10S170), a gene frequently rearranged with RET in papillary thyroid carcinomas: Functional characterization. Oncogene 23: 109–121 [DOI] [PubMed] [Google Scholar]
- Chappuis-Flament S, Pasini A, De Vita G, Ségouffin-Cariou C, Fusco A, Attié T, Lenoir GM, Santoro M, Billaud M 1998. Dual effect on the RET receptor of MEN 2 mutations affecting specific extracytoplasmic cysteines. Oncogene 17: 2851–2861 [DOI] [PubMed] [Google Scholar]
- Collins BJ, Chiappetta G, Schneider AB, Santoro M, Pentimalli F, Fogelfeld L, Gierlowski T, Shore-Freedman E, Jaffe G, Fusco A 2002. RET expression in papillary thyroid cancer from patients irradiated in childhood for benign conditions. J Clin Endocrinol Metab 87: 3941–3946 [DOI] [PubMed] [Google Scholar]
- De Falco V, Buonocore P, Muthu M, Torregrossa L, Basolo F, Billaud M, Gozgit JM, Carlomagno F, Santoro M 2013. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J Clin Endocrinol Metab 10.1210/jc.2012-2672 [DOI] [PubMed] [Google Scholar]
- de Graaff E, Srinivas S, Kilkenny C, D’Agati V, Mankoo BS, Costantini F, Pachnis V 2001. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 15: 2433–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot JW, Links TP, Plukker JT, Lips CJ, Hofstra RM 2006. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocr Rev 27: 535–560 [DOI] [PubMed] [Google Scholar]
- Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens PJ, Ross J, Miller VA, Ginsberg MS, Zakowski MF, et al. 2013. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 10.1158/2159-8290.CD-13-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW 2007. Chromosome fragile sites. Annu Rev Genet 41: 169–192 [DOI] [PubMed] [Google Scholar]
- Encinas M, Crowder RJ, Milbrandt J, Johnson EM Jr 2004. Tyrosine 981, a novel ret autophosphorylation site, binds c-Src to mediate neuronal survival. J Biol Chem 279: 18262–18269 [DOI] [PubMed] [Google Scholar]
- Esseghir S, Todd SK, Hunt T, Poulsom R, Plaza-Menacho I, Reis-Filho JS, Isacke CM 2007. A role for glial cell derived neurotrophic factor induced expression by inflammatory cytokines and RET/GFR-α 1 receptor up-regulation in breast cancer. Cancer Res 67: 11732–11741 [DOI] [PubMed] [Google Scholar]
- Fazioli F, Piccinini G, Appolloni G, Bacchiocchi R, Palmonella G, Recchioni R, Pierpaoli E, Silvetti F, Scarpelli M, Bruglia M, et al. 2008. A new germline point mutation in Ret exon 8 (cys515ser) in a family with medullary thyroid carcinoma. Thyroid 18: 775–782 [DOI] [PubMed] [Google Scholar]
- Gandhi M, Medvedovic M, Stringer JR, Nikiforov YE 2006. Interphase chromosome folding determines spatial proximity of genes participating in carcinogenic RET/PTC rearrangements. Oncogene 25: 2360–2366 [DOI] [PubMed] [Google Scholar]
- Gandhi M, Evdokimova V, Nikiforov YE 2010a. Mechanisms of chromosomal rearrangements in solid tumors: The model of papillary thyroid carcinoma. Mol Cell Endocrinol 321: 36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M, Dillon LW, Pramanik S, Nikiforov YE, Wang YH 2010b. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene 29: 2272–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP, Carlson DL, Shah JP, Fong Y, Wong RJ 2010. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J Natl Cancer Inst 102: 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gild ML, Bullock M, Robinson BG, Clifton-Bligh R 2011. Multikinase inhibitors: A new option for the treatment of thyroid cancer. Nat Rev Endocrinol 7: 617–624 [DOI] [PubMed] [Google Scholar]
- Greco A, Miranda C, Pierotti MA 2010. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol 321: 44–49 [DOI] [PubMed] [Google Scholar]
- Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G 1990. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 60: 557–563 [DOI] [PubMed] [Google Scholar]
- Hamatani K, Eguchi H, Ito R, Mukai M, Takahashi K, Taga M, Imai K, Cologne J, Soda M, Arihiro K, et al. 2008. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res 68: 7176–7182 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Heinlein CA, Ting HJ, Yeh S, Chang C 1999. Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor γ. J Biol Chem 274: 16147–16152 [DOI] [PubMed] [Google Scholar]
- Ito T, Seyama T, Iwamoto KS, Hayashi T, Mizuno T, Tsuyama N, Dohi K, Nakamura N, Akiyama M 1993. In vitro irradiation is able to cause RET oncogene rearrangement. Cancer Res 53: 2940–2943 [PubMed] [Google Scholar]
- Ito S, Iwashita T, Asai N, Murakami H, Iwata Y, Sobue G, Takahashi M 1997. Biological properties of Ret with cysteine mutations correlate with multiple endocrine neoplasia type 2A, familial medullary thyroid carcinoma, and Hirschsprung’s disease phenotype. Cancer Res 57: 2870–2872 [PubMed] [Google Scholar]
- Jain S 2009. The many faces of RET dysfunction in kidney. Organogenesis 5: 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Watson MA, DeBenedetti MK, Hiraki Y, Moley JF, Milbrandt J 2004. Expression profiles provide insights into early malignant potential and skeletal abnormalities in multiple endocrine neoplasia type 2B syndrome tumors. Cancer Res 64: 3907–3913 [DOI] [PubMed] [Google Scholar]
- Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, Kim YT, Kim JI, Kang JH, Seo JS 2012. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res 22: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. 2010. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 466: 869–873 [DOI] [PubMed] [Google Scholar]
- Kang J, Qian PX, Pandey V, Perry JK, Miller LD, Liu ET, Zhu T, Liu DX, Lobie PE 2010. Artemin is estrogen regulated and mediates antiestrogen resistance in mammary carcinoma. Oncogene 29: 3228–3240 [DOI] [PubMed] [Google Scholar]
- Kawamoto Y, Takeda K, Okuno Y, Yamakawa Y, Ito Y, Taguchi R, Kato M, Suzuki H, Takahashi M, Nakashima I 2004. Identification of RET autophosphorylation sites by mass spectrometry. J Biol Chem 279: 14213–14224 [DOI] [PubMed] [Google Scholar]
- Khetchoumian K, Teletin M, Tisserand J, Mark M, Herquel B, Ignat M, Zucman-Rossi J, Cammas F, Lerouge T, Thibault C, et al. 2007. Loss of Trim24 (Tif1α) gene function confers oncogenic activity to retinoic acid receptor α. Nat Genet 39: 1500–1506 [DOI] [PubMed] [Google Scholar]
- Kim DW, Jo YS, Jung HS, Chung HK, Song JH, Park KC, Park SH, Hwang JH, Rha SY, Kweon GR, et al. 2006. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab 91: 4070–4076 [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA 2000. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat Genet 26: 89–92 [DOI] [PubMed] [Google Scholar]
- Kjaer S, Kurokawa K, Perrinjaquet M, Abrescia C, Ibáñez CF 2006. Self-association of the transmembrane domain of RET underlies oncogenic activation by MEN2A mutations. Oncogene 25: 7086–7095 [DOI] [PubMed] [Google Scholar]
- Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, Pacini F, Ringel MD, Schlumberger M, et al. 2009. Medullary thyroid cancer: Management guidelines of the American Thyroid Association. Thyroid 19: 565–612 [DOI] [PubMed] [Google Scholar]
- Klugbauer S, Rabes HM 1999. The transcription coactivator HTIF1 and a related protein are fused to the RET receptor tyrosine kinase in childhood papillary thyroid carcinomas. Oncogene 18: 4388–4393 [DOI] [PubMed] [Google Scholar]
- Knowles PP, Murray-Rust J, Kjaer S, Scott RP, Hanrahan S, Santoro M, Ibáñez CF, McDonald NQ 2006. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem 281: 33577–33587 [DOI] [PubMed] [Google Scholar]
- Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, et al. 2012. KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18: 375–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollara A, Kahn HJ, Marks A, Brown TJ 2001. Loss of androgen receptor associated protein 70 (ARA70) expression in a subset of HER2-positive breast cancers. Breast Cancer Res Treat 67: 245–253 [DOI] [PubMed] [Google Scholar]
- Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, Pfister DG, Cohen EE, Janisch L, Nauling F, et al. 2011. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol 29: 2660–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone V, Mansueto G, Pierantoni GM, Tornincasa M, Merolla F, Cerrato A, Santoro M, Grieco M, Scaloni A, Celetti A, et al. 2010. CCDC6 represses CREB1 activity by recruiting histone deacetylase 1 and protein phosphatase 1. Oncogene 29: 4341–4351 [DOI] [PubMed] [Google Scholar]
- Li P, Yu X, Ge K, Melamed J, Roeder RG, Wang Z 2002. Heterogeneous expression and functions of androgen receptor co-factors in primary prostate cancer. Am J Pathol 161: 1467–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Feng Y, Fang R, Fang Z, Xia J, Han X, Liu XY, Chen H, Liu H, Ji H 2012. Identification of RET gene fusion by exon array analyses in “pan-negative” lung cancer from never smokers. Cell Res 22: 928–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligr M, Li Y, Zou X, Daniels G, Melamed J, Peng Y, Wang W, Wang J, Ostrer H, Pagano M, et al. 2010. Tumor suppressor function of androgen receptor coactivator ARA70α in prostate cancer. Am J Pathol 176: 1891–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor NM, Honchel R, Khosla S, Thibodeau SN 1995. Mutations in the RET protooncogene in sporadic pheochromocytomas. J Clin Endocrinol Metab 80: 627–629 [DOI] [PubMed] [Google Scholar]
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, et al. 2012. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 18: 382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Vega QC, Decker RA, Pandey A, Worby CA, Dixon JE 1996. Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. J Biol Chem 271: 5309–5312 [DOI] [PubMed] [Google Scholar]
- Manié S, Santoro M, Fusco A, Billaud M 2001. The RET receptor: Function in development and dysfunction in congenital malformation. Trends Genet 17: 580–589 [DOI] [PubMed] [Google Scholar]
- Margraf RL, Crockett DK, Krautscheid PM, Seamons R, Calderon FR, Wittwer CT, Mao R 2009. Multiple endocrine neoplasia type 2 RET protooncogene database: Repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum Mutat 30: 548–556 [DOI] [PubMed] [Google Scholar]
- Mehlen P, Bredesen DE 2011. Dependence receptors: From basic research to drug development. Sci Signal 4: mr2. [DOI] [PubMed] [Google Scholar]
- Melillo RM, Carlomagno F, De Vita G, Formisano P, Vecchio G, Fusco A, Billaud M, Santoro M 2001a. The insulin receptor substrate (IRS)-1 recruits phosphatidylinositol 3-kinase to Ret: Evidence for a competition between Shc and IRS-1 for the binding to Ret. Oncogene 20: 209–218 [DOI] [PubMed] [Google Scholar]
- Melillo RM, Santoro M, Ong SH, Billaud M, Fusco A, Hadari YR, Schlessinger J, Lax I 2001b. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol Cell Biol 21: 4177–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo RM, Castellone MD, Guarino V, De Falco V, Cirafici AM, Salvatore G, Caiazzo F, Basolo F, Giannini R, Kruhoffer M, et al. 2005. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and fitogeni phenotype of thyroid cancer cells. J Clin Invest 115: 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Melillo RM, Guarino V, Avilla E, Galdiero MR, Liotti F, Prevete N, Rossi FW, Basolo F, Ugolini C, de Paulis A, et al. 2010. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 29: 6203–6215 [DOI] [PubMed] [Google Scholar]
