

Abstract
Recombination-dependent DNA replication, often called break-induced replication (BIR), was initially invoked to explain recombination events in bacteriophage but it has recently been recognized as a fundamentally important mechanism to repair double-strand chromosome breaks in eukaryotes. This mechanism appears to be critically important in the restarting of stalled and broken replication forks and in maintaining the integrity of eroded telomeres. Although BIR helps preserve genome integrity during replication, it also promotes genome instability by the production of loss of heterozygosity and the formation of nonreciprocal translocations, as well as in the generation of complex chromosomal rearrangements.
Break-induced replication restarts stalled replication forks and may help maintain telomeres. But it can also promote genome instability (e.g., by promoting loss of heterozygosity and the formation of nonreciprocal translocations).
The break-copy mode of recombination (as opposed to break-join), was initially proposed by Meselson and Weigle (1961). Break-copy recombination, now more commonly known as recombination-dependent DNA replication or break-induced replication (BIR), is believed to account for restarting replication at broken replication forks and may also play a central role in the maintenance of telomeres in the absence of telomerase. BIR has been studied in various model systems and has been invoked to explain chromosome rearrangements in humans. This review focuses primarily on mechanistic studies in Escherichia coli and its bacteriophages, T4 and λ, in the budding yeasts Saccharomyces cerevisiae and Kluyveromyces lactis and on apparently similar, but less well-documented, mechanisms in mammalian cells.
Homology-dependent repair of DNA double-strand breaks (DSBs) occur by three major repair pathways (Pâques and Haber 1999) (Fig. 1). When both ends of the DNA share substantial homology with a donor template (a sister chromatid, a homologous chromosome, or an ectopically located segment), repair occurs almost exclusively by gene conversion (GC). If the DSB is flanked by direct repeats, then a second repair process, single-strand annealing (SSA), can occur as 5′ to 3′ resection of the DSB ends exposes complementary sequences that can anneal to each other and repair the break by the formation of a deletion. However, when only one DSB end shares homology with a donor sequence, repair occurs by BIR. There are two BIR pathways, one dependent on Rad51 recombinase and the other independent of Rad51.
Figure 1.

Three major repair pathways of homology-dependent recombination. Noncrossover (NCO) and crossover (CO) events are indicated. Black triangles represent resolution of Holliday junctions (HJs). Dashed lines represent new DNA synthesis. GC, gene conversion; SSA, single-strand annealing; BIR, break-induced replication.
BIR IN BACTERIA AND BACTERIOPHAGES
Recombination-Dependent Replication in Phage T4
Recombination-dependent replication (RDR) is an intrinsic part of the T4 life cycle (reviewed in Mosig 1987; Kreuzer 2000). Early studies of this process, both in vivo and in vitro, provided a molecular framework and paved the way for the study of similar processes in bacterial and eukaryotic cells. T4 is a linear dsDNA bacteriophage; its packaged nucleic acid is slightly more than one genome’s worth, causing its ends to be direct repeats. Because of the 5′ to 3′ directionality of replication and a requirement for RNA primers to initiate DNA synthesis, the T4 genome, like all linear chromosomes, has a problem with replication of its termini. RDR provides a solution to complete terminal replication.
The initial phase of T4 replication begins at one of several discrete origins of replication. This initiation requires host RNA polymerase, involves the formation of RNA:DNA hybrid molecules (R-loops), and is blocked by the RNAP inhibitor rifampicin (reviewed in Kreuzer and Brister 2010). Later, T4 proteins modify the specificity of RNAP, and the later phase is a rifampicin-resistant mode of recombination-dependent replication (RDR) (Luder and Mosig 1982). Mutations in T4’s recombination functions not only lead to poor recovery of genetic recombinants, but also block the secondary, late pathway of replication (reviewed in Mosig 1987; Kreuzer 2000). One of these recombination functions, UvsX, operates much like the E. coli RecA protein in the catalysis of strand-pairing, invasion, and strand-transfer reactions (reviewed in Liu and Morrical 2010) (Table 1). Another recombination function, UvsY, acts as a mediator protein in promoting UvsX binding and replacement of the single-strand DNA binding protein gp32 on single-strand DNA substrates, similar to E. coli’s RecFOR and eukaryotic Rad52. A third recombination function, UvsW, is a DNA helicase that promotes stability of D-loops by branch migration of the junction between the two homologous molecules (Carles-Kinch et al. 1997), analogous to the E. coli RecG helicase. In vivo, recombination is initiated at T4 ends, at which 3′ ssDNA should be persistent because of the end-replication problem. These ends appear to be bound or further processed by gp46/47 nuclease, proteins related to the SbcCD complex of bacteria and the Mre11 Rad50 complexes of eukaryotes (Sharples and Leach 1995). Mutants in gp46/47 exhibit a profound arrest of late DNA replication (RDR) (Mosig 1998). Replication is initiated at these recombination intermediates, leading to a network of branched molecules (Kemper and Brown 1976) that are resolved to linear packageable DNA molecules by the gp49 resolvase function (Mizuuchi et al. 1982).
Table 1.
Recombination-dependent replication functions in T4 bacteriophage, E. coli, and S. cerevisiae
| Function | T4 | E. coli | Budding yeast |
|---|---|---|---|
| Strand invasion and exchange | UvsX | RecA | Rad51 |
| Mediator | UvsY | RecFOR RecBCD |
Rad52 Rad55/57 |
| Single-strand DNA binding protein | gp32 | SSB | RPA |
| End processing nuclease | gp46/47 | SbcCD RecBCD |
Mre11/Rad50/Xrs2 Sae2 Dna2/Sgs1 Exo1 |
| Replicative helicase | gp41 | DnaB | Mcm2-7 complex |
| Helicase reloader | gp59 | PriA, PriB PriC DnaC DnaT |
? |
| Branch migration helicase | gp41, UvsW | RuvAB RecG |
Sgs1 (RecQ helicase family) Srs2 |
| Branched molecule cleavage | gp49 (Endo VII) | RuvC | Mus81/Mms4 Yen1 |
By the 1980s, Mosig had codified a mechanism for late replication, initiated by homologous recombination (Mosig 1987). The key intermediate in this mechanism is the displacement loop (D-loop) (Fig. 2), a heteroduplex joint molecule formed by the strand invasion of a 3′ tailed linear molecule, catalyzed by UvsX, UvsY, and gp32. Such D-loops would be recognized as substrates for the recruitment of the replicative helicase/primase complex, gp61/41, and loading of the DNA polymerase/clamp complex. This model has been further validated by in vitro reconstitution reactions and by in vivo experiments monitoring the processing of T4 DSBs.
Figure 2.

Scheme for recombination-dependent replication of T4. The end-replication problem leaves 3′ ssDNA ends on dsDNA (blue), further processed by gp46/47 nuclease. Mediator UvsY assists UvsX to form a presynaptic filament, competent for strand invasion with a homolgous duplex (red) to form D-loop intermediates. UvsW helicase stabilizes this structure. Full assembly of the replisome, consisting of gp43 (DNA polymerase), gp32 (single-strand DNA binding protein), gp45 (clamp) and gp44/62 (clamp loader), primase (gp41), and (gp61) fork helicase and helicase loader (gp59), establishing bidirectional replication.
Formosa and Alberts (1986) reconstituted an in vitro system for replication initiated by recombination between a single-strand linear molecule and a homologous covalently closed circle. These reactions required UvsX, gp32, gp43 (DNA polymerase), and gp45 and gp44/62 (clamp and clamp loader complex). The products of such reactions were long, single-stranded DNA concatemers. UvsX/gp32 could not be replaced with their cognate functions from E. coli, RecA/SSB, indicating that there must be a species-specific interaction between the replisome and the recombination machinery. In these reactions, the nature of the products and the absence of any requirement for topoisomerase activity led investigators to propose the bubble migration model, a conservative mechanism of DNA synthesis, by which DNA is polymerized and then displaced as a single strand from its template (much like the mechanism of RNA transcription from DNA) (Fig. 3). The bubble migration mechanism corresponds to the priming of leading- but not lagging-strand synthesis, which likely does not reflect the natural mechanism (see below).
