

Abstract
The mammalian target of rapamycin (mTOR) pathway is an important integrator of nutrient-sensing signals in all mammalian cells, and acts to coordinate the cell proliferation with the availability of nutrients such as glucose, amino acids and energy (oxygen and ATP). A large part of the immune response depends on the proliferation and clonal expansion of antigen-specific T cells, which depends on mTOR activation, and the pharmacological inhibition of this pathway by rapamycin is therefore potently immunosuppressive. It is only recently, however, that we have started to understand the more subtle details of how the mTOR pathway is involved in controlling the differentiation of effector versus memory CD8+ T cells and the decision to generate different CD4+ helper T-cell subsets. In particular, this review will focus on how nutrient sensing via mTOR controls the expression of the master transcription factor for regulatory T cells in order to maintain the balance between tolerance and inflammation.
Keywords: FOXP3, metabolism, nutrient sensing, regulatory T cells
T cells need to co-ordinate their activation and metabolism
All cells need to be able to coordinate their proliferation and differentiation with their metabolic demands and the availability of essential nutrients. The mammalian target of rapamycin (mTOR) signalling pathway acts as an important integrator of nutrient-sensing pathways, which in turn control and coordinate the metabolism of the cell according to its need to proliferate or functionally differentiate.1 T-cell activation is intimately coupled to metabolism and energy generation, with a switch from primarily oxidative phosphorylation in resting T cells to an aerobic form of glycolysis, known as the ‘Warburg effect’,2 during activation and proliferation.3 Although the differentiating effector T cell needs to generate ATP as an energy source, which is most efficient via mitochondrial oxidative phosphorylation, this needs to be balanced by maintaining glycolysis (which is more conventionally associated with anaerobic conditions), because this pathway can use glucose as the basic source of carbon to generate many of the fundamental building blocks of the proliferating cell, such as amino acids, lipids, complex carbohydrates and ribonucleotides.4 Although the details of how this switch occurs in T cells remain unclear, the mTOR pathway is strongly implicated, because its activation up-regulates the surface expression of the glucose transporter, Glut1, probably as a result of T-cell receptor and CD28 signalling through phosphatidylinositide 3-kinase (PI3K) and protein kinase B (PKB also known as AKT).5 AKT signalling via mTOR also leads to higher expression of amino acid and other nutrient transporters, such as the transferrin receptor.6
Nutrient sensing and the mTOR pathway in T cells
The mTOR pathway acts in all cells to coordinate many other aspects of cell growth and metabolism, including the response to hypoxia and the biogenesis and oxidative capacity of mitochondria.7 mTOR forms two structurally distinct complexes (TORC1 and TORC2).8 The core components of TORC1, which is thought to represent the main nutrient-sensing complex, are the serine/threonine kinase mTOR itself, the scaffolding protein Raptor, the positive accessory proteins FKB12, Deptor and mLST8, plus a regulatory subunit PRAS40, which is a target of AKT downstream of PI3K signalling.9 The immunosuppressive drug rapamycin (which gave mTOR its name as the mammalian target of rapamycin) actually binds to FKB12 and disrupts the formation and function of the TORC1 complex.10 A critical activator of the TORC1 complex is the ras homologue expressed in brain (Rheb), which is localized within the cell in a Rab7+ lysosomal compartment. Rheb is in turn controlled by the tuberous sclerosis (TSC) 1/2 complex, which acts downstream of many different signalling pathways, including AMP-activated protein kinase, PI3K and AKT.11 AMP kinase can act as a sensor of increasing AMP/ATP ratios during hypoxia, while PI3K provides signals from growth factor receptors and co-stimulatory molecules such as CD28 and programmed death-1 during T-cell receptor activation. The interaction between TORC1 and Rheb is entirely dependent on the sensing of sufficient amino acids, and although the molecular sensor has yet to be identified in mammals, downstream signalling requires the four ras-related GTP binding (or RAG GTPase: RRAG) proteins (A–D) together with the ragulator complex,12,13 so that a lack of available amino acids acts as a potent inhibitor of TORC1 activity. Conversely, activation of TORC1 drives protein synthesis via phosphorylation of S6K1, which in turn phosphorylates the ribosomal protein S6, which is required for the initiation of translation. At the same time, 4E-BP1, an inhibitor of protein translation, is also deactivated by mTOR-mediated phosphorylation. Much less is known about how the TORC2 complex is regulated: in the short term (i.e. minutes) it is thought to be negatively regulated by TORC1 activity, but chronic long-term inhibition (over hours to days) of TORC1 with rapamycin14 or by amino acid starvation15 seems to eventually reduce the activity of TORC2. TORC2 is thought to control spatial aspects of cell growth, in particular cell polarity and responses to chemotactic signals via G-protein-coupled activation of RAS.16