- Merolla F, Pentimalli F, Pacelli R, Vecchio G, Fusco A, Grieco M, Celetti A 2007. Involvement of H4(D10S170) protein in ATM-dependent response to DNA damage. Oncogene 26: 6167–6175 [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kyoizumi S, Suzuki T, Iwamoto KS, Seyama T 1997. Continued expression of a tissue specific activated oncogene in the early steps of radiation-induced human thyroid carcinogenesis. Oncogene 15: 1455–1460 [DOI] [PubMed] [Google Scholar]
- Mizuno T, Iwamoto KS, Kyoizumi S, Nagamura H, Shinohara T, Koyama K, Seyama T, Hamatani K 2000. Preferential induction of RET/PTC1 rearrangement by X-ray irradiation. Oncogene 19: 438–443 [DOI] [PubMed] [Google Scholar]
- Monaco C, Visconti R, Barone MV, Pierantoni GM, Berlingieri MT, De Lorenzo C, Mineo A, Vecchio G, Fusco A, Santoro M 2001. The RFG oligomerization domain mediates kinase activation and re-localization of the RET/PTC3 oncoprotein to the plasma membrane. Oncogene 20: 599–608 [DOI] [PubMed] [Google Scholar]
- Moura MM, Cavaco BM, Pinto AE, Leite V 2011. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab 96: E863–E868 [DOI] [PubMed] [Google Scholar]
- Murakami H, Iwashita T, Asai N, Shimono Y, Iwata Y, Kawai K, Takahashi M 1999. Enhanced phosphatidylinositol 3-kinase activity and high phosphorylation state of its downstream signalling molecules mediated by ret with the MEN 2B mutation. Biochem Biophys Res Commun 262: 68–75 [DOI] [PubMed] [Google Scholar]
- Myers SM, Eng C, Ponder BA, Mulligan LM 1995. Characterization of RET proto-oncogene 3′ splicing variants and polyadenylation sites: A novel C-terminus for RET. Oncogene 11: 2039–2045 [PubMed] [Google Scholar]
- Narita N, Tanemura A, Murali R, Scolyer RA, Huang S, Arigami T, Yanagita S, Chong KK, Thompson JF, Morton DL, et al. 2009. Functional RET G691S polymorphism in cutaneous malignant melanoma. Oncogene 28: 3058–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov YE, Nikiforova MN 2011. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol 7: 569–580 [DOI] [PubMed] [Google Scholar]
- Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE 2000. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 290: 138–141 [DOI] [PubMed] [Google Scholar]
- Pasini B, Hofstra RM, Yin L, Bocciardi R, Santamaria G, Grootscholten PM, Ceccherini I, Patrone G, Priolo M, Buys CH, et al. 1995. The physical map of the human RET proto-oncogene. Oncogene 11: 1737–1743 [PubMed] [Google Scholar]
- Plaza-Menacho I, Morandi A, Robertson D, Pancholi S, Drury S, Dowsett M, Martin LA, Isacke CM 2010. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene 29: 4648–4657 [DOI] [PubMed] [Google Scholar]
- Powell DJ Jr, Russell J, Nibu K, Li G, Rhee E, Liao M, Goldstein M, Keane WM, Santoro M, Fusco A, et al. 1998. The RET/PTC3 oncogene: Metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res 58: 5523–5528 [PubMed] [Google Scholar]
- Puxeddu E, Knauf JA, Sartor MA, Mitsutake N, Smith EP, Medvedovic M, Tomlinson CR, Moretti S, Fagin JA 2005. RET/PTC-induced gene expression in thyroid PCCL3 cells reveals early activation of genes involved in regulation of the immune response. Endocr Relat Cancer 12: 319–334 [DOI] [PubMed] [Google Scholar]
- Rhoden KJ, Unger K, Salvatore G, Yilmaz Y, Vovk V, Chiappetta G, Qumsiyeh MB, Rothstein JL, Fusco A, Santoro M, et al. 2006. RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: Follicular cells of Hashimoto’s thyroiditis share low-level recombination events with a subset of papillary carcinoma. J Clin Endocrinol Metab 91: 2414–2423 [DOI] [PubMed] [Google Scholar]