Figure 3.

Key intermediates of replication initiated by homologous recombination. The double-stranded DNA end is resected, exposing the single-stranded 3′ end. Strand invasion results in a D-loop intermediate. Bubble migration involves priming of leading- but not lagging-strand DNA synthesis (left), whereas full fork establishment involves coupling of leading- and lagging-strand synthesis (right). The solid line with arrowhead represents the invading 3′ end. Dashed lines represent new DNA synthesis.
Full establishment of bona fide replication forks from recombination intermediates requires helicase loading via gp59. Because gene 59 mutants display a late replication arrest phenotype (Wu and Yeh 1974), gp59 must play an essential role in RDR, presumably in the loading of gp41 onto recombination-generated D-loops, in a manner reminiscent of the primosome PriA protein of E. coli (see below). Inclusion of gp59/41/61 to the reconstituted system promotes establishment of full leading- and lagging-strand semiconservative DNA synthesis from UvsX-generated D-loops (Barry and Alberts 1994) in contrast to the conservative replication mode of bubble migration seen in the absence of gp59/41/61 (Formosa and Alberts 1986). Gp59 promotes the coupling of lagging-strand DNA synthesis with that of the leading strand (Dudas and Kreuzer 2005) by acting as a gatekeeper to block engagement of the 3′ invading strand by DNA polymerase. Indicative of this gatekeeper property of gp59, specific interactions between gp59 and DNA polymerase gp43 (Xi et al. 2005) inhibit 3′ end extension (Nossal et al. 2001) in vitro. This gatekeeper function presumably prevents premature leading-strand synthesis prior to assembly of the helicase/primase complex that will provoke coupled lagging/leading-strand replication.
Insights into the RDR mechanism have been provided by systems in which site-specific DSBs induce coupled recombination and replication of a homologous partner molecule in vivo (Kreuzer et al. 1995; George and Kreuzer 1996; George et al. 2001). Such reactions require the suite of T4 recombination proteins (UvsX, UvsY, UvsW, gp46/47) and T4 replication functions (gp43, gp45, gp44/62), gp59, primase (gp41), and helicase (gp61). Consistent with the extensive chromosome replication mechanism of RDR, the two broken ends of a plasmid molecule can engage two separate homologous partners, independently provoking their replication (Stohr and Kreuzer 2002).
Break-Induced Replication in E. coli
Even before elucidation of T4 RDR, connections between replication and recombination in bacteria had been suspected. Lederberg (1955) proposed a break-copy model of recombination, whereby a broken chromosome would stimulate replication by recombination. Skalka (1974), through her studies of bacteriophage lambda, suggested that convergence of replication forks onto preexisting nicks in the template strand leads to DSBs, which might be repaired by recombination with a homologous chromosome through a D-loop intermediate.
By the mid-1980s, the homologous recombination pathways in E. coli had been defined genetically: the RecBCD helicase/nuclease, in conjunction with the RecA strand transfer protein, constituted a mechanism for DSB repair (Wang and Smith 1983; Clark and Sandler 1994). The sequence of the E. coli chromosome revealed that Chi sites (GCTGGTGG) that stimulate RecBCD-mediated recombination and attenuate its nuclease activity (reviewed in Persky and Lovett 2008) are found overrepresented in the E. coli genome, in the orientation that protects broken forks from extensive degradation (Burland et al. 1993) and promotes their processing into D-loops that could restore a replication fork. The preferential orientation of Chi sites suggests that repair of collapsed replication forks, rather than simple repair of DSBs, is the primary role of the RecBCD repair system (Kuzminov 1995). Smith (1991) proposed that even the classical recombination processes in E. coli (involving DNA transfer via conjugation or dsDNA introduced by phage transduction) set up obligate replication forks to mediate inheritance of genetic markers.
Despite earlier suggestions (Asai et al. 1994), it took several years to establish conclusively that DNA DBSs could prime replication in E. coli (Kuzminov and Stahl 1999). When recombination between bacteriophage lambda is catalyzed by the E. coli recombination systems, the break-copy mechanism accounts for approximately one-half of the detected recombinants, and the nonreplicative break-join mechanism accounts for the remainder (Motamedi et al. 1999). These two mechanisms are distinguished by the sedimentation properties of recombinant phage grown in light (14N) medium from heavy (15N)-labeled parents. Break-join phage remain heavy (HH), depend on HJ resolvase activity, and can appear in the absence of DNA polymerase III activity, whereas break-copy outcomes show extensive replication with an HL density, are independent of resolvase, and are dependent on DNA polymerase III.
Ascertainment of recombination-associated replication in E. coli in vivo has been aided by its requirement for a special set of replication restart or primosome assembly factors that are not required for normal replication initiation at the origin of replication, oriC. During normal replication, the DnaA protein, a AAA+ ATPase, is required, along with DnaC protein, to load the replicative helicase for initiation at oriC. In contrast, for replication restart, an alternative set of factors (PriA, PriB, PriC, and DnaT), along with DnaC, loads DnaB. These factors were initially characterized for the role in the initiation of replication of phiX174 bacteriophage (Shlomai and Kornberg 1980a,b) at an origin consisting of a region of DNA secondary structure. Phage phiX174 has evidently commandeered this special restart system to initiate its replication.
The PriA protein, an AAA+ ATPase and 3′ to 5′ helicase, initiates the assembly of replication forks from recombinational D-loop structures (Liu et al. 1999; Xu and Marians 2000). PriA’s binding to DNA substrates appears to be enhanced by PriB, potentially through an interaction with SSB; it then recruits DnaT, which aids the loading of DnaB helicase from a DnaBC complex and concomitantly the DnaG primase (reviewed in Gabbai and Marians 2010). In addition to orchestrating the primosome assembly process through protein recruitment, PriA 3′ helicase activity may process intermediates to provide a single-strand assembly site on the lagging-strand template for DnaB loading. Interestingly, like its counterpart gp59 of bacteriophage T4, PriA binds and occludes the 3′ invading strand from DNA polymerase extension, potentially ensuring that a full fork supporting both leading and lagging strand is established (Xu and Marians 2003). PriA can also support restart from some forklike structures, suggesting a role for restart both with and without associated replication. For forks with a leading-strand gap, an alternative pathway involving PriC and Rep appears to be more efficient (Heller and Marians 2006).
In vivo, mutants in priA have reduced viability, especially under fast growth conditions that support multiple ongoing replication forks. In priA mutants, a subpopulation of cells is filamentous and is induced for the SOS response to DNA damage, indicating an abnormal response to spontaneous source of damage (Lee and Kornberg 1991; Nurse et al. 1991). Mutants in priA are sensitive to DNA damaging agents, such as UV and gamma irradiation and mitomycin C (Nurse et al. 1991; Masai et al. 1994; Kogoma et al. 1996). These phenotypes can be fully complemented by the Walker A K230R mutant, which is defective for helicase activity but fully competent for primosome assembly (Zavitz and Marians 1992). In priA mutants, there is a high level of RecFOR-mediated recombination, indicative of persistent DNA gaps (Grompone et al. 2004). PriA mutants also show reduced recombinational inheritance during transduction and conjugation (Kogoma et al. 1996), implicating establishment of replication forks are invoked during these processes as proposed earlier by Smith (1991). In addition to one-ended recombination events involving repair of a broken fork with its sister chromosome, PriA is required for two-ended recombination events that repair a DSB caused by SbcCD nuclease cleavage of secondary structure elements (Eykelenboom et al. 2008).