mTOR is a critical regulator of FOXP3 expression
It has long been known that mTOR inhibition by rapamycin (which is used clinically in organ transplantation under the name Sirolimus) is potently immunosuppressive, partly because it blocks the ability of T cells to respond to interleukin-2 and consequently their ability to proliferate in response to antigen stimulation.17 It is only more recently that is has become clear that the mTOR pathway also controls the differentiation of different T helper cell subsets,18 and in particular, the expression of forkhead box P3 (FOXP3), the ‘master’ transcription factor for regulatory T cells (Fig. 1). Downstream activation by mTOR of the T-cell receptor, CD28 co-stimulation and cytokine-mediated PI3K signalling is generally required for the differentiation of effector T cells but is inhibitory for FOXP3 expression.19,20 Signalling downstream of the sphingomyelin phosphate receptor (S1PR), which is required for lymphocyte trafficking and exit from the lymph nodes, also acts to activate mTOR.21 Interestingly, this pathway is also the target of a relatively new immunosuppressive drug known as Fingolimod/FTY720,22 which therefore might also have the potential to promote regulatory T (Treg) cell development.23 Although the exact mechanism of FOXP3 inhibition by mTOR has not been clarified, there is some evidence for the involvement of a number of different pathways. These include poorly defined effects on FOXP3 translation via phosphorylation of ribosomal protein S6, and mTOR acting either indirectly via suppressor of cytokine signalling 3 (SOCS3)24,25 or directly on signal transducer and activator of transcription 3 (STAT3) downstream of interleukin-6 and the satiety hormone leptin,26 which then competes for the interleukin-2-driven STAT5 enhancement of foxp3 transcription.27 In addition, two transcription factors promoting FOXP3 expression, FOXO3a28,29 and the transforming growth factor-β (TGF-β) signalling component SMAD3, are negatively regulated by AKT downstream of TORC2.30 Evidence from raptor (TORC1) deficient and rictor (TORC2) deficient mice has suggested that TORC1 tends to promote T helper type 1 (Th1) differentiation,18 while TORC2 may bias the response to Th2 via AKT and PKCθ,31 while inhibition of both complexes is required for optimal FOXP3+ Treg cell induction. Th17 cell development seems to be independent of TORC2, but is inhibited by rapamycin in favour of FOXP3+ Treg cells.32
Figure 1.
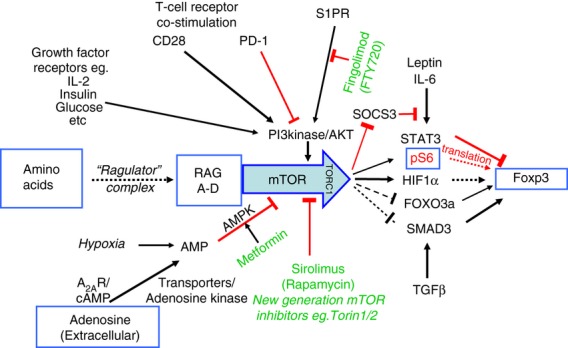
A mammalian target of rapamycin (mTOR) -centric view of nutrient sensing for the induction of forkhead box P3 (FOXP3). The mTOR pathway in T cells integrates antigen receptor signalling through the T-cell receptor and co-stimulatory molecules such as CD28 and programmed death 1 (PD-1) with a range of nutrient-sensing and growth factor signals. The main nutrient sources that are sensed via mTOR are the essential amino acids, glucose and adenosine, while relevant growth factors include insulin and the interleukin-2 family of cytokines. The shingomyelin phosphate receptor (S1PR), which controls lymphocyte exit from the lymph nodes, also acts via this pathway. The availability of amino acids is detected via a unique pathway that involves the ragulator complex and the four Ras-like GTPases (RAGs A–D), activation of which are required before mTOR can form the TORC1 complex and respond to any of the other signals. When the T-cell receptor is stimulated and there are both sufficient nutrients and growth factors available, mTOR is activated, which in turn inhibits FOXP3 expression, so that the differentiation of effector T cells is promoted. When nutrient or growth factor availability is restricted, the resulting mTOR inhibition allows FOXP3 induction and the development of FOXP3+ regulatory T cells. Although the exact mechanism linking mTOR inhibition to FOXP3 expression is not well defined, a number of different pathways have been implicated, as shown. These include direct effects on FOXP3 transcription dependent on hypoxia inducible factor 1α (HIF1α) and forkhead box O3a (FOXO3a), the regulation of FOXP3 mRNA translation by phosphorylation of the ribosomal protein S6 (pS6), and indirect effects on the regulation of FOXP3 expression in response to cytokines such as transforming growth factor-β (TGF-β)and interleukin-6 (IL-6) signalling through SMAD3 or signal transducer and activator of transcription 3 (STAT3), respectively. Positive signalling is indicated by black arrows, while inhibitory signals are indicated by blocked red lines. Broken lines indicate where the details of signalling are still poorly defined or unclear. Approved drugs that are available to manipulate this system in vivo are shown in green.
