

Abstract
β-Arrestins are well-known regulators and mediators of G protein-coupled receptor signalling, and accumulating evidence reveals that they are functionally involved in inflammation and autoimmune diseases. Of the two β-arrestins, β-arrestin 1 is documented to play regulatory roles in an animal model of multiple sclerosis (MS), whereas the role of β-arrestin 2 is less clear. Here, we show that β-arrestin 2-deficient mice displayed the exacerbated and sustained symptoms of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. At the cellular level, deficiency of β-arrestin 2 led to a decreased number of Foxp3+ CD4+ regulatory T (Treg) cells in peripheral lymphoid organs of EAE mice. Consistently, our in vitro observations also revealed that loss of β-arrestin 2 impaired the conversion of Foxp3− CD4+ T cells into Foxp3+ CD4+ inducible Treg cells. Taken together, our data suggest that β-arrestin 2 plays a regulatory role in MS, that is opposite to that of β-arrestin 1, in autoimmune diseases such as MS, which is at least partially through regulation of iTreg cell differentiation.
Keywords: experimental autoimmune encephalomyelitis, regulatory T cells, β-arrestin 2
Introduction
Experimental autoimmune encephalomyelitis (EAE) is a widely used animal model of multiple sclerosis (MS), a human autoimmune disease characterized by chronic inflammatory demyelination and degeneration of the central nervous system (CNS).1–3 The mechanisms involved in the formation of acute inflammatory CNS lesions are to a large extent shared between MS and EAE, and involve pivotal roles played by autoreactive CD4+ T cells, in particular during disease initiation. Of the CD4+ T cells, those expressing interferon-γ [IFN-γ; T helper type 1 (Th1)] or interleukin-17 (IL-17; Th17) are generally regarded to be highly encephalitogenic, whereas the Foxp3+ CD4+ regulatory T (Treg) cells are currently thought to protect against the ongoing autoimmune response.4–7 Both the number and function of Treg cells could be altered in EAE, and this seems to be tightly associated with disease progression and severity.4,8,9 However, the molecular mechanisms underlying the generation of Treg cells and their functions in EAE are still unclear.
β-Arrestins (β-arrestin 1 and β-arrestin 2), the well-defined regulators of G protein-coupled receptor desensitization and endocytosis,10 also play important roles in the regulation of immune activation and inflammatory diseases, such as MS,11 rheumatoid arthritis,12 primary biliary cirrhosis13 and allergic asthma.14 At the cellular level, previous investigations revealed that β-arrestin 2 can regulate lipopolysaccharide-induced cytokine production by polymorphonuclear leucocytes15 and inhibitory signalling in natural killer cells.16 However, the role of β-arrestin 2 in CD4+ T cells and EAE development remains poorly explored, although β-arrestin 1 is well known to regulate EAE severity through promotion of CD4+ T-cell survival.11 In this study, we found that loss of β-arrestin 2 increases the severity of EAE, through a reduction in the population of peripherally derived Treg cells, a finding that is essentially the opposite of that obtained in β-arrestin 1-knockout animals.
Materials and methods
Mice
C57BL/6 mice were obtained from the Shanghai Laboratory Animal Centre (Chinese Academy of Sciences). Arrb2−/− mice on a C57BL/6 background were provided by Robert J. Lefkowitz (Duke University Medical Center, Durham, NC). Foxp3-IRES-GFP (Foxp3egfp) transgenic mice, which contain the enhanced green fluorescence protein (egfp) gene under the control of an internal ribosomal entry site (IRES) inserted downstream of the foxp3 coding region as described elsewhere,17 were provided by Dr Honglin Wang (Shanghai Jiaotong University). Arrb2−/− Foxp3egfp-Tg mice were obtained by crossing Arrb2−/− mice with Foxp3egfp mice. All mice were maintained in pathogen-free conditions. Animal care and use were in accordance with the guidelines of the Shanghai Institute of Biochemistry and Cell Biology.