A phenomenon known as induced stable DNA replication (iSDR) has been studied in some detail and reflects a mode of BIR in E. coli. DNA replication, as detected by radioactive thymine precursor incorporation, normally ceases after inhibition of protein synthesis such as caused by the antibiotic chloramphenicol. However, induction of the SOS response via treatments such as UV irradiation, nalidixic acid treatment, or thymine starvation permits a mode of DnaA-independent, chloramphenicol-resistant replication (reviewed in Kogoma 1997), which depends on the primosomal assembly factor, PriA (Masai et al. 1994), and DNA recombination proteins, RecA and RecBCD (Magee and Kogoma 1990). The requirement for RecBCD, an enzyme specific for recombination initiated at DSBs (see Persky and Lovett 2008), implicates chromosomal breaks in the mechanism although the RecF pathway is required for a minor component of iSDR is (Kogoma 1997). The role of the helicase function activity of PriA in iSDR is unclear since the Walker A K230R mutant has been reported both to restore (Kogoma et al. 1996) and to lack (Tanaka et al. 2003) iSDR function. The above observations support a model by which replication is initiated at RecABCD-dependent D-loops (Kogoma 1997); thus, defects in the proteins that process D-loops, RecG, and RuvABC elevate iSDR in vivo (Asai and Kogoma 1994). iSDR is apparently mutagenic (Lark and Lark 1979) but the molecular nature of these mutations has not been fully investigated.
BIR in Budding Yeast
The best-studied BIR system in eukaryotes is budding yeast S. cerevisiae. Three different assays have been used to explore BIR: (1) transformation of ends-out double-stranded DNA fragments, (2) induction of site-specific HO or I-SceI endonucleases to create chromosomal DSBs that share only one-ended homology, and (3) creation of strains that lack functional telomerase but maintain their telomeres by recombination-dependent replication (Fig. 4). None of these systems directly address the problem of repair of a stalled and broken replication fork where there is a broken chromosome end and an intact sister chromatid. However, ways to address this central question have been developed by Aguilera (Cortes-Ledesma et al. 2007) and by Bjergbaek (Nielsen et al. 2009), using site-specific enzymes to create single-strand nicks in G1 cells, which are converted to DSBs as cells enter S phase. Although all of these events require the Rad52 protein, there are both Rad51-dependent and Rad51-independent BIR processes.
Figure 4.
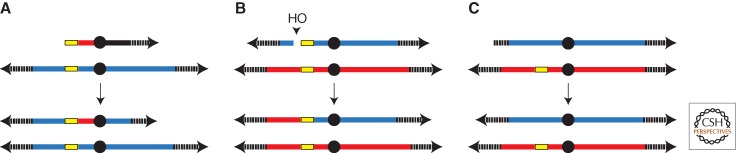
Three different assays used to explore BIR. BIR assays involving (A) a typical chromosome fragmentation vector (CFV), (B) HO-endonuclease-induced DSB where only one end shares homology with the donor (where homology is indicated in yellow), and (C) telomere maintenance in the absence of telomerase by recombination-dependent replication.
Rad51-Dependent BIR during Transformation
Dunn et al. (1984) first showed that a transformed linearized plasmid sharing homology at one end with subtelomeric sequences could acquire new telomeres by Rad52-dependent recombination. Evidence of much more extensive recombination-dependent replication was provided by Morrow et al. (1997) using chromosome fragmentation vectors (CFV), carrying an origin of replication (ARS) and a centromere (Fig. 4A). These studies also found that centromeres pose major barriers to BIR, so that most transformants actually contained half-crossover outcomes most likely representing strand invasion events that branch-migrate into an intermediate that can be resolved as a crossover but cause loss of the template chromosome. Why centromeres are not barriers to bulk replication but block BIR is an unsolved question. Surprisingly, BIR can progress through the entire array of more than 100 9-kb rDNA repeats (B Llorente, pers. comm.).
Another BIR-related phenomenon occurs during gene targeting. In a significant fraction of transformations, one finds hit-and-run events in which one or both ends invade and set up a BIR event that can copy substantial lengths of DNA adjacent to the integration site (Adair et al. 1989). If the copying includes an ARS and the two ends then rejoin, the apparent gene knockout will prove to be an autonomously replicating circle carrying the selectable marker (Kraus et al. 2001).
DSB-Induced Rad51-Dependent BIR
HO endonuclease cleaves a 24-bp site to catalyze mating-type (MAT) switching, an efficient GC event in which both sides of the DSB share homology with a donor (Haber 2012). However, the normal donors can be deleted and the cleavage site inserted at other locations to establish situations in which only the centromere-proximal end of the DSB shares homology with an ectopic sequence (in a haploid) or allelic sequence (in a diploid). HO-induced BIR can occur with as little as 70 bp of homology to create a nonreciprocal translocation (Bosco and Haber 1998; Ira and Haber 2002). Although Rad51-dependent BIR is quite efficient, it represents only a tiny fraction of outcomes in the situation where both ends of the break share homology and GC dominates.
Because HO induction is rapid and highly efficient, it is possible to follow repair kinetics on Southern blots (Malkova et al. 2005) (Fig. 5). Whereas interchromosomal GC is completed in 2–3 h, BIR events are delayed until 4–6 h. This delay is enforced after Rad51 promotes strand invasion (measured by a chromatin immunoprecipitation assay) but before the initiation of new DNA synthesis (assayed by PCR). Once BIR is initiated, its rate of DNA synthesis over the 100 kb to the end of the chromosome, 3–4 kb/min, is comparable to normal replication.
Figure 5.
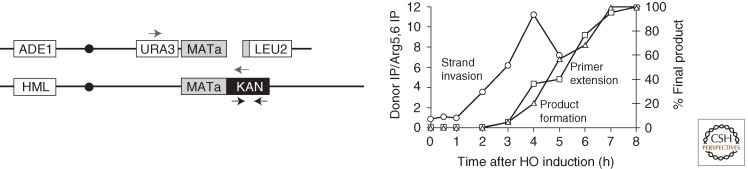
Kinetics of BIR. Schematic representation of a diploid BIR strain used to measure the kinetics of BIR, where there is only homology to the left of the DSB (left). Black arrows represent the position of primers used for ChIP analysis. Gray arrows represent the position of primers used for primer extension assay. The graph (right) shows the kinetics of strand invasion (circles), initiation of new DNA synthesis (squares), and product formation (triangles). (Figure modified from Jain et al. 2009.)
The delay in initiating BIR after strand invasion reflects the action of a recombination execution checkpoint that can sense whether the two ends of a DSB can synapse with the same donor sequence within a critical distance and in the proper orientation to complete repair (Jain et al. 2009). Thus, if the two halves of the donor sequence are separated by about 1 kb, repair is rapid and efficient; but if the two homologous targets are 5 kb or more apart, the initiation of repair DNA synthesis is—like BIR—delayed by several hours. As further evidence that there is a transition from GC to BIR, the large gap repair events become dependent on Pol32 (Jain et al. 2009).
DSB-Induced Rad51-Independent BIR
In a diploid where MAT cleavage on one chromosome can be repaired by allelic recombination, nearly all repair occurs by GC. Without Rad51, there is still surprisingly efficient repair by Rad51-independent BIR, retaining a marker on the opposite arm of the chromosome but losing heterozygosity to the right of the DSB. These events are distinctly different from Rad51-dependent BIR. First, many of the repair events occur after the cells have escaped from arrest imposed by the DNA damage checkpoint so that the colonies are sectored, containing both BIR outcomes and 2n-1 cells that have lost the broken chromosome. Second, repair depends primarily on a pair of inverted Ty retrotransposons elements lying 30 kb proximal to the break. Most repair events prove to be nonreciprocal translocations involving Ty sequences on other chromosomes. This process requires the MRX complex, Rad59, and Rdh54/Tid1, as well as Rad52 (Signon et al. 2001).
A second instance in which Rad51-independent BIR predominates is when both ends of an HO-induced DSB share very limited homology with a donor, for example, in a centromeric plasmid containing sequences in inverted orientation, with only 33 bp homology shared between the donor and recipient (Ira and Haber 2002). Repair significantly increases without rad51. Presumably, Rad51 binds to the ssDNA ends containing the 33 bp homology, but it is incapable of carrying out the next steps in recombination. The repair events that are recovered in the absence of Rad51 are also distinctive in 50% having an apparent reciprocal crossover. As suggested by Symington (Bartsch et al. 2000), these repair events most likely arise by BIR followed by single-strand annealing (Fig. 6).