Modulation of FOXP3 expression by adenosine and hypoxia via AMP kinase
Hypoxia-induced factor (HIF) 1α, another downstream target of TORC1, has also been implicated as both a positive33,34 and a negative35,36 regulator of FOXP3 expression and it is also thought to bind directly to FOXP3 protein to target it for proteosomal degradation.36 HIF1α is a BHLH-Pas transcription factor that has an essential role in the response of cells to hypoxia. The level of HIF1α transcription is controlled by nuclear factor-κβ,37 but its activity is mainly controlled post-translation by an oxygen-mediated ubiquitination and degradation controlled by the Von Hippel–Lindau tumor suppressor complex and by positive regulation via a TORC1-mediated phosphorylation.38 The differentiation of naive T cells under hypoxic conditions has also been suggested to enhance FOXP3 expression and the development of regulatory activity,34 but it is not clear whether this is a direct effect of HIF1α on FOXP3 expression, or whether it is acting indirectly, as HIF1α activation can also inactivate mTOR.39 Hypoxia is associated with raised levels of AMP within the cell, which activates AMP-activated protein kinase and consequently inhibits mTOR via tuberous sclerosis complex 1/2. Other sources of AMP that may activate this pathway are downstream of G protein signalling where the generated cAMP from ATP is subsequently broken down to AMP by cAMP phosphodiesterases. In addition, extracellular adenosine can generate cAMP via activation surface receptors (e.g. the A2AR on T cells40,41) or can be directly taken up by specific transporters42 where, once inside the cell, it will be rapidly converted to AMP by adenosine kinase, one of the most abundant enzymes present in mammalian cells. Adenosine is particularly relevant to immune regulation, as TGF-β is able to induce in a range of haematopoietic cells the co-expression of two ectoenzymes, CD39 and CD73,43 that are constitutively expressed on Treg cells.44 These enzymes act to convert extracellular sources of ATP, which is associated with inflammation and cell necrosis, into the anti-inflammatory product adenosine (Fig. 2). Although there is some evidence that this pathway may be relevant to tumours escaping immune surveillance,45,46 it remains, however, to be resolved just how important adenosine is as a component of the anti-inflammatory microenvironment within tolerated tissues.
Figure 2.
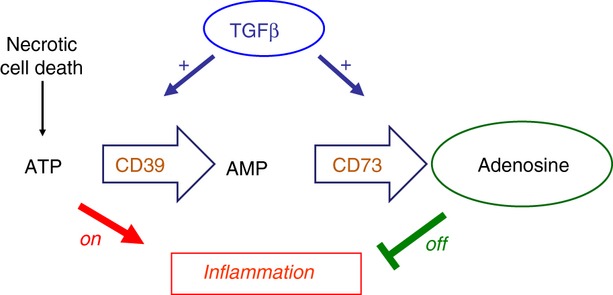
Transforming growth factor-β (TGF-β) regulates the production of extracellular adenosine. Extracellular ATP released from infections and necrotic cell death is potently inflammatory. Regulatory T (Treg) cells constitutively express the two ectoenzymes CD39 and CD73 that can convert ATP, via AMP into adenosine, thereby converting an inflammatory stimulus into an anti-inflammatory one. TGF-β is, however, able to induce the expression of both CD39 and CD73 on the majority of activated T cells, as well as on other cell types such as macrophages and dendritic cells, thereby providing a mechanism to dramatically amplify the anti-inflammatory action of these two enzymes.
