

Abstract
Background
Secretoneurin is a neuropeptide located in nerve fibers along blood vessels, is up-regulated by hypoxia and induces angiogenesis. We tested the hypothesis that secretoneurin gene therapy exerts beneficial effects in a rat model of myocardial infarction and evaluated the mechanism of action on coronary endothelial cells.
Methods and Results
In-vivo secretoneurin improved left ventricular function, inhibited remodeling and reduced scar formation. In the infarct border zone secretoneurin induced coronary angiogenesis as shown by increased density of capillaries and arteries. In-vitro secretoneurin induced capillary tubes, stimulated proliferation, inhibited apoptosis and activated Akt and ERK in coronary endothelial cells. Effects were abrogated by a VEGF-antibody and secretoneurin stimulated VEGF receptors in these cells. Secretoneurin furthermore increased binding of VEGF to endothelial cells and binding was blocked by heparinase indicating that secretoneurin stimulates binding of VEGF to heparan sulfate proteoglycan binding sites. Additionally, secretoneurin increased binding of VEGF to its co-receptor neuropilin 1. In endothelial cells secretoneurin also stimulated FGF receptor-3 and IGF-1 receptor and in coronary vascular smooth muscle cells we observed stimulation of VEGF receptor-1 and FGF receptor-3. Exposure of cardiac myocytes to hypoxia and ischemic heart after myocardial infarction revealed increased secretoneurin m-RNA and protein.
Conclusions
Our data show that secretoneurin acts as an endogenous stimulator of VEGF signaling in coronary endothelial cells by enhancing binding of VEGF to low affinity binding sites and neuropilin 1 and stimulates further growth factor receptors like FGF receptor-3. Our in-vivo findings indicate that secretoneurin might be a promising therapeutic tool in ischemic heart disease.
Keywords: angiogenesis, gene therapy, myocardial infarction
Introduction
Congestive heart failure represents an increasing global health problem that occurs predominantly due to coronary artery disease.1 Cardiac dysfunction after myocardial infarction (MI) results from two major mechanisms: loss of cardiac myocytes in the area of infarction and remodelling of the spared myocardium in the left ventricle (LV). Inadequate structural adaptation of the vascular bed in the area of cardiac myocyte hypertrophy accounts for the progression of LV-dysfunction of the heart. Although ischemia induces endogenous myocardial angiogenesis, vascular growth is insufficient to maintain normal capillary density in the hypertrophied myocardium. Induction of neovascularization is recognized to be a valid approach to modify the pathological changes of ventricular remodeling. Gene transfer of growth factors known to induce angiogenesis like vascular endothelial growth factor (VEGF), hepatocyte growth factor, sonic hedgehog or basic fibroblast growth factor (bFGF) attenuates cardiac dysfunction after myocardial infarction in animal models.2-5 In patients with coronary heart disease randomized phase II and phase III gene therapy studies did not reach the primary endpoint, although in most studies signs of angiogenic bioactivity were observed.6-8 Like in peripheral arterial disease, patient and study endpoint selection as well as determination of transgene expression will be important for planning future clinical studies evaluating treatment of ischemic cardiomyopathy. Additionally, combination of growth factors or growth factors combined with cell therapy might be more promising than therapy with a single factor.9
In this study, we present a plasmid gene therapy approach based on secretoneurin (SN), a neuropeptide of 33 amino acids length, derived from secretogranin-II (SG2), a member of the chromogranin/secretogranin family, which induces angiogenesis.10-12 In a recent study we could show that SN gene therapy restored tissue integrity, function and perfusion in the mouse hindlimb ischemia model by induction of angiogenesis, arteriogenesis and vasculogenesis.13 We therefore hypothesized that SN might also exert beneficial effects in a rat model of myocardial infarction. We provide evidence for improvement of cardiac function after MI by SN gene therapy in-vivo and show a VEGF-dependent mechanism of SN action on human coronary artery endothelial cells (HCAECs) in-vitro.
Methods
Construction of the human SN expression plasmid
Synthetic oligonucleotides encoding SN and a signal peptide were cloned into an expression plasmid vector as has been described previously.13
Rat model of myocardial infarction
Protocols described in this study were approved by the Austrian committee for care and use of laboratory animals. MI was induced by ligation of the left anterior descending coronary artery (LAD) as described previously.14 Animals received 100 μg of SN- or control-plasmid (p-SN or p-CTR) intramyocardially.
Echocardiography
Echocardiograms were recorded under light anesthesia. 2D-directed M-mode and Doppler transthoracic echocardiographic studies were performed 3 days, 2 weeks and 4 weeks post ligation using an Acuson Sequoia Echocardiography system (Acuson Corporation, Mountain View, Calif) with a commercially available 15-MHz linear-array transducer system (AcuNav, Acuson Corporation).15 Data were analyzed using software resident on the ultrasonograph by an experienced researcher, who was blinded to the treatment.
Hemodynamic analysis
Cardiac hemodynamic were measured with a 1.4 F catheter (Millar instruments) as described previously.16 Rats were anaesthetized and placed in supine position. After intubation, rats were mechanically ventilated and the anterior chest wall was opened. During direct insertion of the catheter into the LV, ventilation was discontinued. Recordings of dP/dt and Tau were performed under electrocardiographic monitoring in a stable state during different cardiac cycles.
Cell culture and in vitro assays
Following cell lines were used and purchased from Promo Cell: human coronary artery endothelial cells (HCAECs), human coronary artery smooth muscle (HCASMC) and human cardiac myocytes (HCM). Migration- and tube formation- assays, Western blotting and Immunoprecipitation were performed as described previously or in detail in the supplemental methods.13
RTK Profiler and Immunoprecipitation
Human phospho-receptor tyrosine kinase (RTK) array Kits (R&D, ARY001) were used for investigation of receptors involved in HCAECs and HCASMCs signaling with SN as suggested by the manufacturer.
I125VEGF-binding assays on HCAECs
For binding assays HCAECs were cultured in 24-well plates and incubated with 250.000 cpm I125VEGF (Perkin Elmer) as detailed in the supplements.
Statistical analysis
All results are expressed as mean ± SEM. Statistical comparisons between 2 groups were performed by Student-t test. Multiple groups were analyzed by 1-way ANOVA test followed by appropriate post hoc tests to determine statistical significance. Probability values <0.05 were considered statistically significant. All experiments were repeated at least in triplicate.
*… p<0.001; §… p<0.005; +… p<0.01; # … p<0.05
For further details regarding materials and methods please also see supplemental data.
Results
In-vivo expression of plasmid derived SN
After intramyocardial injection of SN-plasmid, real-time PCR with plasmid specific primers (detecting plasmid-derived SN but not endogenous SN) was performed from muscle extracts. We observed m-RNA of plasmid derived SN, 7 and 14 days after injection but not 28 days after application (Fig. 1A).
Figure 1.