Figure 6.

Schematic of events that involve a combination of BIR and SSA. Inverted repeats are shown as blue and red arrows, respectively, which are separated by marker AB. DSB at one of the inverted repeats is followed by resection and invasion into the other (homologous) inverted repeat. DNA synthesis (dashed line) proceeds until the end of the break. Depending on which strand gets resected, SSA results in different orientation of the marker (AB). (Figure adapted from Bartsch et al. 2000.)
Telomere Maintenance without Telomerase
BIR also allows yeast telomeres to be maintained in the absence of telomerase (Lundblad and Blackburn 1993; McEachern and Blackburn 1996; Teng and Zakian 1999; McEachern and Haber 2006). In mammalian cells, such alternative lengthening of telomeres (ALT) has been shown to involve recombination, but how it occurs is not well defined. In budding yeast, there are two recombination-dependent mechanisms, both requiring Rad52, but one which is Rad51-independent. In cells lacking either telomerase RNA or protein components, telomeres erode progressively until the vast majority of cells undergo senescence and cell death. However, a tiny proportion of cells become survivors and proliferate. There are two distinct pathways for telomere maintenance. One pathway (Type I) involves recombination with and amplification of Y′ subtelomeric sequences (Lundblad and Blackburn 1993; Louis et al. 1994). Type I survivors depend on the canonical recombination proteins including Rad52, Rad51, Rad55/57, and Rad54 (Le et al. 1999; Teng and Zakian 1999). A second mechanism (Type II) leads to long extensions of the telomere repeats themselves; these survivors arise independent of Rad51 but require Rad52, the MRX complex, and Rad59 (Lundblad and Blackburn 1993; McEachern and Blackburn 1996; Teng and Zakian 1999; Teng et al. 2000). Type II survivors also require Sgs1 helicase (not required for HO-induced Rad51-independent BIR) and the checkpoint proteins Mec1 and Tel1 (Cohen and Sinclair 2001; Huang et al. 2001; Johnson et al. 2001; Signon et al. 2001; Tsai et al. 2002). Both pathways require the nonessential DNA polymerase Pol δ, Pol32 (Lydeard et al. 2007), and thus are replicative processes rather than half-crossovers that would elongate one telomere at the expense of a sister chromatid.
In Vivo Biochemical Analysis of Rad51-Dependent BIR
A detailed investigation of the factors needed for Rad51-dependent BIR compared to those for GC conducted by Lydeard et al. (2010b) revealed that BIR requires nearly all the factors for normal replication, except some of those required to establish the prereplication complex at origins.
Recombination-Mediated Establishment of Replication
The initial stages of Rad51-dependent BIR are shared with gene conversion, namely, 5′ to 3′ resection of the DSB, homology searching, and strand invasion. Resection is initiated by the MRX-Sae2 proteins but is then taken over by two different activities, the exonuclease Exo1 or the helicase-endonuclease complex of Sgs1-Rmi1-Top3 and Dna2 (Neale et al. 2005; Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008; Symington and Gautier 2011). Surprisingly, deletion of these different proteins had opposing effects on the efficiencies of BIR; mre11Δ or rad50Δ caused a 50% reduction in BIR, whereas sae2Δ resulted in a slight increase (Lydeard et al. 2010a). This difference could reflect other roles of MRX and Sae2 in the establishment and maintenance of the DNA damage checkpoint that prevents mitosis before BIR is complete or in later steps of recombination. However, a transformation-based assay found no role for MRX in BIR (Davis and Symington 2004).
Deleting Exo1 or Sgs1 results in a nearly 50% increase in BIR; conversely, their overexpression severely inhibits BIR (Lydeard et al. 2010a). The increase in BIR in the absence of Exo1 or Sgs1 is also seen in transformation-based assay (Marrero and Symington 2010). Reducing the extent of ssDNA may ensure that Rad51 is bound to the homologous sequences at the end of the DSB rather than being attracted to more distant unrelated ssDNA. Supporting this idea, overexpressing Rad51 nearly doubles the efficiency of BIR and partially suppresses overexpressing Sgs1 or Exo1 (Lydeard et al. 2010a). Alternatively, Sgs1 may discourage strand invasion and synapsis with short regions of homology, similar to its documented role in dismantling heteroduplex DNA containing mismatches, but how Exo1 would affect this is unclear. Similar to Sgs1, deleting another 3′ to 5′ helicase, Mph1, causes significant increases in BIR efficiency (JE Haber and S Jain, unpubl.). More strikingly, an sgs1 mph1 double mutant additively increases BIR efficiency and unexpectedly also seems to eliminate the delay in initiating new DNA synthesis. How this occurs remains under investigation.
As noted above, the initial steps in recombination appear to be the same for BIR as for GC: 5′ to 3′ resection of DSB ends, loading of RPA, and then its displacement by Rad51, aided by Rad52, Rad55, and Rad57 (Krogh and Symington 2004). However, another set of Rad51-aiding proteins, Psy3-Csm2-Shu1-Shu2 (PCSS), are much more important in BIR than in GC (Lydeard et al. 2010b). The idea that the PCSS proteins might be an alternative Rad51-facilitating pathway is supported by the finding that overexpressing Rad51 suppresses this defect in both shu2Δ rad55Δ and shu2Δ.
Initiation of New DNA Synthesis
One of the least-understood steps in BIR is the mechanism by which the strand-invasion D-loop is converted into a replication fork with leading- and lagging-strand synthesis. There is no homolog of bacterial PriA. A striking finding is that nearly all BIR events require Pol32, a nonessential subunit of DNA Pol δ, whereas GC is not affected by pol32Δ (Lydeard et al. 2007). It is possible that Pol32, similar to PriA in prokaryotes, acts in some way to recruit a replicative helicase and assemble an origin-independent replication fork.
A second novel requirement for BIR that has little effect on normal replication or GC was found as a suppressor of pol32Δ’s cold-sensitivity for growth. Surprisingly, two pol30 mutations that suppress pol32Δ’s cold sensitivity do not suppress pol32Δ’s BIR defect; in fact, these pol30 mutations, by themselves, are defective in BIR (Lydeard et al. 2010b). Moreover, these mutations are dominant, suggesting that even one such mutant subunit of the PCNA trimer may be sufficient to disrupt BIR. These PCNA mutations could be defective in the steps of establishing a full replication fork. The defect of pol30-FF248, 249AA is much more severe than the 50% reduction seen in a PCNA mutation that cannot be ubiquitylated or SUMOylated (pol30-K164R) or when the ubiquitylation and SUMOylation pathways are disrupted (rad18Δ siz1Δ)(Lydeard et al. 2010b).
BIR requires virtually all of the components of normal DNA replication, including the replicative helicase complex consisting of the Mcm2-7 heterohexamer, Cdc45, and the GINS complex (Lydeard et al. 2010b). BIR also requires the DDK kinase complex, Cdc7-Dbf4. These components are not needed for GC. Although Cdc7 can be bypassed by mutations in Mcm proteins, these suppressors do not relieve the requirement for Cdc7 in BIR. BIR involves both leading- and lagging-strand synthesis; consequently, BIR requires Pol α-primase and two factors that help Pol α act processively: Mcm10 and Ctf4. BIR also requires components that were previously identified as prereplication origin-loading factors: Dpb11-Sld2-Sld3 and Cdt1. Interestingly, without these factors, even the initial presumably leading-strand extension from the 3′ end of the invading strand does not occur. That no leading-strand synthesis occurs without Pol α or its cofactors again suggests that leading- and lagging-strand synthesis are tightly coordinated and that there is not an initial step of primer extension (as in GC) that is later converted into a full replication fork.