Immune regulation and tolerance are associated with a nutrient-depleted microenvironment
It has only recently become clear that tolerance can be maintained by Treg cells acting within a highly localized microenvironment to induce a state of acquired immune privilege.47,48 This can best be demonstrated in experiments where donor alloantigen-specific tolerance has been induced to a skin graft (e.g. by a short period of co-receptor blockade with anti-CD4 and anti-CD8 monoclonal antibodies), and then that tolerated graft is removed and re-transplanted onto a secondary recipient with no T cells of its own (e.g. a recombinase activating gene 1 knockout mouse). As expected, this skin graft is accepted by the secondary recipient because it has no T cells to cause rejection. If, however, we treat the recipient at the time of grafting with monoclonal antibodies that deplete or inactivate FOXP3+ Treg cells (e.g. anti-CD25, or anti-hCD2, if the original recipient carries the hCD2.FOXP3 knock-in reporter), the grafts are rapidly rejected.48 This demonstrates that the tolerated, re-transplanted skin graft carried over within it perfectly functional effector T cells, but that FOXP3+ Treg cells were actively blocking their ability to reject and so maintained the tolerant state within the graft. By studying the changes in gene expression of dendritic cells when they interact with Treg cells,49,50 it was found that in addition to the known down-regulation of co-stimulatory ligands and antigen presentation, there was up-regulation of a number of enzymes that either catabolize or use essential amino acids51 (Fig. 3). In the context of a microenvironment with a restricted availability of nutrients, the local depletion of essential amino acids by these enzymes would be an effective mechanism to control the immune response via the mTOR nutrient sensing pathway. It has also been shown that the intracellular availability of leucine and consequently mTOR activation is controlled by T-cell-receptor-induced expression of the neutral amino acid transporter slc7a5 in Th1 and Th2 cells, where it is essential for their activation and differentiation, while Treg cells seem not to require this particular transporter.52
Figure 3.
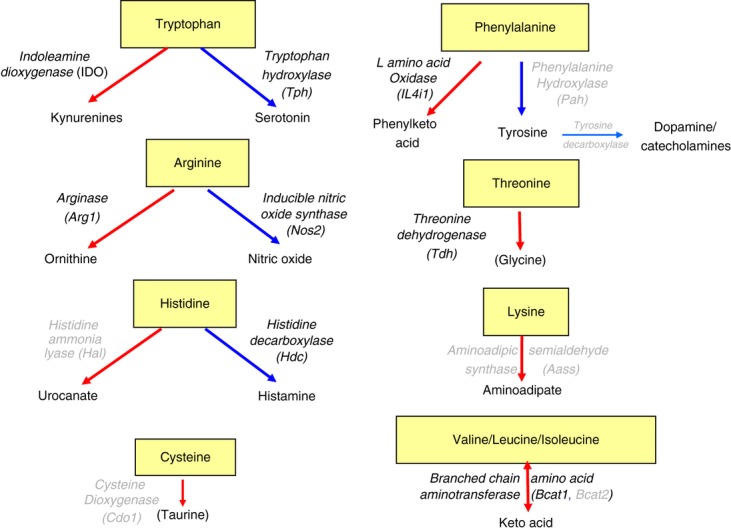
Enzymes that catabolize or use essential amino acids. Each of the amino acids that are considered essential for mammalian cells, because they are unable to synthesize them, are either catabolized (red arrows) or used to synthesize various products (blue arrows) by specific enzymes. Many of these enzymes are up-regulated in dendritic cells in response to inflammation and cytokines (both pro- and anti-inflammatory) or by the action of regulatory T (Treg) cells (highlighted in bold face). For example, arginase 1 (Arg1) and inducible nitric oxide synthase (iNOS/NOS2) can both consume arginine, the availability of which is sensed through the RAG/mammalian target of rapamycin (mTOR) pathway, leading to the inhibition of T-cell proliferation and the promotion of forkhead box P3 (FOXP3) expression (Fig. 1).