SN gene therapy improves myocardial function after MI. A Plasmid derived SN expression in rat myocardium after MI. Real time PCR analysis for plasmid derived SN in myocardium after 7, 14 and 28 days revealed high SN expression after 7 and 14 days. Values are arbitrary units (n=4). B Effect of SN gene therapy on cardiac function. Echocardiographic assessment of myocardial function showed improved left ventricular ejection fraction and fractional shortening (LVEF and LVFS) 4 weeks after MI and treatment with SN-plasmid (p-SN) compared to control plasmid (p-CTR). Results are shown as mean ± SEM (n=12 per group; *p<0.001; #p<0.05 p-SN vs. p-CTR). C Effects of SN gene therapy on left ventricular remodeling. Echocardiographic analysis of left ventricular end diastolic and systolic diameter (LVEDD and LVESD) revealed inhibition of left ventricular dilatation 4 weeks after MI by SN gene therapy compared to treatment with control vector. Results are shown as mean ± SEM (n=12 per group; #p<0.05; §p<0.005 p-SN vs. p-CTR). D Cardiac Hemodynamic data reveal improved ventricular function after SN gene therapy. 4 weeks after MI, left ventricular maximum positive (+dP/dt) and negative (−dP/dt) pressure development were improved in the SN-treated animal group. Results are shown as mean ± SEM (n=16; §p<0.005 and #p<0.05 p-SN vs. p-CTR).
SN improves cardiac function after myocardial infarction
To test whether SN gene therapy of ischemic myocardium improves cardiac function, echocardiographic investigations were performed 4 weeks after LAD ligation and treatment (n=12 per group). We observed significant improvement of left ventricular ejection fraction (LVEF: p-SN 64.4±3.1 % vs. p-CTR 40.3±2.7 %, p=0.00001; sham operation 61.9±3.5%) and left ventricular fractional shortening (FS: p-SN 39.8±4.6 % vs. p-CTR 25.1±4.2 %, p=0.04; sham operation 41.2±5.1%) in the SN-treated group, compared to the control group (Fig. 1B). Additionally, left ventricular end diastolic diameter (LVEDD: p-SN 9.1±0.6 mm vs. p-CTR 10.5±0.3 mm, p=0.04), as well as left ventricular end systolic diameter (LVESD: p-SN 6.5±0.6 mm vs. p-CTR 9.1±0.5 mm, p=0.002) were significantly reduced in rats receiving SN-plasmid compared to control treatment (Fig. 1C). In addition we performed cardiac hemodynamic measurements 4 weeks after myocardial infarction. Both, left ventricular +dP/dt and −dP/dt were significantly improved in the SN treated animal group (+dP/dt: 5726±458; −dP/dt: −3830±435 mmHg/s) vs. p-CTR (+dP/dt: 3875±365 mmHg/s; p=0.004 p-SN vs. p-CTR; −dP/dt: −2656±357 mmHg/s, p=0.04 p-SN vs. p-CTR; n=16 each group; Fig. 1D; sham: +dP/dt: 6790±367). Additionally, Tau values were determined and revealed significant improvement in the p-SN treated animal group (22±1.9, p=0.002 vs. p-CTR) compared to control (33±1.8; sham operation: 19±1.2, n=5 each group). These data indicate that intramyocardial administration of SN-plasmid exerts favourable impact on LV-function and -remodeling in rats after MI.
Additionally, SN therapy inhibited organ congestion as shown by reduced heart and lung weight compared to body weight (suppl. Table. 1) and in a second in-vivo model (myocardial ischemia reperfusion) SN reduced number of apoptotic cells (suppl. Fig. 1 A,B).
SN induces myocardial angiogenesis and arteriogenesis in-vivo
Vascular structures in ischemic myocardium (n=15 per group) were visualized by immunofluorescent staining for rat endothelial cell antigen (RECA; Fig. 2A) and alpha smooth muscle actin (α-SMA; Fig. 2B). RECA-positive capillaries showed significantly higher blood vessel density (number of capillaries/HPF) in peri-infarct regions in SN-treated animals compared with control animals (p-SN 381.7±100.1 vs. p-CTR 277.9±54.9, p=0.0005) and staining with anti α-SMA showed higher density for arteries and arterioles in the SN-treatment group (p-SN 9.9±3.0 vs. p-CTR 5.2±1.2, p=0.00004) (Fig. 2C). These observations indicate induction of angiogenesis and arteriogenesis in the infarct border zone by SN gene therapy.
Figure 2.

Effects of SN gene therapy on angiogenesis and arteriogenesis in-vivo. Immunofluorescence staining for RECA (A) and for α-SMA (B) in rat myocardium of the infarct border zone treated with SN-plasmid or control plasmid 4 weeks after MI. (C) Values are expressed as RECA positive cells (capillaries) or as α-SMA positive cells (arteries/arterioles) per high power field (HPF, n=15; *p<0.001 p-SN vs. p-CTR).
SN reduces fibrosis after MI
Figure 3A shows Masson's-trichrome-stained myocardial sections from control- and SN-plasmid treated rats 4 weeks after LAD ligation. Treatment with SN-plasmid resulted in remarkable reduction of fibrosis in the LV after MI compared with controls. Figure 3B shows the percentage of fibrosis in hearts after permanent LAD occlusion. Hearts receiving SN gene-therapy exhibited significant reduction in fibrosis (p-SN 21.7±1.6%) compared to controls (p-CTR 35.4±2.8%, p=0.0007, n=13).
Figure 3.
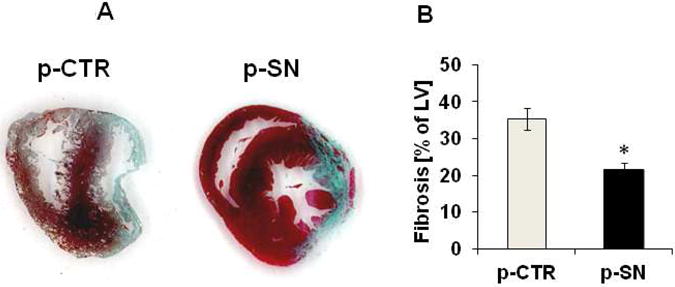
Effects of SN gene therapy on ventricular fibrosis after MI. A SN prevents anterior wall fibrosis after MI. Representative Masson's trichrome-stained sections in the area of infarction, 4 weeks after MI (blue=collagen). B Quantification of ventricular fibrosis. Quantification of Masson's trichrome-stained sections, 4 weeks after MI showed significantly reduced fibrosis in animals treated with SN-plasmid compared to control animals (n=13; * p<0.001 p-SN vs. p-CTR).
In-vitro effects of SN on migration, angiogenesis and apoptosis of HCAECs
SN caused dose dependent induction of chemotaxis in HCAECs with a maximum effect at 100 ng/ml (relative chemotactic index (CI) 2.4±0.2, p=0.0009 vs. Control; Fig. 4A). Blockade of SN with a specific neutralizing antibody (SN-Ab) completely inhibited SN-mediated HCAEC migration indicating specificity of the observed effect (relative CI: SN 100 ng/ml + SN-Ab 1.4±0.1, p=0.009 vs. SN 100 ng/ml). Interestingly, the chemotactic effect of SN was also inhibited by a VEGF- antibody (VEGF-Ab) (relative CI: SN 100 ng/ml + VEGF-Ab 1.4±0.1, p=0.006 vs. SN 100 ng/ml) and the mitogen-activated protein kinase (MAPK) inhibitor PD98.059 (PD) (relative CI: SN 100 ng/ml + PD 1.3±0.01, p=0.005 vs. SN 100 ng/ml).