One remarkable difference between normal replication and BIR is that the initiation of BIR requires Pol δ, but proceeds without Pol ε (Lydeard et al. 2007). However Pol ε is required for replication of the template to continue after the first several kilobases. This separation of functions between Pol δ and Pol ε is strikingly different from what has been seen in replication, where Pol ε appears to direct leading-strand synthesis, and Pol δ is concerned with lagging-strand copying. However, it is known that S-phase replication can be completed in the absence of catalytic activity of Pol ε so long as the carboxy-terminal portion of the Pol 2 (Pol ε) protein is present (Kesti et al. 1999). The nonessential subunits of Pol ε (Dpb3 and Dpb4) are not required for BIR (Lydeard et al. 2010b).
Replication during BIR Is Far More Mutagenic than Normal Replication
The idea that there is a transition between Pol ε-independent and Pol ε-dependent replication complexes after the first several kilobases of new DNA synthesis resonates with the finding from the Symington lab that there are frequent template switches when a linearized fragment can initiate BIR with either of two polymorphic homologous chromosomes of a diploid (Smith et al. 2007). These template switches are confined to the first several kilobases, after which copying is apparently processive. This observation could be explained if initial DNA synthesis involved only the primer-extended leading strand, which then matured into a full and more processive replication fork. However, as noted above, physical monitoring of BIR does not support this hypothesis, as there was no primer extension when Pol α primase was inactivated. It is possible that there are mechanistic differences in CFV events compared to a chromosomal DSB.
Despite the fact that BIR uses the complete replication machinery that is employed in normal S phase, the rate of mutation accompanying BIR is as much as 2800 times higher than spontaneous events. By placing a lys2 gene containing an out-of-frame run of As at several locations along the template chromosome arm, Deem et al. (2011) demonstrated that frameshift mutations increased both close to the point of strand invasion and much further down the template chromosome. Another set of reversion events involved a template jump between quasi-palindromic sequences. Mutations were independent of the error-prone DNA polymerase Pol ς or another translesion polymerase, Pol η. However, 50% or more of possible frameshift mutations were corrected by the mismatch repair machinery because the rate was two- to 11-fold higher in an msh2Δ mutant. Similarly, in a Pol δ proofreading mutant (pol3-5DV), there is a significantly higher level of BIR-associated frameshifts, suggesting that Pol δ is a major player in creating, but then correcting, errors. One contribution to the high mutation rate is a DNA damage checkpoint regulated, Dun1-dependent increase in dNTP pools, causing DNA polymerases to be error prone.
These results are quite different from the high level of mutations seen in GC, where the high rate of frameshift mutagenesis and base pair substitutions were independent of mismatch repair (Hicks et al. 2010). Moreover, a different Pol δ mutation, pol3-01, had a dramatically opposite effect on mutations, namely eliminating essentially all the template jumps, be they –1 frameshifts, quasipalindrome events, or interchromosomal template switches.
Resolution of BIR
A major unsolved question concerns the resolution of Holliday junctions (HJ) and the final disposition of newly synthesized strands during BIR. If BIR proceeds by formation of a unidirectional replication fork inside a D-loop, there should be an HJ near the point of strand invasion. If it is cleaved by an HJ resolvase, the newly synthesized DNA should remain base paired to its template. In this case, one should find a new DNA strand on both the donor and the new translocated region. If this branch migrates toward the terminus, the two strands of the donor will be re-paired, as will the two newly made strands, so that the replication will appear to be conservative, as was found for short patch DNA synthesis in GC (Ira et al. 2006).
Whether BIR leads to semiconservative or conservative DNA synthesis can be answered by DNA combing or by methods in which base analogs such as BrdU are incorporated into DNA and detected using immunofluorescence. By analyzing the location of the Lys+ revertants in the BIR assay described above, Malkova (A Malkova, pers. comm.) has preliminary evidence that they are all in the recipient (and hence conservatively replicated) chromosome.
Budding yeast has at least four HJ resolvases. Two of these—Mus81-Mms4 and Yen1—appear to play the major role in generating crossovers in interchromosomal mitotic recombination (Ho et al. 2010); however, even the mus81Δ yen1Δ double mutant did not affect the success of BIR.
BIR in Other Organisms
Kluyveromyces lactis
Studies in K. lactis have provided useful mechanistic details of telomere maintenance in the absence of telomerase (McEachern and Blackburn 1995; McEachern and Iyer 2001; Natarajan and McEachern 2002; Groff-Vindman et al. 2005; Cesare et al. 2008) and also helped in establishing the generality of telomere maintenance by means of BIR (McEachern and Haber 2006). Unlike in S. cerevisiae, all K. lactis survivors are of Type II, except when telomeric sequences are placed internally to the chromosome ends, when Type I survivors are also observed (Natarajan et al. 2003). However, the unit telomere repeat in K. lactis is 25 nt, compared to the degenerate TG1-3 repeats in S. cerevisiae. A key intermediate that enables telomere maintenance in the absence of telomerase is a telomeric circle, which can be excised by recombination from its original location. By a mechanism termed roll and spread, the telomeric circle is copied onto the telomere at the chromosome end by rolling circle replication, involving BIR. Two main observations support this hypothesis: visualization of telomeric circles in strains having a dysfunctional telomerase and the spreading of a marker from a transformed plasmid containing telomeric sequences into a majority of the telomeric ends (Natarajan and McEachern 2002). However, an alternative mechanism—the reinsertion of amplified circles back into the telomeric ends by recombination—has not been ruled out.
Schizosaccharomyces pombe
BIR has been documented in S. pombe by Humphrey’s lab, using an HO-induced DSB on a minichromosome that was derived from Chr III (Cullen et al. 2007). In addition to gene conversion events, it was possible to recover repair events in which the distal portion of the minichromosome was lost, resulting in loss of heterozygosity (LOH). Some of these events must have arisen by BIR. These events required the fission yeast homologs of Rad51, Rad52, Rad55-Rad57, Rad54, and the MRX proteins. Unlike budding yeast, BIR in fission yeast also needs the branched structure resolvase, Mus81.
Drosophila
Drosophila deals with the end-replication problem by means of a pathway utilizing transpositions of retrotransposons Het-A and TART to the chromosome ends. However, it has been proposed that a terminal break induced by P-element excision can result in copying of sequences from the homologous chromosomes and proceeding to the end of the chromosome. BIR has not yet been studied at sites of P-element excision or when site-specific endonucleases create DSBs, although this should be possible. Data from the Golic lab suggest that a few gene-targeting outcomes could involve BIR (Engels 2000; Rong and Golic 2000).
BIR in Mammals
BIR has not been formally demonstrated in mammals. One possible example is the nonreciprocal translocation that is often associated with facioscapularhumeral muscular dystrophy (FSHD) in humans. This involves recombination between sets of diverged 3.3-kb repeated sequences that are located within 100 kb of the ends of chromosomes 4 and 10 (Tawil and Van Der Maarel 2006). Whether these occur by BIR is presently unknown, but the nonreciprocal nature of the events and their asymmetry (more often chromosome 10 repeats transferred to chromosome 4 than vice versa) are compatible with such a mechanism.
Recently, Halazonetis (TD Halazonetis, pers. comm.) has found that when mammalian cells are placed under replication stress by overexpression of an oncogene, cell growth is severely impaired if the Pol32 homolog, Pol3D, is ablated. There is no effect in untransformed cells.