Immune regulation as a result of indoleamine 2,3 dioxygenase-mediated tryptophan catabolism
The first example of such amino acid catabolism being able to control the immune response was the expression of indoleamine 2,3 dioxygenase (IDO) in the placenta during pregnancy, which acts locally to deplete the essential amino acid tryptophan in order to block the maternal immune response to paternal alloantigens.53 This tryptophan-depleted microenvironment is sensed by general control non-repressed 2 (GCN2), which is one of the initiators of the integrated stress response, and leads to a block in the proliferation of CD8 effector T cells,54 and is required for the survival of T cells, including CD4+ Treg cells, during periods of amino acid starvation.51 GCN2, however, was not essential for T cells to sense the absence of essential amino acids in vitro,51 neither is it required for the induction of tolerance to skin grafts in mice by co-receptor blockade (S. Cobbold, E. Adams and H. Waldmann, unpublished results). The induction of FOXP3 by stimulating naive CD4+ T cells in the presence of low doses of TGF-β in vitro was also unaffected by stimulating the GCN2 pathway with histidinol; whereas, inhibition of the mTOR pathway gave a synergistic increase in FOXP3 induction.51 It has also now been shown that 1-methyltryptophan mediated blocking of IDO and tryptophan sensing can act via mTOR and PKCθ signalling.55
Depletion of essential amino acids as a fundamental mechanism of immune regulation
Indoleamine 2,3 dioxygenase may have been recognized as the first example of immune regulation due to amino acid catabolism because, of all the essential amino acids, tryptophan is thought to be present at the lowest concentration. Recently, it has been shown that mast cells in tolerated skin grafts express the enzyme tryptophan hydroxylase (TPH1),56 which can also deplete tryptophan, using it to synthesize serotonin. Tolerance was abrogated in TPH1 knockout mice, and this could be reconstituted with wild-type mast cells, but not by providing 5-hydroxytryptophan to bypass TPH1 and allow normal serotonin synthesis.57 In a similar manner, arginase (ARG1) expression has often been associated with protective, type 2, macrophages within tissues,58 and like IDO, has been implicated in regulating the immune response during pregnancy.59,60 Arginine is also the substrate for the inducible form of nitric oxide synthase (iNOS), which is normally associated with a Th1 effector cell response, but under limiting concentrations of arginine in vitro, both arginase and iNOS can cause sufficient depletion of this essential amino acid to cause mTOR inhibition and block T-cell proliferation.51 Interleukin-4-induced 1 (IL4i1) was named for its induction in myeloid cells under Th2 conditions, and is also an enzyme that catabolizes amino acids, but with preference for those with a hydrophobic side chain such as phenylalanine.61
Regulatory T cells were able to induce many of these essential amino acid consuming enzymes in dendritic cells in vitro and within skin grafts in vivo,51 whereas the enzymes that catabolize threonine (threonine dehydrogenase: TDH) and the branched chain amino acids (branched chain amino acid aminotransferase: BCAT1) were more closely associated with innate inflammation or wound healing,51 suggesting that tissues have a built-in mechanism for protecting themselves against immune attack under these circumstances. Intriguingly, long-term surviving, fully healed syngeneic skin grafts also had higher levels of these particular enzymes, as well as increased infiltration by FOXP3+ Treg cells, suggesting that self tolerance and allo-tolerance within tissues may use similar mechanisms that depend on the availability of nutrients to T cells.62
Coordinating metabolism and T-cell differentiation
T-cell activation is primarily associated with glucose metabolism, even under aerobic conditions, as this not only provides a source of ATP for energy and effector cell activity, it generates the precursors for nucleotide synthesis and lipogenesis that are required for cell proliferation.4 Under conditions of nutrient restriction and mTOR inhibition, however, it would be expected that T cells would switch to the more efficient pathways of ATP generation, such as oxidative phosphorylation and long-chain fatty acid oxidation, both of which require active mitochondria. Indeed, it has been shown that Treg cells have high levels of AMP kinase activity, which leads to mTOR inhibition, reduced levels of Glut1 and preferential lipid oxidation, effects that can be reversed in Glut1 over-expressing transgenic mice.63
Evidence is now beginning to emerge that the metabolic pathways active in a T-cell are not only a response to activation and differentiation, but can actually be the trigger to determine their differentiation and cell fate. For example, it has recently been shown that the glycolytic enzyme glyceraldehyde phosphate dehydrogenase (GAPDH) has a secondary function as a component of the interferon-γ-activated inhibitor of translation (GAIT) complex.64 This binds to AU-rich elements in the 3′ untranslated region of the interferon-γ mRNA and blocks its translation, but only if the substrate for GAPDH, glyceraldehyde 3-phosphate, is unavailable. If activated T cells are deprived of glucose, and instead provided with galactose, then glycolysis cannot take place, and yet the T cells still activate and proliferate (because galactose provides alternative precursors for nucleotide synthesis via the pentose phosphate pathway), but now because GAPDH has no substrate, it blocks the translation of interferon-γ. Under these conditions the T cells also then express other markers of T-cell exhaustion such as programmed death 1.64 The corollary of this is that inducing glycolysis, for example by mTOR activation, will tend to promote effector cell differentiation. There are also suggestions that there may be other examples where metabolic enzymes, for example hexokinase65 and IDO,26 can have a secondary, signalling role in dendritic cell differentiation.