Figure 4.
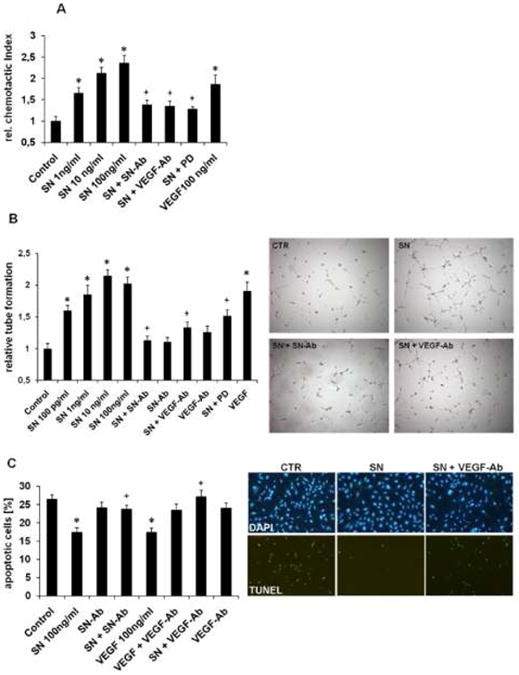
Effects of SN on HCAECs in cell culture. A SN induces HCAEC migration. HCAEC migration is expressed as chemotactic index relative to control. SN induced cell migration with a maximum effect at 100 ng/ml could be blocked with neutralizing SN-antibody, VEGF-antibody and the MAPK inhibitor PD98.059 (PD). VEGF served as positive control (n=4; *p<0.001 vs. Control; +p<0.01 vs. SN 100 ng/ml). B SN induces capillary tube formation in-vitro. HCAECs were seeded on matrigel for 6 hours and capillary tubes were counted. SN induced capillary tube formation with a maximum effect at 10 ng/ml. Neutralizing SN-antibody, VEGF-antibody and PD blocked SN-induced tube formation. VEGF served as positive control. (n=6; *p<0.001 vs. Control; +p<0.01 vs. SN 100 ng/ml). Representative images of findings with control medium, SN 100ng/ml, SN and neutralizing SN-antibody, SN and VEGF-antibody are shown. C SN inhibits HCAEC apoptosis. HCAECs were starved and stained for TUNEL and DAPI (apoptotic cells are given as % of TUNEL positive cells of all DAPI positive cells). SN reduced HCAEC apoptosis with a maximum at 100 ng/ml concentration whereas co-incubation with SN-antibody and VEGF-antibody inhibited SN-induced protective effects. VEGF served as positive control. (n=4; *p<0.001 vs. Control; +p<0.01 vs. SN 100 ng/ml). Representative images of TUNEL and DAPI stains with control medium, SN 100ng/ml, SN and VEGF-antibody are shown.
SN induced angiogenesis in-vitro was demonstrated by increase in capillary-like tube formation in a matrigel assay with HCAECs with a maximum effect at 10 ng/ml (relative tube formation 2.2±0.1, p=0.00002 vs. Control; Fig. 4B). Addition of SN-Ab abolished SN-induced tube formation (1.1±0.1, p=0.001 vs. SN 100 ng/ml) indicating specificity of observed effects. Confirmatively to the observation in the migration assay, SN-induced tube formation was also impaired by VEGF-Ab (relative tube formation 1.3±0.1, p=0.005 vs. SN 100 ng/ml). PD also inhibited SN-induced effects (relative tube formation 1.5±0.1, p=0.007 vs. SN 100 ng/ml), indicating a MAPK dependent signalling of SN.
For investigating the effect of SN on HCAEC apoptosis, cells were starved and stained with TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) and DAPI. SN significantly inhibited HCAEC apoptosis (% TUNEL positive cells of DAPI positive cells: Control 26.4±1.2 vs. SN 100 ng/ml 17.4±1.3, p=0.0008; Fig. 4C). SN-Ab and VEGF-Ab abrogated the anti-apoptotic effect of SN (% TUNEL positive cells of DAPI positive cells: SN + SN-Ab 24.1±1.6, p=0.004 vs. SN; SN + VEGF-Ab 26.5±1.4, p=0.005 vs. SN).
SN signalling in HCAECs
SN signalling was evaluated by Western blotting, which revealed activation of MAPK and PI3-kinase/Akt as judged by phosphorylation of extracellular signal-regulated kinase (ERK) and Akt. In HCAECs, SN stimulated MAPK phosphorylation at 100 ng/ml concentration, starting after 20 minutes with a continuing, long lasting stimulation until 4 hours. Akt was activated by SN 100 ng/ml after 40 minutes with 4 hours duration (Fig. 5A). As observed in tube formation and migration assays, SN-induced effects on ERK activation were again blocked by VEGF-Ab (Fig. 5B).
Figure 5.
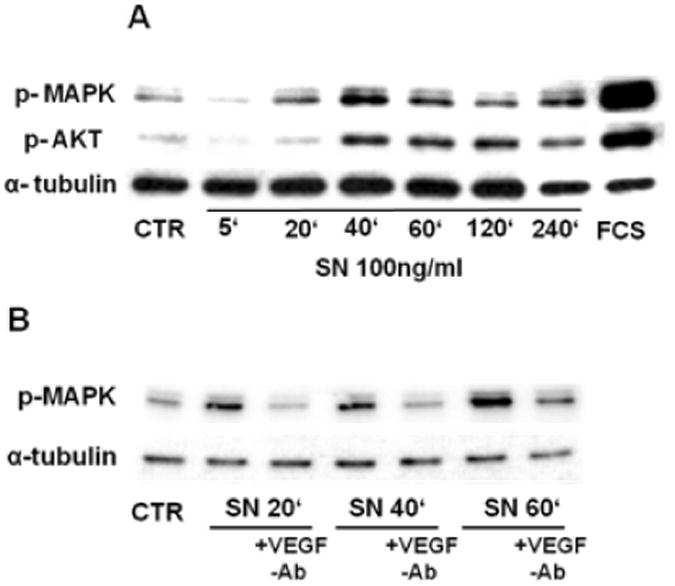
SN stimulates intracellular signaling pathways in HCAECs. A SN stimulates MAPK (ERK) and Akt activation in HCAECs. HCAECs were incubated with SN 100 ng/ml for different time points, cell lysates were collected and further treated for Western blotting. SN activated MAPK after 20 minutes and Akt after 40 minutes, lasting until 4 hours. B SN induced MAPK (ERK) phosphorylation is mediated by VEGF. HCAECs were incubated with SN 100 ng/ml ± VEGF-antibody and extracts were analyzed for MAPK phosphorylation by Western blotting. Co-incubation with a VEGF-antibody inhibits SN-induced MAKP activation.