The main focus of attention in thinking about BIR in humans involves the maintenance of telomeres by ALT (Scheel and Poremba 2002; Reddel 2003). A majority of human tumor cells escape senescence by reactivating telomerase, but many transformed cell lines and some cancers avoid cell death by telomere shortening by a recombination-dependent mechanism (Dunham et al. 2000). There are multiple pieces of evidence, direct and indirect, supporting recombination in telomere maintenance and ALT. Cells deficient in Rad51D or Rad54 exhibit telomere shortening (Jaco et al. 2003; Tarsounas et al. 2004). Telomeres in ALT cells are associated with ALT-associated promyoleucytic leukemia (PML) bodies (APBs) that contain—in addition to telomere DNA sequences—Rad51, the MRN proteins, and the BLM and WRN helicases (Henson et al. 2002). Overexpressing Sp100, a constituent of PML bodies but not APBs, cause the relocalization of MRN proteins away from APBs and result in the inactivation of ALT and the progressive shortening of telomeres (Jiang et al. 2005). Because Mre11, Rad50, and Xrs2 are essential for type II telomere maintenance in budding yeast, it is of course tempting to imagine these proteins are carrying out a similar role in mammalian ALT. In addition, depletion of the replication-associated flap endonuclease FEN1 causes the senescence of ALT cells, except those expressing telomerase (Saharia and Stewart 2009). Similar to results in K. lactis, a marker integrated into a telomeric region can be found at multiple chromosomal sites in ALT cells (Dunham et al. 2000). There is also a significantly elevated rate of telomere sister-chromatid exchange (T-SCE) relative to sister-chromatid exchange (SCE) rates elsewhere in the genome (Londono-Vallejo et al. 2004). This high rate is not seen in telomerase-positive cells or in BLM syndrome cells that exhibit ∼10-fold elevation in SCE rates. SCEs could arise in BIR by resolution of an HJ. Mus81 HJ resolvase appears to be required for ALT (Zeng et al. 2009), analogous to BIR dependence on Mus81 in S. pombe. Thus, although it is clear that recombination is involved in ALT, the implication of BIR awaits further experimentation.
VARIATIONS ON A THEME: MM-BIR
Recent human-genome-sequencing studies attribute 12% of the variation among genomes to copy number variation (CNV) (Redon et al. 2006; Zhang et al. 2009; Conrad et al. 2010). These studies have revealed many examples of duplications and triplications, often in quite very complex rearrangements, the most dramatic of which are the apparent shattering and rejoining within a single chromosome, termed chromothripsis (Stephens et al. 2011). These rearrangements are partially explained by forkstalling and template switching (FoSTeS), now renamed microhomology-mediated BIR (MM-BIR) (Hastings et al. 2009). Although some nonrecurrent CNV events may represent multiple breakages and rejoinings by nonhomologous end joining (NHEJ), those where the copy number is increased to three or more copies must have involved some type of replication, presumably MM-BIR. The initial steps that trigger MM-BIR remain speculative, but recent evidence suggests that replicative stress leading to broken forks might be an important factor. Indeed, agents that perturb replication, such as HU and aphidicolin, are potent inducers of CNVs (Durkin et al. 2008; Arlt et al. 2009, 2011).
The most direct study of nonrecurrent CNV has been performed in budding yeast, where the slow growth caused by deleting one of two copies of an essential ribosomal protein gene, RPL20A, provides a strong selection for duplications of RPL20B (Koszul et al. 2004; Payen et al. 2008). These rearrangements can be tandem duplications of the chromosomal region containing RPL20B or segmental duplications (SDs), in which part of the chromosome containing RPL20B is inserted into another chromosome. SDs arise spontaneously at a rate of 1 × 10−7 cells; but defects in DNA replication increases the incidence of SD 700 fold. Sequences that naturally stall replication fork progression, such as the tRNA genes, are also hotspots of chromosome breakage leading to SDs. Most tandem duplications in wild-type cells are recurrent, arising by what was first assumed to be unequal crossing over between two divergent 300-bp-long terminal repeat (LTR) sequences flanking RPL20B. Surprisingly, even these tandem duplication events are Pol32 dependent, as if they have not occurred by a simple unequal crossover but involve some kind of BIR (Payen et al. 2008). Most nonrecurrent intrachromosomal and interchromosomal SDs do not involve LTR sequences, have junctions containing microhomologies and are Pol32 dependent. Most likely they involve MM-BIR.
Analysis of SDs in various mutant backgrounds has revealed other distinct mechanisms of SD formation. Deletion of Rad51 leads to an increase in interchromosomal BIR events that appear to occur between the LTR centromere proximal to RPL20B and divergent LTRs elsewhere in the genome. This finding fits a pattern of observations reviewed above that the great majority of Rad51-independent BIR events use these diverged sequences.
An even more striking discovery is that there is a remarkably efficient Rad52-independent pathway of SD formation because both Rad51-dependent and Rad51-independent BIR require Rad52. Deleting Rad52 completely eliminates intrachromosomal and interchromosomal rearrangements involving the LTR repeats, but in their place are found very short regions of microhomology located at both intrachromosomal tandem duplications and interchromosomal insertions (Payen et al. 2008). Moreover, these events are Pol32 dependent. These outcomes seem most compatible with some sort of perturbation of DNA replication that may involve MM-BIR.
The key feature of MM-BIR is that recombination-induced replication is not processive and may dissociate a partially replicated strand and invade and anneal at another location where replication proceeds. These exits and entries may happen many times. The exact causes of the jumps and whether these dissociations and invasions involve still-attached DNA polymerases or would involve a homologous recombination step (recognizing unusually tiny lengths of homology) have yet to be answered.
ACKNOWLEDGMENTS
Research in the Haber and Lovett Laboratories are supported by National Institutes of Health grants GM20056, GM61766, GM76020, 2T32GM007122, GM51753, and earlier grants from the National Science Foundation and Department of Energy. R.P.A. is grateful to the Ruth L. Kirschstein National Research Service Award 1F32GM096690-01.
Footnotes
Editors: Stephen D. Bell, Marcel Méchali, and Melvin L. DePamphilis
Additional Perspectives on DNA Replication available at www.cshperspectives.org
REFERENCES
- Adair GM, Nairn RS, Wilson JH, Seidman MM, Brotherman KA, MacKinnon C, Scheerer JB 1989. Targeted homologous recombination at the endogenous adenine phosphoribosyltransferase locus in Chinese hamster cells. Proc Natl Acad Sci 86: 4574–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, Warren ST, Glover TW 2009. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet 84: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Ozdemir AC, Birkeland SR, Wilson TE, Glover TW 2011. Hydroxyurea induces de novo copy number variants in human cells. Proc Natl Acad Sci 108: 17360–17365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Kogoma T 1994. Roles of ruvA, ruvC and recG gene functions in normal and DNA damage-inducible replication of the Escherichia coli chromosome. Genetics 137: 895–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Bates DB, Kogoma T 1994. DNA replication triggered by double-stranded breaks in E. coli: Dependence on homologous recombination functions. Cell 78: 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry J, Alberts B 1994. Purification and characterization of bacteriophage T4 gene 59 protein: A DNA helicase assembly protein involved in DNA replication. J Biol Chem 269: 33049–33062 [PubMed] [Google Scholar]
- Bartsch S, Kang LE, Symington LS 2000. RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates. Mol Cell Biol 20: 1194–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Haber JE 1998. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150: 1037–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burland V, Plunkett G III, Daniels DL, Blattner FR 1993. DNA sequence and analysis of 136 kilobases of the Escherichia coli genome: Organizational symmetry around the origin of replication. Genomics 16: 551–561 [DOI] [PubMed] [Google Scholar]
- Carles-Kinch K, George JW, Kreuzer KN 1997. Bacteriophage T4 UvsW protein is a helicase involved in recombination, repair and the regulation of DNA replication origins. EMBO J 16: 4142–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Groff-Vindman C, Compton SA, McEachern MJ, Griffith JD 2008. Telomere loops and homologous recombination-dependent telomeric circles in a Kluyveromyces lactis telomere mutant strain. Mol Cell Biol 28: 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ, Sandler SJ 1994. Homologous genetic recombination: The pieces begin to fall into place. Crit Rev Microbiol 20: 125–142 [DOI] [PubMed] [Google Scholar]