Differing roles of mTOR during the differentiation and function of Treg cells
Inhibition of mTOR therefore seems to be associated with tolerance and FOXP3+ Treg cell induction, and this appeared to be confirmed by T-cell-specific mTOR knockout mice, which develop an excess of FOXP3+ Treg cells over Th1 and Th2 effector cells.18 Recent data, however, from FOXP3-Cre.Raptorfl/fl mice where TORC1 activity has been specifically knocked out in FOXP3+ Treg cells, indicates that TORC1 activation is still required for Treg cells to function, as evidenced by the development of an autoinflammatory condition very similar to scurfy or FOXP3-deficient mice.66 CD4-Cre.Raptorfl/fl mice, lacking TORC1 activity in all T cells, however, did not develop disease, presumably because this also compromised the effector T cells. This raises the possibility that the optimal induction and expansion of FOXP3+ Treg cells takes place in the nutrient-depleted microenvironments associated with tolerance, but the Treg cells only become fully active and proliferative when there is inflammation that needs to be controlled, which requires a re-activation of their mTOR pathway. Interestingly, it had previously been postulated that the optimal functional induction of FOXP3+ Treg cells required alternate cycles or oscillations of mTOR inhibition, first to promote induction, and subsequently mTOR activation to promote proliferation.67
mTOR regulates the differentiation of memory T cells
CD8+ effector T cells also need to rapidly proliferate and expand, particularly in response to viral infection, and so would be expected to require mTOR activation, but perhaps surprisingly, it has been shown that mTOR inhibition with rapamcyin actually promotes a better protective response during vaccination.68,69 Under physiological circumstances this seems to be due to AMP-kinase-α1-mediated sensing of glucose deprivation and subsequent mTOR inhibition70 that favours the development of long-lived central memory CD8+ T cells that provide better protection with later viral challenges, rather than short-term memory effector cells that target only the initial infection. There is some, perhaps rather controversial, evidence that CD8+ T cells, when first activated to proliferate, require an asymmetric cell division to provide one daughter that will generate the effector cell lineage while the other daughter gives rise to memory cells.71 If that is true, it is tempting to speculate that TORC2, which seems to have an evolutionary conserved function in controlling cell shape and polarity,16,72 may regulate asymmetric cell divisions and the subsequent lineage decisions of both CD4+ and CD8+ T cells in ways we do not yet understand.
Summary and conclusions
The mTOR pathway can therefore be thought of as the fulcrum that balances the different requirements of T cells in tolerance compared with inflammation (Fig. 4). During inflammation, effector T-cell differentiation dominates, which is associated with extracellular ATP and a ready availability of amino acids that, in turn, drive mTOR activation, cell proliferation and glucose metabolism. In contrast, tolerance is maintained by an excess of regulatory T cells, associated with a TGF-β-induced expression of CD39 and CD73, and conversion of extracellular ATP to adenosine. Tolerance within tissues is also associated with the up-regulation of many different enzymes that consume many, if not all, of the essential amino acids. Under these conditions, mTOR is inhibited, FOXP3 induction is promoted in naive T cells (i.e. infectious tolerance), and both iTreg and nTreg cells may have a competitive advantage to accumulate relative to effector T cells. However, under conditions of mTOR inhibition, Treg cells may not be optimally functional, and it may only be in response to inflammation and mTOR activating conditions that the Treg cells acquire the full suppressive potential.
Figure 4.
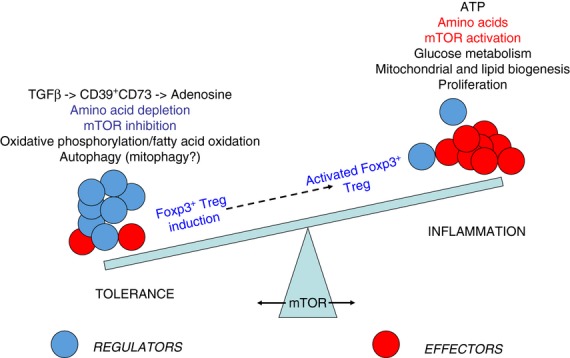
Mammalian target of rapamycin (mTOR) at the fulcrum of the immunoregulatory balance. The immune system has to maintain a delicate balance between the inflammation required to protect the body from infection while limiting the potential pathology and risk of autoimmunity. Tolerance is maintained primarily by ensuring that the relative frequency of regulatory T (Treg) cells (blue) is in excess of effector T cells (red), but when an inflammatory response is required, the generation of effector T cells is favoured. The decision to preferentially generate effector T cells rather than Treg cells is dependent on the availability of glucose and essential amino acids, and this activates mTOR, which then coordinates the switch in metabolism from primarily fatty acid oxidation to glucose metabolism. Conversely, the local depletion of essential amino acids from the tolerogenic microenvironment inhibits mTOR and encourages induction of Treg cells over effector cells. Committed induced Treg cells may, however, still be able to respond to mTOR activation by proliferating and/or increasing their suppressive function if they are required to limit the development of any inflammatory pathology.