SN stimulates VEGF-receptors
As SN-mediated in-vitro effects were blocked by a VEGF-Ab, SN-induced signalling in HCAECs was further investigated using RTK profiler assays (R&D). Results show an activation of VEGF receptor-1 and -2 (VEGFR1, VEGFR2) after 40 minutes treatment with SN 100 ng/ml (Fig. 6A). This effect again could be blocked by VEGF-Ab (1:500 dilution; Fig. 6B) indicating that SN-induced VEGF receptor activation depends on endogenous VEGF.
Figure 6.
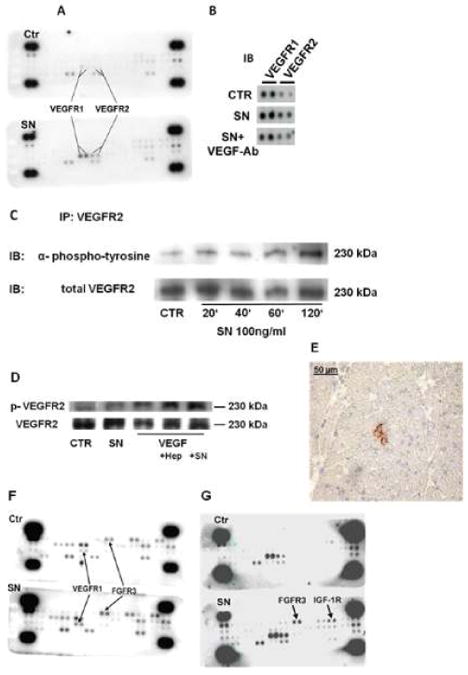
SN stimulates VEGF- and FGF-receptors. A Treatment of HCAECs with SN induces phosphorylation of VEGFR1 and 2. Profiler assays were used to investigate the role of receptor tyrosine kinases in SN stimulated cells. HCAECs were stimulated with SN 100 ng/ml for 40 min and further treated as recommended by the manufacturer. In comparison to untreated cells, SN induced phosphorylation of VEGFR1 and 2. B SN-induced VEGF receptor activation is mediated by endogenous VEGF. For receptor tyrosine kinase Profiler assays, HCAECs were treated with control medium or SN 100 ng/ml with or without VEGF-antibody. SN-mediated VEGF receptor activation was blocked by the VEGF-antibody. C SN induces phosphorylation of VEGFR2 (Immunoprecipitaton). HCAECs were stimulated with SN 100 ng/ml for different time periods, cell lysates were collected and immunoprecipitated for VEGFR2. After blotting, membranes were probed with anti-phospho-tyrosine antibody. SN induced activation of VEGFR2 after 120 minutes. D SN effects on VEGF-mediated VEGFR2 activation are comparable to those of heparin. HCAECs were treated with control medium, SN 100 ng/ml and VEGF 50 ng/ml in combination with either heparin 1 μg/ml or SN 100 ng/ml. Levels of VEGFR2 phosphorylation were determined by Western blot analysis using a specific phospho-VEGFR2 antibody. SN, like heparin stimulated VEGF mediated VEGFR2 stimulation. E VEGFR2 phosphorylation in vivo. Frozen Sections of rat heart 3 days after MI and SN gene therapy showed positive staining for phospho-VEGFR2. F Treatment of HCASMCs with SN induced phosphorylation of VEGFR1 and FGFR3. HCASMCs were stimulated with SN 100 ng/ml for 40 min and cell lysates were used for profiler assays. In comparison to untreated cells, SN induced phosphorylation of VEGFR1 and FGFR3. G SN also activates FGFR3 and IGF-1R in HCAECs. In unstarved HCAECs, profiler assays revealed stimulation of FGFR3 and IGF-1 receptor with SN 100 ng/ml after 40 minutes, beside the already observed phosphorylation of VEGFR1 and 2.
Immunoprecipitation of HCAEC lysates with VEGFR2-antibody and immuno-blotting for phospho-tyrosine confirmed findings of the RTK profiler and revealed SN-induced VEGFR2 phosphorylation after 120 minutes (Fig. 6C).
Western Blot analysis of phosphorylated VEGFR2 additionally showed an increase of VEGF (50 ng/ml) induced VEGFR2 activation by SN, comparable to the already described effect of heparin (1 μg/ml) as shown in Figure 6D. These data indicate that SN stimulates VEGF receptor activation via a VEGF-dependent mechanism.
Immunohistochemical staining for phospho-VEGFR2 could only be found in ischemic rat hearts treated with p-SN 3 days after MI (Figure 6E), but not in control animals (data not shown).
In HCASMCs we found activation of VEGFR1 and fibroblast growth factor receptor-3 (FGFR3) by SN 100 ng/ml after 40 minutes (Fig. 6F). In contrast to HCAECs however, we were not able to block SN-mediated effects by a neutralizing VEGF-Ab (as demonstrated with a chemotaxis assay in HCASMCs, suppl. Fig. 2A).
Without starvation of HCAECs before stimulation with SN, a different picture was observed in RTK profiler assays: VEGFR1 and VEGFR2 still were stimulated by SN but also activation of FGFR3 and insulin-like growth factor-1 receptor (IGF-1R) were detectable (Fig. 6G). To evaluate if FGF also plays a role in SN-mediated effects on HCAECs, we inhibited bFGF with a neutralizing antibody but were not able to block SN-induced in-vitro angiogenesis in contrast to inhibition of VEGF (suppl. Fig. 2B). Therefore SN-induced VEGF signalling in HCAECs was further evaluated.
In order to test the hypothesis that VEGFR2, which has been reported to be mainly responsible for angiogenic effects within the VEGF receptor family, is involved in SN-signalling in HCAECs, a specific VEGFR2 inhibitor, SU1498 was tested. SU1498 (40 μM) showed inhibition of SN induced MAPK activation after 20, 40 and 60 minutes, indicating, that SN-induced MAPK activation is mediated by VEGFR2 (Fig. 7A).
Figure 7.
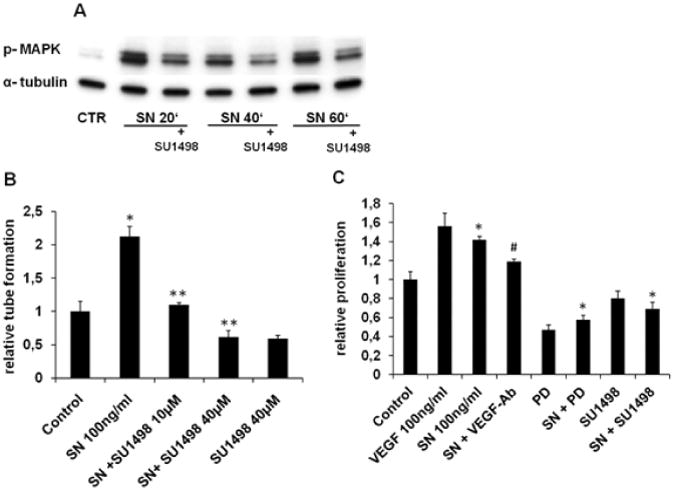
VEGFR2 mediates SN-induced effects on HCAECs. A SN-induced MAPK activation is blocked by SU1498. HCAECs were preincubated with SU1498 (40 μM) for 60 min. Thereafter cells were stimulated with SN 100 ng/ml and further processed for Western blotting. The specific VEGFR2 inhibitor SU1498 abrogated SN-induced ERK activation at different time-points. B SN mediated tube formation is impaired with SU1498. HCAECs were seeded on matrigel and after 6 hours, capillary tubes were counted. SN 100 ng/ml induced capillary tube formation was blocked with SU1498 (10 and 40 μM). (n=8; *p<0.001 SN vs. Control, **p<0.001 SN vs. SN + SU1498). C Proliferation of HCAECs is modulated by ERK and VEGFR2. HCAECs were cultured and stimulated with SN 100 ng/ml with or without VEGF-antibody, PD and SU1498 (10μM). Both PD and SU1498 abolished SN-induced cell proliferation (n=6; **p<0.001 SN vs. SN + SU1498, SN + PD; SN vs. Control *p<0.001; SN vs. SN + VEGF-Ab +p<0.01).