- Cohen H, Sinclair DA 2001. Recombination-mediated lengthening of terminal telomeric repeats requires the Sgs1 DNA helicase. Proc Natl Acad Sci 98: 3174–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ledesma F, Tous C, Aguilera A 2007. Different genetic requirements for repair of replication-born double-strand breaks by sister-chromatid recombination and break-induced replication. Nucleic Acids Res 35: 6560–6570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen JK, Hussey SP, Walker C, Prudden J, Wee BY, Dave A, Findlay JS, Savory AP, Humphrey TC 2007. Break-induced loss of heterozygosity in fission yeast: Dual roles for homologous recombination in promoting translocations and preventing de novo telomere addition. Mol Cell Biol 27: 7745–7757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Symington LS 2004. RAD51-dependent break-induced replication in yeast. Mol Cell Biol 24: 2344–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A 2011. Break-induced replication is highly inaccurate. PLoS Biol 9: e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas KC, Kreuzer KN 2005. Bacteriophage T4 helicase loader protein gp59 functions as gatekeeper in origin-dependent replication in vivo. J Biol Chem 280: 21561–21569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham MA, Neumann AA, Fasching CL, Reddel RR 2000. Telomere maintenance by recombination in human cells. Nat Genet 26: 447–450 [DOI] [PubMed] [Google Scholar]
- Dunn B, Szauter P, Pardue ML, Szostak JW 1984. Transfer of yeast telomeres to linear plasmids by recombination. Cell 39: 191–201 [DOI] [PubMed] [Google Scholar]
- Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, Glover TW 2008. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc Natl Acad Sci 105: 246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels WR 2000. Reversal of fortune for Drosophila geneticists? Science 288: 1973–1975 [DOI] [PubMed] [Google Scholar]
- Eykelenboom JK, Blackwood JK, Okely E, Leach DR 2008. SbcCD causes a double-strand break at a DNA palindrome in the Escherichia coli chromosome. Mol Cell 29: 644–651 [DOI] [PubMed] [Google Scholar]
- Formosa T, Alberts BM 1986. DNA synthesis dependent on genetic recombination: Characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell 47: 793–806 [DOI] [PubMed] [Google Scholar]
- Gabbai CB, Marians KJ 2010. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 9: 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JW, Kreuzer KN 1996. Repair of double-strand breaks in bacteriophage T4 by a mechanism that involves extensive DNA replication. Genetics 143: 1507–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JW, Stohr BA, Tomso DJ, Kreuzer KN 2001. The tight linkage between DNA replication and double-strand break repair in bacteriophage T4. Proc Natl Acad Sci 98: 8290–8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP 2008. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22: 2767–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groff-Vindman C, Cesare AJ, Natarajan S, Griffith JD, McEachern MJ 2005. Recombination at long mutant telomeres produces tiny single- and double-stranded telomeric circles. Mol Cell Biol 25: 4406–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grompone G, Sanchez N, Dusko Ehrlich S, Michel B 2004. Requirement for RecFOR-mediated recombination in priA mutant. Mol Microbiol 52: 551–562 [DOI] [PubMed] [Google Scholar]
- Haber JE 2012. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191: 33–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR 2009. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Marians KJ 2006. Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439: 557–562 [DOI] [PubMed] [Google Scholar]
- Henson JD, Neumann AA, Yeager TR, Reddel RR 2002. Alternative lengthening of telomeres in mammalian cells. Oncogene 21: 598–610 [DOI] [PubMed] [Google Scholar]
- Hicks WM, Kim M, Haber JE 2010. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CK, Mazon G, Lam AF, Symington LS 2010. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell 40: 988–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Pryde FE, Lester D, Maddison RL, Borts RH, Hickson ID, Louis EJ 2001. SGS1 is required for telomere elongation in the absence of telomerase. Curr Biol 11: 125–129 [DOI] [PubMed] [Google Scholar]
- Ira G, Haber JE 2002. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol Cell Biol 22: 6384–6392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Satory D, Haber JE 2006. Conservative inheritance of newly synthesized DNA in double-strand break-induced gene conversion. Mol Cell Biol 26: 9424–9429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaco I, Munoz P, Goytisolo F, Wesoly J, Bailey S, Taccioli G, Blasco MA 2003. Role of mammalian Rad54 in telomere length maintenance. Mol Cell Biol 23: 5572–5580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Lydeard J, Vaze M, Tanguy Le Gac N, Haber JE 2009. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23: 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang WQ, Zhong ZH, Henson JD, Neumann AA, Chang AC, Reddel RR 2005. Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol Cell Biol 25: 2708–2721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, Guarente L 2001. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J 20: 905–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper B, Brown DT 1976. Function of gene 49 of bacteriophage T4: II. Analysis of intracellular development and the structure of very fast-sedimenting DNA. J Virol 18: 1000–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesti T, Flick K, Keranen S, Syvaoja JE, Wittenberg C 1999. DNA polymerase epsilon catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol Cell 3: 679–685 [DOI] [PubMed] [Google Scholar]
- Kogoma T 1997. Stable DNA replication: Interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev 61: 212–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T, Cadwell GW, Barnard KG, Asai T 1996. The DNA replication priming protein, PriA, is required for homologous recombination and double-strand break repair. J Bacteriol 178: 1258–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koszul R, Caburet S, Dujon B, Fischer G 2004. Eucaryotic genome evolution through the spontaneous duplication of large chromosomal segments. EMBO J 23: 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus E, Leung WY, Haber JE 2001. Break-induced replication: A review and an example in budding yeast. Proc Natl Acad Sci 98: 8255–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer KN 2000. Recombination-dependent DNA replication in phage T4. Trends Biochem Sci 25: 165–173 [DOI] [PubMed] [Google Scholar]
- Kreuzer KN, Brister JR 2010. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: A review in the Virology Journal series on bacteriophage T4 and its relatives. Virol J 7: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzer KN, Saunders M, Weislo LJ, Kreuzer HW 1995. Recombination-dependent DNA replication stimulated by double-strand breaks in bacteriophage T4. J Bacteriol 177: 6844–6853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS 2004. Recombination proteins in yeast. Annu Rev Genet 38: 233–271 [DOI] [PubMed] [Google Scholar]
- Kuzminov A 1995. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol 16: 373–384 [DOI] [PubMed] [Google Scholar]
- Kuzminov A, Stahl FW 1999. Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev 13: 345–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lark KG, Lark CA 1979. recA-dependent DNA replication in the absence of protein synthesis: Characteristics of a dominant lethal replication mutation, dnaT, and requirement for recA+ function. Cold Spring Harb Symp Quant Biol 43: 537–549 [DOI] [PubMed] [Google Scholar]
- Le S, Moore JK, Haber JE, Greider CW 1999. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152: 143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J 1955. Genetic recombination in bacteria. Science 122: 920. [DOI] [PubMed] [Google Scholar]
- Lee EH, Kornberg A 1991. Replication deficiencies in priA mutants of Escherichia coli lacking the primosomal replication n′ protein. Proc Natl Acad Sci 88: 3029–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Morrical SW 2010. Assembly and dynamics of the bacteriophage T4 homologous recombination machinery. Virol J 7: 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu L, Sandler SJ, Marians KJ 1999. Replication fork assembly at recombination intermediates is required for bacterial growth. Proc Natl Acad Sci 96: 3552–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londono-Vallejo JA, Der-Sarkissian H, Cazes L, Bacchetti S, Reddel RR 2004. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res 64: 2324–2327 [DOI] [PubMed] [Google Scholar]