Glossary
- AKT
protein kinase B
- EAA
essential amino acid
- FOXP3
forkhead box P3
- GAPDH
glyceraldehyde phosphate dehydrogenase
- IDO
indoleamine 2,3 dioxygenase
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositide 3-kinase
- TGF-β
transforming growth factor-β
- Th1
T helper type 1
- Treg
regulatory T
Disclosures
The author has no conflict of interests.
References
- 1.Peter C, Waldmann H, Cobbold SP. mTOR signalling and metabolic regulation of T cell differentiation. Curr Opin Immunol. 2010;22:655–61. doi: 10.1016/j.coi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 3.Buttgereit F, Brand MD, Muller M. ConA induced changes in energy metabolism of rat thymocytes. Biosci Rep. 1992;12:381–6. doi: 10.1007/BF01121501. [DOI] [PubMed] [Google Scholar]
- 4.Caro-Maldonado A, Gerriets VA, Rathmell JC. Matched and mismatched metabolic fuels in lymphocyte function. Semin Immunol. 2012;24:405–13. doi: 10.1016/j.smim.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–46. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, Powell JD. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol. 2007;178:2163–70. doi: 10.4049/jimmunol.178.4.2163. [DOI] [PubMed] [Google Scholar]
- 7.Schieke SM, Phillips D, McCoy JP, Jr, Aponte AM, Shen RF, Balaban RS, Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–52. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 8.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–68. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 9.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 11.Heikamp EB, Powell JD. Sensing the immune microenvironment to coordinate T cell metabolism, differentiation & function. Semin Immunol. 2012;24:414–20. doi: 10.1016/j.smim.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 15.Tato I, Bartrons R, Ventura F, Rosa JL. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem. 2011;286:6128–42. doi: 10.1074/jbc.M110.166991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charest PG, Shen Z, Lakoduk A, Sasaki AT, Briggs SP, Firtel RA. A Ras signaling complex controls the RasC-TORC2 pathway and directed cell migration. Dev Cell. 2010;18:737–49. doi: 10.1016/j.devcel.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–3. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 18.Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–44. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauer S, Bruno L, Hertweck A, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci USA. 2008;105:7797–802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+ Foxp3+ cells. J Exp Med. 2008;205:565–74. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, Chi H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10:769–77. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 23.Sehrawat S, Rouse BT. Anti-inflammatory effects of FTY720 against viral-induced immunopathology: role of drug-induced conversion of T cells to become Foxp3+ regulators. J Immunol. 2008;180:7636–47. doi: 10.4049/jimmunol.180.11.7636. [DOI] [PubMed] [Google Scholar]
- 24.Qin H, Wang L, Feng T, et al. TGF-β promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JH, Kim JE, Liu HY, Cao W, Chen J. Regulation of interleukin-6-induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3-SOCS3 pathway. J Biol Chem. 2008;283:708–15. doi: 10.1074/jbc.M708568200. [DOI] [PubMed] [Google Scholar]
- 26.Pallotta MT, Orabona C, Volpi C, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870–8. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 27.Yang XP, Ghoreschi K, Steward-Tharp SM, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–54. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harada Y, Harada Y, Elly C, Ying G, Paik JH, DePinho RA, Liu YC. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med. 2010;207:1381–91. doi: 10.1084/jem.20100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–27. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, Cui F, Fang L, Hong J, Zheng B, Zhang JZ. TNF-α impairs differentiation and function of TGF-β-induced Treg cells in autoimmune diseases through Akt and Smad3 signaling pathway. J Mol Cell Biol. 2013;5:85–98. doi: 10.1093/jmcb/mjs063. [DOI] [PubMed] [Google Scholar]
- 31.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–53. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol. 2007;7:1819–24. doi: 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clambey ET, McNamee EN, Westrich JA, et al. Hypoxia-inducible factor-1α-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA. 2012;109:E2784–93. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+ CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1α. Eur J Immunol. 2008;38:2412–8. doi: 10.1002/eji.200838318. [DOI] [PubMed] [Google Scholar]