- Robledo M, Gil L, Pollán M, Cebrián A, Ruíz S, Azañedo M, Benitez J, Menárguez J, Rojas JM 2003. Polymorphisms G691S/S904S of RET as genetic modifiers of MEN 2A. Cancer Res 63: 1814–1817 [PubMed] [Google Scholar]
- Romei C, Elisei R, Pinchera A, Ceccherini I, Molinaro E, Mancusi F, Martino E, Romeo G, Pacini F 1996. Somatic mutations of the ret protooncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J Clin Endocrinol Metab 81: 1619–1622 [DOI] [PubMed] [Google Scholar]
- Russell JP, Shinohara S, Melillo RM, Castellone MD, Santoro M, Rothstein JL 2003. Tyrosine kinase oncoprotein, RET/PTC3, induces the secretion of myeloid growth and chemotactic factors. Oncogene 22: 4569–4577 [DOI] [PubMed] [Google Scholar]
- Salvatore D, Melillo RM, Monaco C, Visconti R, Fenzi G, Vecchio G, Fusco A, Santoro M 2001. Increased in vivo phosphorylation of ret tyrosine 1062 is a potential pathogenetic mechanism of multiple endocrine neoplasia type 2B. Cancer Res 61: 1426–1431 [PubMed] [Google Scholar]
- Santoro M, Melillo RM, Grieco M, Berlingieri MT, Vecchio G, Fusco A 1993. The TRK and RET tyrosine kinase oncogenes cooperate with ras in the neoplastic transformation of a rat thyroid epithelial cell line. Cell Growth Differ 4: 77–84 [PubMed] [Google Scholar]
- Santoro M, Dathan NA, Berlingieri MT, Bongarzone I, Paulin C, Grieco M, Pierotti MA, Vecchio G, Fusco A 1994. Molecular characterization of RET/PTC3; a novel rearranged version of the RET proto-oncogene in a human thyroid papillary carcinoma. Oncogene 9: 509–516 [PubMed] [Google Scholar]
- Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH, et al. 1995. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 267: 381–383 [DOI] [PubMed] [Google Scholar]
- Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, Picone A, Portella G, Santelli G, Vecchio G, et al. 1996. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene 12: 1821–1826 [PubMed] [Google Scholar]
- Sawai H, Okada Y, Kazanjian K, Kim J, Hasan S, Hines OJ, Reber HA, Hoon DS, Eibl G 2005. The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Cancer Res 65: 11536–11544 [DOI] [PubMed] [Google Scholar]
- Schilling T, Bürck J, Sinn HP, Clemens A, Otto HF, Höppner W, Herfarth C, Ziegler R, Schwab M, Raue F 2001. Prognostic value of codon 918 (ATG→ACG) RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. Int J Cancer 95: 62–66 [DOI] [PubMed] [Google Scholar]
- Schlumberger M, Carlomagno F, Baudin E, Bidart JM, Santoro M 2008. New therapeutic approaches to treat medullary thyroid carcinoma. Nat Clin Pract Endocrinol Metab 4: 22–32 [DOI] [PubMed] [Google Scholar]
- Schuetz G, Rosário M, Grimm J, Boeckers TM, Gundelfinger ED, Birchmeier W 2004. The neuronal scaffold protein Shank3 mediates signaling and biological function of the receptor tyrosine kinase Ret in epithelial cells. J Cell Biol 167: 945–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segouffin-Cariou C, Billaud M 2000. Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. J Biol Chem 275: 3568–3576 [DOI] [PubMed] [Google Scholar]
- Songyang Z, Carraway KL III, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, et al. 1995. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature 373: 536–539 [DOI] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, et al. 2007. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318: 287–290 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Asai N, Iwashita T, Isomura T, Miyazaki K, Matsuyama M 1993. Characterization of the ret proto-oncogene products expressed in mouse L cells. Oncogene 8: 2925–2929 [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et al. 2012. RET, ROS1 and ALK fusions in lung cancer. Nat Med 18: 378–381 [DOI] [PubMed] [Google Scholar]