- Louis EJ, Naumova ES, Lee A, Naumov G, Haber JE 1994. The chromosome end in yeast: Its mosaic nature and influence on recombinational dynamics. Genetics 136: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder A, Mosig G 1982. Two alternative mechanisms for initiation of DNA replication forks in bacteriophage T4: Priming by RNA polymerase and by recombination. Proc Natl Acad Sci 79: 1101–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad V, Blackburn EH 1993. An alternative pathway for yeast telomere maintenance rescues est1 senescence. Cell 73: 347–360 [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE 2007. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448: 820–823 [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Lipkin-Moore Z, Jain S, Eapen VV, Haber JE 2010a. Sgs1 and Exo1 redundantly inhibit break-induced replication and de novo telomere addition at broken chromosome ends. PLoS Genet 6: e1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Lipkin-Moore Z, Sheu YJ, Stillman B, Burgers PM, Haber JE 2010b. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 24: 1133–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee TR, Kogoma T 1990. Requirement of RecBC enzyme and an elevated level of activated RecA for induced stable DNA replication in Escherichia coli. J Bacteriol 172: 1834–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE 2005. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol 25: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero VA, Symington LS 2010. Extensive DNA end processing by Exo1 and Sgs1 inhibits break-induced replication. PLoS Genet 6: e1001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Asai T, Kubota Y, Arai K, Kogoma T 1994. Escherichia coli PriA protein is essential for inducible and constitutive stable DNA replication. EMBO J 13: 5338–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH 1995. Runaway telomere elongation caused by telomerase RNA gene mutations. Nature 376: 403–409 [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Blackburn EH 1996. Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes Dev 10: 1822–1834 [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE 2006. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135 [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Iyer S 2001. Short telomeres in yeast are highly recombinogenic. Mol Cell 7: 695–704 [DOI] [PubMed] [Google Scholar]
- Meselson M, Weigle JJ 1961. Chromosome breakage accompanying genetic recombination in bacteriophage. Proc Natl Acad Sci 47: 857–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuuchi K, Kemper B, Hays J, Weisberg RA 1982. T4 endonuclease VII cleaves Holliday structures. Cell 29: 357–365 [DOI] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P 1997. “Break copy” duplication: A model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig G 1987. The essential role of recombination in phage T4 growth. Annu Rev Genet 21: 347–371 [DOI] [PubMed] [Google Scholar]
- Mosig G 1998. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu Rev Genet 32: 379–413 [DOI] [PubMed] [Google Scholar]
- Motamedi MR, Szigety SK, Rosenberg SM 1999. Double-strand-break repair recombination in Escherichia coli: Physical evidence for a DNA replication mechanism in vivo. Genes Dev 13: 2889–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S, McEachern MJ 2002. Recombinational telomere elongation promoted by DNA circles. Mol Cell Biol 22: 4512–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan S, Groff-Vindman C, McEachern MJ 2003. Factors influencing the recombinational expansion and spread of telomeric tandem arrays in Kluyveromyces lactis. Eukaryot Cell 2: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale MJ, Pan J, Keeney S 2005. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436: 1053–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen I, Bentsen IB, Lisby M, Hansen S, Mundbjerg K, Andersen AH, Bjergbaek L 2009. A Flp-nick system to study repair of a single protein-bound nick in vivo. Nat Methods 6: 753–757 [DOI] [PubMed] [Google Scholar]
- Nossal NG, Dudas KC, Kreuzer KN 2001. Bacteriophage T4 proteins replicate plasmids with a preformed R loop at the T4 ori(uvsY) replication origin in vitro. Mol Cell 7: 31–41 [DOI] [PubMed] [Google Scholar]
- Nurse P, Zavitz KH, Marians KJ 1991. Inactivation of the Escherichia coli priA DNA replication protein induces the SOS response. J Bacteriol 173: 6686–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâques F, Haber JE 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63: 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen C, Koszul R, Dujon B, Fischer G 2008. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet 4: e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persky NS, Lovett ST 2008. Mechanisms of recombination: Lessons from E. coli. Crit Rev Biochem Mol Biol 43: 347–370 [DOI] [PubMed] [Google Scholar]
- Reddel RR 2003. Alternative lengthening of telomeres, telomerase, and cancer. Cancer Lett 194: 155–162 [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YS, Golic KG 2000. Gene targeting by homologous recombination in Drosophila. Science 288: 2013–2018 [DOI] [PubMed] [Google Scholar]
- Saharia A, Stewart SA 2009. FEN1 contributes to telomere stability in ALT-positive tumor cells. Oncogene 28: 1162–1167 [DOI] [PubMed] [Google Scholar]
- Scheel C, Poremba C 2002. Telomere lengthening in telomerase-negative cells: The ends are coming together. Virchows Arch 440: 573–582 [DOI] [PubMed] [Google Scholar]
- Sharples GJ, Leach DR 1995. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol Microbiol 17: 1215–1217 [DOI] [PubMed] [Google Scholar]
- Shlomai J, Kornberg A 1980a. A prepriming DNA replication enzyme of Escherichia coli. I. Purification of protein n′: A sequence-specific, DNA-dependent ATPase. J Biol Chem 255: 6789–6793 [PubMed] [Google Scholar]
- Shlomai J, Kornberg A 1980b. A prepriming DNA replication enzyme of Escherichia coli. II. Actions of protein n′: A sequence-specific, DNA-dependent ATPase. J Biol Chem 255: 6794–6798 [PubMed] [Google Scholar]
- Signon L, Malkova A, Naylor ML, Klein H, Haber JE 2001. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol 21: 2048–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalka AM 1974. A replicator’s view of recombination (and repair). In Mechanism and recombination (ed. RF Grell), pp. 421–432 Plenum, New York [Google Scholar]
- Smith GR 1991. Conjugational recombination in E. coli: Myths and mechanisms. Cell 64: 19–27 [DOI] [PubMed] [Google Scholar]
- Smith CE, Llorente B, Symington LS 2007. Template switching during break-induced replication. Nature 447: 102–105 [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohr BA, Kreuzer KN 2002. Coordination of DNA ends during double-strand-break repair in bacteriophage T4. Genetics 162: 1019–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Taniyama C, Arai K, Masai H 2003. ATPase/helicase motif mutants of Escherichia coli PriA protein essential for recombination-dependent DNA replication. Genes Cells 8: 251–261 [DOI] [PubMed] [Google Scholar]
- Tarsounas M, Munoz P, Claas A, Smiraldo PG, Pittman DL, Blasco MA, West SC 2004. Telomere maintenance requires the RAD51D recombination/repair protein. Cell 117: 337–347 [DOI] [PubMed] [Google Scholar]
- Tawil R, Van Der Maarel SM 2006. Facioscapulohumeral muscular dystrophy. Muscle Nerve 34: 1–15 [DOI] [PubMed] [Google Scholar]
- Teng SC, Zakian VA 1999. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19: 8083–8093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng SC, Chang J, McCowan B, Zakian VA 2000. Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol Cell 6: 947–952 [DOI] [PubMed] [Google Scholar]
- Tsai YL, Tseng SF, Chang SH, Lin CC, Teng SC 2002. Involvement of replicative polymerases, Tel1p, Mec1p, Cdc13p, and the Ku complex in telomere-telomere recombination. Mol Cell Biol 22: 5679–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TC, Smith KC 1983. Mechanisms for recF-dependent and recB-dependent pathways of postreplication repair in UV-irradiated Escherichia coli uvrB. J Bacteriol 156: 1093–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Yeh YC 1974. DNA arrested mutants of gene 59 of bacteriophage T4. II. Replicative intermediates. Virology 59: 108–122 [DOI] [PubMed] [Google Scholar]
- Xi J, Zhang Z, Zhuang Z, Yang J, Spiering MM, Hammes GG, Benkovic SJ 2005. Interaction between the T4 helicase loading protein (gp59) and the DNA polymerase (gp43): Unlocking of the gp59-gp43-DNA complex to initiate assembly of a fully functional replisome. Biochemistry 44: 7747–7756 [DOI] [PubMed] [Google Scholar]
- Xu L, Marians KJ 2000. Purification and characterization of DnaC810, a primosomal protein capable of bypassing PriA function. J Biol Chem 275: 8196–8205 [DOI] [PubMed] [Google Scholar]
- Xu L, Marians KJ 2003. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell 11: 817–826 [DOI] [PubMed] [Google Scholar]
- Zavitz KH, Marians KJ 1992. ATPase-deficient mutants of the Escherichia coli DNA replication protein PriA are capable of catalyzing the assembly of active primosomes. J Biol Chem 267: 6933–6940 [PubMed] [Google Scholar]
- Zeng S, Xiang T, Pandita TK, Gonzalez-Suarez I, Gonzalo S, Harris CC, Yang Q 2009. Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol 11: 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gu W, Hurles ME, Lupski JR 2009. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 10: 451–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]