Inhibition of SN mediated in-vitro angiogenesis by SU1498 was demonstrated by matrigel assays: after addition of SU1498 (10 or 40 μM, Fig. 7B), SN induced capillary tube formation was completely blocked (SN + SU1498 10 μM 1.1±0.03 vs. SN: 2.1±0.2, p=0.0001), indicating that also SN-induced in-vitro angiogenesis is mediated by VEGFR2. BrdU (5-bromo-2′-deoxyuridine) assays performed with HCAECs identified VEGFR2 and the MAPK pathway as essential modulators for SN-induced HCAECs proliferation. Both PD and SU1498 abolished SN-induced relative proliferation compared to SN 100 ng/ml (SN 1.42±0.04 vs. SN + PD 0.58±0.04, p=0.00007; SN vs. SN + SU1498 0.69±0.07, p=0.00001; Fig. 7C).
SN increases VEGF-binding to HCAECs, heparin and neuropilin 1
To determine the mechanism of SN-induced VEGF receptor stimulation, we hypothesized that SN might interact with VEGFR2 and its co-receptor neuropilin 1, or with heparan sulfate proteoglycans, shown to be necessary for VEGFR2 activation.17 However, we couldn't find direct binding of SN neither to VEGFR2 nor to neuropilin 1 (data not shown), nor to heparin using heparin coated sepharose columns (data not shown). In order to test the hypothesis that SN increases the binding of VEGF to HCAECs, we performed binding studies with VEGF labelled by radioactive iodine (I125VEGF) on HCAECs. Incubation with SN resulted in increased binding of I125VEGF to HCAECs with a maximum effect at 100 ng/ml SN (I125VEGF 1.7±0.1 fmol vs. SN 100 ng/ml + I125VEGF 2.1±0.1 fmol/105 cells, p=0.031; Fig. 8A). Based on the effect of SN at a concentration of 100 ng/ml, we performed saturation studies in the presence or absence of SN, with increasing amounts of I125VEGF starting from 2.6 fmol/ml to a maximum concentration of 523 fmol/ml. Bmax and Kd for VEGF-binding to HCAECs was 4.7 fmol/105 cells and 290.3 fmol, respectively, whereas binding of VEGF in presence of SN 100 ng/ml to HCAECs reached values of Bmax of 5.2 fmol/105 cells and Kd of 228.2 fmol. These findings indicate that SN induces increased binding and increased affinity of HCAECs to VEGF.
Figure 8.
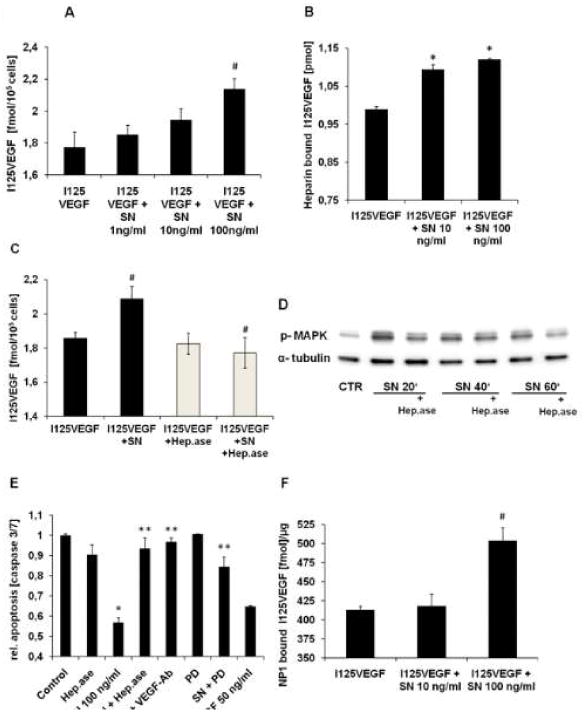
SN stimulates binding of VEGF to heparan sulfate proteoglycans and neuropilin 1. A SN increases I125VEGF-binding to HCAECs. For binding assays 10 ng/ml I125VEGF with or without increasing amounts of SN (1, 10 and 100 ng/ml), was added to HCAECs for 2 hours at 4°C. Binding of I125VEGF was significantly increased in the presence of SN 100ng/ml (I125VEGF vs. SN 100 ng/ml + I125VEGF, # p<0.05 SN; n=4). B SN enhances binding of I125VEGF to heparin. Maxisorb tubes were coated with heparin-BSA complex and incubated with 0.5 ng/ml I125 VEGF ± 10 or 100 ng/ml SN for 2 hours at room temperature. In the presence of SN, binding of I125VEGF to heparin is significantly increased compared to I125VEGF alone (I125VEGF vs. SN + I125VEGF, *p<0.001; n=5). C Heparinase pre-treatment blocks SN mediated I125VEGF-binding to HCAECs. To investigate the influence of heparan sulfate proteoglycans on I125VEGF-binding, HCAECs were pre-incubated with heparinase for 4 hours at 37°C. Thereafter binding assays were performed with I125VEGF ± SN 100ng/ml. Pre-incubation with heparinase diminished SN-induced increase of I125VEGF-binding to HCAECs (I125VEGF vs. SN+I125VEGF and I125VEGF + SN vs. I125VEGF + SN + Heparinase, #p<0.05; n=3). D Heparinase inhibits SN mediated MAPK activation. After preincubation of HCAECs with heparinase, cells were stimulated with SN 100 ng/ml for different time periods and lysates were processed for Western blotting. E VEGF-Ab, PD and heparinases abrogate SN-induced anti-apoptotic effects. HCAECs were starved over night in medium without supplements ± SN 100 ng/ml, PD (10 μM), VEGF-antibody and heparinase. VEGF 50 ng/ml served as positive control (SN vs. Control, *p<0.001; SN + Heparinase, SN + VEGF-Ab and SN + PD vs. SN 100 ng/ml, ** p<0.001; n=4). F SN increases binding of I125VEGF to neuropilin 1. Maxisorb tubes were coated with recombinant, human neuropilin 1 and binding assays in the presence or absence of SN 10 and 100 ng/ml were performed. SN 100 ng/ml significantly increased I125VEGF-binding to its co-receptor neuropilin 1 (I125VEGF vs. I125VEGF + SN, #p<0.05; n=3).