- 35.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–76. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dang EV, Barbi J, Yang HY, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–84. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1β-mediated up-regulation of HIF-1α via an NFκB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–7. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 38.Safran M, Kaelin WG., Jr HIF hydroxylation and the mammalian oxygen-sensing pathway. J Clin Invest. 2003;111:779–83. doi: 10.1172/JCI18181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A. Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br J Pharmacol. 2008;153(Suppl. 1):S457–64. doi: 10.1038/bjp.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohtsuka T, Changelian PS, Bouis D, Noon K, Harada H, Lama VN, Pinsky DJ. Ecto-5′-nucleotidase (CD73) attenuates allograft airway rejection through adenosine 2A receptor stimulation. J Immunol. 2010;185:1321–9. doi: 10.4049/jimmunol.0901847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Apasov SG, Sitkovsky MV. The extracellular versus intracellular mechanisms of inhibition of TCR-triggered activation in thymocytes by adenosine under conditions of inhibited adenosine deaminase. Int Immunol. 1999;11:179–89. doi: 10.1093/intimm/11.2.179. [DOI] [PubMed] [Google Scholar]
- 43.Regateiro FS, Howie D, Nolan KF, Agorogiannis EI, Greaves ER, Cobbold SP, Waldmann H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-β. Eur J Immunol. 2011;41:2955–65. doi: 10.1002/eji.201141512. [DOI] [PubMed] [Google Scholar]
- 44.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–65. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. 2011;187:676–83. doi: 10.4049/jimmunol.1003884. [DOI] [PubMed] [Google Scholar]
- 46.Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, Murakami T, Robson SC, et al. CD39/ENTPD1 expression by CD4+ Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. 2010;139:1030–40. doi: 10.1053/j.gastro.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev. 2006;212:301–13. doi: 10.1111/j.0105-2896.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- 48.Kendal AR, Chen Y, Regateiro FS, Ma J, Adams E, Cobbold SP, Hori S, Waldmann H. Sustained suppression by Foxp3+ regulatory T cells is vital for infectious transplantation tolerance. J Exp Med. 2011;208:2043–53. doi: 10.1084/jem.20110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farquhar CA, Paterson AM, Cobbold AP, et al. Tolerogenicity is not an absolute property of a dendritic cell. Eur J Immunol. 2010;40:1728–37. doi: 10.1002/eji.200939974. [DOI] [PubMed] [Google Scholar]
- 50.Cobbold SP, Nolan KF, Graca L, et al. Regulatory T cells and dendritic cells in transplantation tolerance: molecular markers and mechanisms. Immunol Rev. 2003;196:109–24. doi: 10.1046/j.1600-065x.2003.00078.x. [DOI] [PubMed] [Google Scholar]
- 51.Cobbold SP, Adams E, Farquhar CA, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci USA. 2009;106:12055–60. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14:500–8. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 54.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–42. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Metz R, Rust S, Duhadaway JB, Mautino MR, Munn DH, Vahanian NN, Link CJ, Prendergast GC. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology. 2012;1:1460–8. doi: 10.4161/onci.21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zelenika D, Adams E, Humm S, Lin CY, Waldmann H, Cobbold SP. The role of CD4+ T-cell subsets in determining transplantation rejection or tolerance. Immunol Rev. 2001;182:164–79. doi: 10.1034/j.1600-065x.2001.1820113.x. [DOI] [PubMed] [Google Scholar]
- 57.Nowak EC, de Vries VC, Wasiuk A, Ahonen C, Bennett KA, Le Mercier I, Ha DG, Noelle RJ. Tryptophan hydroxylase-1 regulates immune tolerance and inflammation. J Exp Med. 2012;209:2127–35. doi: 10.1084/jem.20120408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chabtini L, Mfarrej B, Mounayar M, et al. TIM-3 regulates innate immune cells to induce fetomaternal tolerance. J Immunol. 2013;190:88–96. doi: 10.4049/jimmunol.1202176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kropf P, Baud D, Marshall SE, et al. Arginase activity mediates reversible T cell hyporesponsiveness in human pregnancy. Eur J Immunol. 2007;37:935–45. doi: 10.1002/eji.200636542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boulland ML, Marquet J, Molinier-Frenkel V, et al. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood. 2007;110:220–7. doi: 10.1182/blood-2006-07-036210. [DOI] [PubMed] [Google Scholar]
- 62.Cobbold SP, Adams E, Waldmann H. Biomarkers of transplantation tolerance: more hopeful than helpful? Front Immunol. 2011;2:9. doi: 10.3389/fimmu.2011.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang CH, Curtis JD, Maggi LB, Jr, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Majewski N, Nogueira V, Bhaskar P, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–30. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 66.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T-cell function. Nature. 2013;499:485–90. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Procaccini C, De Rosa V, Galgani M, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–41. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–12. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKα1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol. 2013;43:889–96. doi: 10.1002/eji.201243008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang JT, Palanivel VR, Kinjyo I, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–91. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 72.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]