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, et al. 2007. High-throughput oncogene mutation profiling in human cancer. Nat Genet 39: 347–351 [DOI] [PubMed] [Google Scholar]
- Tozlu S, Girault I, Vacher S, Vendrell J, Andrieu C, Spyratos F, Cohen P, Lidereau R, Bieche I 2006. Identification of novel genes that co-cluster with estrogen receptor α in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr Relat Cancer 13: 1109–1120 [DOI] [PubMed] [Google Scholar]
- Unger K, Zitzelsberger H, Salvatore G, Santoro M, Bogdanova T, Braselmann H, Kastner P, Zurnadzhy L, Tronko N, Hutzler P, et al. 2004. Heterogeneity in the distribution of RET/PTC rearrangements within individual post-Chernobyl papillary thyroid carcinomas. J Clin Endocrinol Metab 89: 4272–4279 [DOI] [PubMed] [Google Scholar]
- Veit C, Genze F, Menke A, Hoeffert S, Gress TM, Gierschik P, Giehl K 2004. Activation of phosphatidylinositol 3-kinase and extracellular signal-regulated kinase is required for glial cell line-derived neurotrophic factor-induced migration and invasion of pancreatic carcinoma cells. Cancer Res 64: 5291–5300 [DOI] [PubMed] [Google Scholar]
- Verbeek HH, Alves MM, de Groot JW, Osinga J, Plukker JT, Links TP, Hofstra RM 2011. The effects of four different tyrosine kinase inhibitors on medullary and papillary thyroid cancer cells. J Clin Endocrinol Metab 96: E991–E995 [DOI] [PubMed] [Google Scholar]
- Viglietto G, Chiappetta G, Martinez-Tello FJ, Fukunaga FH, Tallini G, Rigopoulou D, Visconti R, Mastro A, Santoro M, Fusco A 1995. RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 11: 1207–1210 [PubMed] [Google Scholar]
- Vizioli MG, Possik PA, Tarantino E, Meissl K, Borrello MG, Miranda C, Anania MC, Pagliardini S, Seregni E, Pierotti MA, et al. 2011. Evidence of oncogene-induced senescence in thyroid carcinogenesis. Endocr Relat Cancer 18: 743–757 [DOI] [PubMed] [Google Scholar]
- Wang J, Knauf JA, Basu S, Puxeddu E, Kuroda H, Santoro M, Fusco A, Fagin JA 2003. Conditional expression of RET/PTC induces a weak oncogenic drive in thyroid PCCL3 cells and inhibits thyrotropin action at multiple levels. Mol Endocrinol 17: 1425–1436 [DOI] [PubMed] [Google Scholar]
- Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, Luo X, Wang L, Li H, Zhang Y, et al. 2012. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol 30: 4352–4359 [DOI] [PubMed] [Google Scholar]
- Wells SA Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, et al. 2012. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J Clin Oncol 30: 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D 2008. Radiation carcinogenesis: Lessons from Chernobyl. Oncogene 27: S9–S18 [DOI] [PubMed] [Google Scholar]
- Wong N, Lai P, Lee SW, Fan S, Pang E, Liew CT, Sheng Z, Lau JW, Johnson PJ 1999. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: Relationship to disease stage, tumor size, and cirrhosis. Am J Pathol 154: 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. 2007. The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113 [DOI] [PubMed] [Google Scholar]
- Xing M 2005. BRAF mutation in thyroid cancer. Endocr Relat Cancer 12: 245–262 [DOI] [PubMed] [Google Scholar]
- Yeh S, Chang C 1996. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc Natl Acad Sci 93: 5517–5521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS 2009. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9: 28–39 [DOI] [PubMed] [Google Scholar]
- Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE 2006. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: Effects of the detection methods and genetic heterogeneity. J Clin Endocrinol Metab 91: 3603–3610 [DOI] [PubMed] [Google Scholar]