As SN did not affect binding of VEGF to VEGFR2 (suppl. Fig. 3A), we further hypothesized, that SN might stimulate binding of VEGF to heparan sulfate proteoglycans of the extracellular matrix. Therefore a heparin-BSA complex as described previously18 was used for analyzing the binding of VEGF to heparin in the presence of SN. Figure 8B illustrates a significant increase in I125VEGF-binding to heparin, when SN 10 or 100 ng/ml was added to the binding buffer (SN 10 ng/ml 1.09±0.01 pmol and SN 100 ng/ml 1.12±0.001 pmol vs. I125VEGF 0.99±0.01 pmol, p=0.0003 and p=0.00007, respectively). A similar effect was observed when heparin-coated sepharose beads were used (suppl. Fig. 3B).
In order to ascertain whether binding to heparan sulfate proteoglycans is responsible for increased SN-induced VEGF-binding to HCAECs and SN-induced effects on HCAECs, we used heparinase I and heparinase III, which in combination digest heparin as well as heparan sulfates. Pre-incubation of cells with the mixture of heparinases resulted in complete loss of SN-mediated increase in VEGF-binding (I125VEGF + SN + Heparinase 1.77±0.09 vs. I125VEGF + SN 2.09±0.08, p=0.027; Fig. 8C). The finding that SN-induced effects depend on heparan sulfate proteoglycans was confirmed by the observations that pre-treatment of HCAECs with heparinases I and III inhibited SN-mediated MAPK activation (Fig. 8D) and anti-apoptotic effects (SN relative to Control 0.57±0.02 vs. SN + Heparinase 0.93±0.05, p=0.0002; SN vs. SN + VEGF-Ab 0.97±0.02, p=0.00003; SN vs. SN + PD 0.85±0.05, p=0.0008; Fig. 8E).
Although we found no effect of SN on binding of VEGF to its receptor VEGFR2 (suppl. data Fig. 3A), significantly increased VEGF-binding to its co-receptor neuropilin 1 was observed with SN (I125VEGF 413.4±4.5 fmol/μg vs. SN + I125VEGF 504±16.7 fmol/μg, p=0.012; Fig. 8F).
Discussion
The main finding of our work is that gene therapy with the angiogenic factor SN improves outcome in a rat MI-model in terms of LV-function (evaluated by echocardiography and invasively by cardiac catheterization), -remodeling and scar formation. These effects were accompanied by an increase of capillary and arterial density in the infarct border zone and we therefore hypothesized that this beneficial effect was largely due to induction of angiogenesis by SN as shown before in other in-vivo models,10 especially in the hindlimb ischemia model.13 Like in this previous work, gene therapy was performed after ligation of the artery by intramuscular plasmid injection and plasmid derived SN was detectable for 2 weeks. Although we cannot exclude direct effects of SN on cardiac myocytes, we didn't observe regulation of classical angiogenic factors like VEGF by SN on these cells neither under normoxic nor under hypoxic conditions (data not shown). First experiments also showed no effect of SN on RTKs in cardiac myocytes as observed in this study for HCAECs and HCASMCs. Additionally, an intermediate time point after MI (14 days) showed a trend to, but no significant, increase of myocardial function and of capillary density by SN indicating that full induction of angiogenesis is necessary for effects of SN on myocardial function (suppl. Fig. 4A-C). Like in skeletal muscle cells 19 however, we also found in cardiac myocytes (but not in coronary endothelial cells) that prolonged hypoxia increases SN at m-RNA and protein levels (suppl. Fig. 5A-C) what might indicate that SN like other angiogenic factors is increased as a physiological response to hypoxia in these cells to counteract decrease of oxygen supply by growth of new blood vessel. We also observed increased SN m-RNA and protein in-vivo after MI by PCR and immunofluorescence (suppl. Fig. 6A and B). Regarding the reduction in scar formation/fibrosis we didn't find effects of SN on human cardiac fibroblasts in-vitro (regulation of SN by hypoxia, influence on cell proliferation or apoptosis by SN, suppl. Fig. 7A-C), therefore we hypothesize that reduction of fibrosis is due to less damage of cardiac myocytes in the SN-group rather than an inhibitory effect of SN on this process.
An interesting finding of our in-vitro studies was that SN-induced effects on HCAEC migration, tube formation, proliferation, anti-apoptosis and ERK activation was blocked by a neutralizing VEGF-antibody whereas a bFGF-antibody had no effect, at least on in-vitro tube formation (suppl. Fig. 2B). This VEGF-dependent effect is in contrast to our data in human umbilical vein endothelial cells (HUVECs)10 (suppl. Fig. 8), where we didn't observe stimulation of VEGF-receptors by SN, indicating specificity of this observed effect for coronary arterial ECs. The reason for this difference has to be evaluated in future studies, and as we found that SN stimulates binding of VEGF to heparan sulfate proteoglycans maybe the composition of the extracellular matrix and/or differences in growth factors stored in the matrix is responsible for these observations. Nevertheless, to investigate if SN stimulates RTKs we used a commercially available profiler and found that SN stimulates phosphorylation of VEGFR1, VEGFR2, FGFR3 and IGF-1 receptor in coronary ECs as well as VEGFR1 and FGFR3 in coronary smooth muscle cells. Additionally, stimulation of VEGFR2 was observed after MI and SN gene therapy in-vivo by immunohistochemistry. These findings indicate that the angiogenic neuropeptide SN stimulates several RTKs in coronary vascular cells what might explain the robust effects observed in our study. Especially the fact that different RTKs shown to be important for angiogenesis like VEGF-and FGF-receptors were activated by SN might be important as a recently published work was able to show that combination of angiogenic growth factors, i.e. FGF and HGF produced a more potent and sustained effect in the corneal angiogenesis and the myocardial infarction model compared to the single factors.9,20 Similar effects were reported previously with the combination of bFGF and platelet-derived growth factor (PDGF).21 Additionally, a close interaction between VEGF and FGF pathways (FGF-dependent regulation of VEGFR2) was shown to be necessary for post-ischemic neovascularization.22 Therefore it is conceivable that combinations of growth factors or stimulation of several different RTKs exert additive, long-lasting effects on therapeutic angiogenesis. This is also of particular interest for clinical studies of therapeutic angiogenesis where application of a single growth factor did not show positive results in phase III studies.23,24
As VEGFR2 is considered to be the pivotal receptor for EC survival and proliferation and for angiogenesis 25 and we found that effects of SN on ECs were blocked by a neutralizing VEGF-antibody and by a VEGFR2 blocker (SU1498), we further characterized the mechanism of SN-induced action regarding VEGFR2 signalling. Activation of VEGFR2 by VEGF is dependent on heparan sulfate proteoglycans 20 and matrix bound VEGF was reported to activate more sustained VEGFR2 stimulation.26 VEGF signalling via VEGFR2 also is enhanced by a co-receptor, i.e. neuropilin 1.27 We therefore hypothesized that SN might bind to one of these VEGF receptors but we were not successful to demonstrate direct binding of SN to VEGF itself, to VEGFR2, to neuropilin or to heparin (data not shown). We therefore analyzed if SN increases binding of VEGF to its receptors, like shown for heparin which stimulates VEGF-binding to VEGFR2.28 Indeed we found that SN stimulated binding of VEGF to coronary ECs and this effect was abolished when cells were pre-incubated with heparinase indicating that SN stimulates binding of VEGF to heparan sulfate proteoglycans. This finding was corroborated by the fact that also SN-induced ERK activation and anti-apoptotic effects were blocked by heparinase. SN did not increase binding of VEGF to VEGFR2 (suppl. Fig. 3A) but stimulated binding of VEGF to its co-receptors heparin and neuropilin 1 indicating that SN-induced effects are mediated by increased VEGF-binding to neuropilin 1 and heparan sulphate proteoglycans. It is known that C-terminal basic amino acids of VEGF are responsible for binding of VEGF-165 to heparin. As SN contains several acidic amino acids we plan to synthesize different SN-derivates to characterize the responsible sequence of SN necessary for increased binding of VEGF to heparin in future studies.
In contrast to ECs we were not able to block SN-induced effects in HCASMCs by a neutralizing VEGF-Ab (suppl. Fig. 2A), allowing the assumption that SN acts via a different mechanism on these cells. As FGFR3 was also activated by SN in HCASMCs we are planning to analyze the role of FGF in SN-mediated effects in SMCs as well as in ECs.
Summarizing our data we found that SN acts as an endogenous enhancer of VEGF-binding to its co-receptor neuropilin 1 and to heparan sulfate proteoglycan binding sites. We would like to propose SN as a novel agent in the treatment of ischemic cardiomyopathy due to several reasons: SN acts as stimulator of several growth factor RTKs known to play essential roles in angiogenesis, i.e. VEGFR2 and FGFR3 in the coronary vasculature. Furthermore, SN also affects coronary artery SMCs as was shown in this and previous studies 10,13 to induce growth of SMC-covered blood vessels known to be more stable than capillaries. Finally, SN improved LV-function and -remodeling in an in-vivo model of MI. Future studies also should be able to determine appropriate vectors for efficient delivery of this peptide in large animal models and in human trials.
Supplementary Material
Clinical Summary.
Our work shows that the angiogenic factor Secretoneurin improves left ventricular function in an animal model of myocardial infarction. Secretoneurin stimulates angiogenesis and arteriogenesis in the infarct border zone and reduces scar size after the infarct. Mechanistically, Secretoneurin exerts beneficial effects on coronary endothelial cells via stimulation of growth factor receptors for angiogenic cytokines like vascular endothelial growth factor (VEGF) or fibroblast growth factor (FGF). Secretoneurin thereby stimulates binding of VEGF to its co-receptors neuropillin-1 and heparansulfate-proteoglycans. Despite promising preclinical and early clinical data for the application of angiogenic cytokines in the treatment of ischemic heart or limb disease (a therapeutic strategy called “therapeutic angiogenesis”) randomized phase III clinical trials (AGENT trial for coronary heart disease, TAMARIS for critical limb ischemia) did not show expected results. Beside patient and study-endpoint selection and pharmacokinetic issues it is also conceivable that administration of a single angiogenic factor is not sufficient to orchestrate a complex biological event like the growth of a new blood vessel. Indeed, combination of angiogenic growth factors (like basic-FGF and platelet-derived growth factor or basic-FGF and hepatocyte growth factor) exerted more potent and long-lasting angiogenic responses and beneficial effects in ischemic diseases compared to administration of single factors. In this context it is of particular interest that secretoneurin, as a single compound, stimulated receptors for several of these cytokines like VEGF or FGF. We therefore think that Secretoneurin might be a promising candidate for therapeutic angiogenesis.
Acknowledgments
We thank Erin Lambers and Veronica Ramirez (Feinberg Cardiovascular Research Institute, Northwestern University, Chicago, USA) for technical support.
Funding Sources: This work was funded by the Austrian Science Fund project P-22808-B18 to Rudolf Kirchmair. Parts of this work were supported by National Institute of Health grants HL091983, HL105597, HL095874 and HL053354 to Raj Kishore.
Footnotes
Conflict of Interest Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gheorghiade M, Bonow RO. Chronic heart failure in the united states: A manifestation of coronary artery disease. Circulation. 1998;97:282–289. doi: 10.1161/01.cir.97.3.282. [DOI] [PubMed] [Google Scholar]
- 2.Kawamoto A, Murayama T, Kusano K, Ii M, Tkebuchava T, Shintani S, Iwakura A, Johnson I, von Samson P, Hanley A, Gavin M, Curry C, Silver M, Ma H, Kearney M, Losordo DW. Synergistic effect of bone marrow mobilization and vascular endothelial growth factor-2 gene therapy in myocardial ischemia. Circulation. 2004;110:1398–1405. doi: 10.1161/01.CIR.0000141563.71410.64. [DOI] [PubMed] [Google Scholar]
- 3.Kondo I, Ohmori K, Oshita A, Takeuchi H, Fuke S, Shinomiya K, Noma T, Namba T, Kohno M. Treatment of acute myocardial infarction by hepatocyte growth factor gene transfer: The first demonstration of myocardial transfer of a “functional” gene using ultrasonic microbubble destruction. J Am Coll Cardiol. 2004;44:644–653. doi: 10.1016/j.jacc.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 4.Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, Thorne T, Takenaka H, Aikawa R, Goukassian D, von Samson P, Hamada H, Yoon YS, Silver M, Eaton E, Ma H, Heyd L, Kearney M, Munger W, Porter JA, Kishore R, Losordo DW. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat Med. 2005;11:1197–1204. doi: 10.1038/nm1313. [DOI] [PubMed] [Google Scholar]
- 5.Yanagisawa-Miwa A, Uchida Y, Nakamura F, Tomaru T, Kido H, Kamijo T, Sugimoto T, Kaji K, Utsuyama M, Kurashima C, Ito H. Salvage of infarcted myocardium by angiogenic action of basic fibroblast growth factor. Science. 1992;257:1401–1403. doi: 10.1126/science.1382313. [DOI] [PubMed] [Google Scholar]
- 6.Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, Schatz RA, Asahara T, Isner JM, Kuntz RE. Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation. 2002;105:2012–2018. doi: 10.1161/01.cir.0000015982.70785.b7. [DOI] [PubMed] [Google Scholar]
- 7.Hedman M, Hartikainen J, Syvanne M, Stjernvall J, Hedman A, Kivela A, Vanninen E, Mussalo H, Kauppila E, Simula S, Narvanen O, Rantala A, Peuhkurinen K, Nieminen MS, Laakso M, Yla-Herttuala S. Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and instent restenosis and in the treatment of chronic myocardial ischemia: Phase ii results of the kuopio angiogenesis trial (kat) Circulation. 2003;107:2677–2683. doi: 10.1161/01.CIR.0000070540.80780.92. [DOI] [PubMed] [Google Scholar]
- 8.Kastrup J, Jorgensen E, Ruck A, Tagil K, Glogar D, Ruzyllo W, Botker HE, Dudek D, Drvota V, Hesse B, Thuesen L, Blomberg P, Gyongyosi M, Sylven C. Direct intramyocardial plasmid vascular endothelial growth factor-a165 gene therapy in patients with stable severe angina pectoris a randomized double-blind placebo-controlled study: The euroinject one trial. J Am Coll Cardiol. 2005;45:982–988. doi: 10.1016/j.jacc.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 9.Krichavsky MZ, Losordo DW. Prevention and recovery of hibernating myocardium by microvascular repair. Circulation. 2011;124:998–1000. doi: 10.1161/CIRCULATIONAHA.111.047746. [DOI] [PubMed] [Google Scholar]
- 10.Kirchmair R, Gander R, Egger M, Hanley A, Silver M, Ritsch A, Murayama T, Kaneider N, Sturm W, Kearny M, Fischer-Colbrie R, Kircher B, Gaenzer H, Wiedermann CJ, Ropper AH, Losordo DW, Patsch JR, Schratzberger P. The neuropeptide secretoneurin acts as a direct angiogenic cytokine in vitro and in vivo. Circulation. 2004;109:777–783. doi: 10.1161/01.CIR.0000112574.07422.C1. [DOI] [PubMed] [Google Scholar]
- 11.Kirchmair R, Egger M, Walter DH, Eisterer W, Niederwanger A, Woell E, Nagl M, Pedrini M, Murayama T, Frauscher S, Hanley A, Silver M, Brodmann M, Sturm W, Fischer-Colbrie R, Losordo DW, Patsch JR, Schratzberger P. Secretoneurin, an angiogenic neuropeptide, induces postnatal vasculogenesis. Circulation. 2004;110:1121–1127. doi: 10.1161/01.CIR.0000139884.81390.56. [DOI] [PubMed] [Google Scholar]
- 12.Fischer-Colbrie R, Kirchmair R, Kähler CM, Wiedermann CJ, Saria A. Secretoneurin: a new player in angiogenesis and chemotaxis linking nerves, blood vessels and the immune system. Curr Protein Pept Sci. 2005;6:373–385. doi: 10.2174/1389203054546334. [DOI] [PubMed] [Google Scholar]
- 13.Schgoer W, Theurl M, Jeschke J, Beer AG, Albrecht K, Gander R, Rong S, Vasiljevic D, Egger M, Wolf AM, Frauscher S, Koller B, Tancevski I, Patsch JR, Schratzberger P, Piza-Katzer H, Ritsch A, Bahlmann FH, Fischer-Colbrie R, Wolf D, Kirchmair R. Gene therapy with the angiogenic cytokine secretoneurin induces therapeutic angiogenesis by a nitric oxide-dependent mechanism. Circ Res. 2009;105:994–1002. doi: 10.1161/CIRCRESAHA.109.199513. [DOI] [PubMed] [Google Scholar]
- 14.Liu YH, Yang XP, Nass O, Sabbah HN, Peterson E, Carretero OA. Chronic heart failure induced by coronary artery ligation in Lewis inbred rats. Am J Physiol. 1997;272:H722–727. doi: 10.1152/ajpheart.1997.272.2.H722. [DOI] [PubMed] [Google Scholar]
- 15.Bonaros N, Rauf R, Wolf D, Margreiter E, Tzankov A, Schlechta B, Kocher A, Ott H, Schachner T, Hering S, Bonatti J, Laufer G. Combined transplantation of skeletal myoblasts and angiopoietic progenitor cells reduces infarct size and apoptosis and improves cardiac function in chronic ischemic heart failure. J Thorac Cardiovasc Surg. 2006;132:1321–1328. doi: 10.1016/j.jtcvs.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 16.Sumida A, Horiba M, Ishiguro H, Takenaka H, Ueda N, Ooboshi H, Opthof T, Kadomatsu K, Kodama I. Midkine gene transfer after myocardial infarction in rats prevents remodelling and ameliorates cardiac dysfunction. Cardiovasc Res. 2010;86:113–121. doi: 10.1093/cvr/cvp386. [DOI] [PubMed] [Google Scholar]
- 17.Jakobsson L, Kreuger J, Holmborn K, Lundin L, Eriksson I, Kjellén L, Claesson-Welsh L. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell. 2006;10:625–634. doi: 10.1016/j.devcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Soulié P, Héroult M, Bernard I, Kerros ME, Milhiet PE, Delbé J, Barritault D, Caruelle D, Courty J. Immunoassay for measuring the heparin-binding growth factors HARP and MK in biological fluids. J Immunoassay Immunochem. 2002;23:33–48. doi: 10.1081/IAS-120002273. [DOI] [PubMed] [Google Scholar]
- 19.Egger M, Schgoer W, Beer AG, Jeschke J, Leierer J, Theurl M, Frauscher S, Tepper OM, Niederwanger A, Ritsch A, Kearney M, Wanschitz J, Gurtner GC, Fischer-Colbrie R, Weiss G, Piza-Katzer H, Losordo DW, Patsch JR, Schratzberger P, Kirchmair R. Hypoxia up-regulates the angiogenic cytokine secretoneurin via an HIF-1alpha- and basic FGF-dependent pathway in muscle cells. FASEB J. 2007;21:2906–2917. doi: 10.1096/fj.06-7440com. [DOI] [PubMed] [Google Scholar]
- 20.Banquet S, Gomez E, Nicol L, Edwards-Lévy F, Henry JP, Cao R, Schapman D, Dautreaux B, Lallemand F, Bauer F, Cao Y, Thuillez C, Mulder P, Richard V, Brakenhielm E. Arteriogenic therapy by intramyocardial sustained delivery of a novel growth factor combination prevents chronic heart failure. Circulation. 2011;124:1059–1069. doi: 10.1161/CIRCULATIONAHA.110.010264. [DOI] [PubMed] [Google Scholar]
- 21.Cao R, Bråkenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med. 2003;9:604–613. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- 22.Murakami M, Nguyen LT, Hatanaka K, Schachterle W, Chen PY, Zhuang ZW, Black BL, Simons M. FGF-dependent regulation of VEGF receptor 2 expression in mice. J Clin Invest. 2011;121:2668–78. doi: 10.1172/JCI44762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henry TD, Grines CL, Watkins MW, Dib N, Barbeau G, Moreadith R, Andrasfay T, Engler RL. Effects of Ad5FGF-4 in patients with angina: an analysis of pooled data from the AGENT-3 and AGENT-4 trials. J Am Coll Cardiol. 2007;50:1038–1046. doi: 10.1016/j.jacc.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Belch J, Hiatt WR, Baumgartner I, Driver IV, Nikol S, Norgren L, Van Belle E TAMARIS Committees and Investigators. Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet. 2011;377:1929–1937. doi: 10.1016/S0140-6736(11)60394-2. [DOI] [PubMed] [Google Scholar]
- 25.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 2010;188:595–609. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staton CA, Kumar I, Reed MW, Brown NJ. Neuropilins in physiological and pathological angiogenesis. J Pathol. 2007;212:237–248. doi: 10.1002/path.2182. [DOI] [PubMed] [Google Scholar]
- 28.Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J Biol Chem. 1992;267:6093–6098. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.