Reagents
Myelin oligodendrocyte glycoprotein peptide (MOG35–55, MEVGWYRSPFSRVVHLYRNGK) with purity of > 95% was purchased from GLBiochem (Shanghai, China). Moloney murine leukaemia virus reverse transcriptase and RNasin RNase inhibitor were from Promega (Madison, WI). SYBR Green JumpStart Taq Ready-Mix kit was from Sigma-Aldrich (St Louis, MO). Percoll was from GE Healthcare (Little Chalfont, UK). FITC anti-mouse CD45 (clone: 30-F11), phycoerythrin (PE) -conjugated anti-mouse CD8a (clone: 53-6.7), allophycocyanin (APC) anti-mouse CD11b (clone: M1/70), PE-Cy7 anti-mouse CD4 (clone: GK1.5), PE anti-mouse CD25 (clone: PC61.5), APC anti-mouse IFN-γ (clone: XMG1.2), PE anti-mouse Foxp3 (clone: FJK16s), APC anti-mouse/rat Foxp3 (clone: FJK16s) and Foxp3 staining set were purchased from eBioscience (San Diego, CA). The PE anti-mouse CD45R (B220; clone: RA3-6B2), PE anti-mouse IL17a (clone: 2B8), and BD Cytofix/Cytoperm kit were purchased from BD Biosciences (San Jose, CA). Dynal Mouse CD4 Negative Isolation Kit (Cat. No. 114.15D) was from Invitrogen (Carlsbad, CA). Mouse CD4 (L3T4, Cat. No.130-049-201) and CD25 MicroBeads Kit (Cat. No. 130-091-072) were from Miltenyi Biotec (Bergisch Gladbach, Germany).
EAE
Mice of age 8–12 weeks were immunized by subcutaneous injection of the myelin peptide MOG35–55 (150 μg) emulsified in complete Freund's adjuvant containing heat-killed Mycobacterium tuberculosis (H37Ra strain, 5 mg/ml; Difco, Detroit, MI). In addition, 200 ng of pertussis toxin (CalBiochem, Darmstadt, Germany) was administered intravenously on the day of immunization and 2 days after. Mice were examined daily for clinical signs by researchers blinded to experimental conditions and were assigned scores on a scale of 0–5 as follows: 0, no clinical signs; 1, paralysed tail; 2, paresis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of both hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund state or death. For analysis of CNS infiltrates, spinal cords were collected from perfused mice and mononuclear cells were prepared by 37–70% Percoll gradient centrifugation.
Histological analysis
For histological staining, mice were anaesthetized and perfused with PBS (pH 7·4) followed by 4% (weight/volume) paraformaldehyde. Lumbar regions of spinal cords were dissected and further fixed in paraformaldehyde overnight. Paraffin-embedded sections were stained with haematoxylin & eosin (H&E) or Luxol fast blue to examine the leucocyte infiltration or demyelination, respectively.
Immunofluorescence of frozen sections
After fixation in 4% paraformaldehyde, tissues were cryoprotected sequentially in 10%, 20%, 30% sucrose solution (weight/volume) in 1 × phosphate buffer and embedded in Optimal Cutting Temperature compound (Tissue-Tek; Sakura, Torrance, CA). Then, 15-μm-thick cryosections were cut from the lumbar region of spinal cords. Sections were allowed to thaw at 20–24°, rehydrated in PBS for 10 min and incubated with blocking buffer (1 × PBS containing 10% normal goat serum (volume/volume) at room temperature for 1 hr. Primary antibody (diluted in 1 × PBS containing 0·5% goat serum) staining was performed at 4° overnight. For primary antibodies, we used antibody against mouse CD45 (rat anti-mouse CD45 purified, clone 30-F11; eBioscience) as a marker of bone marrow-derived leucocytes, and antibody to CD4 as a T helper cell marker (clone L3T4, eBiosciences). For secondary antibodies, we used Alexa 488- or Cy3-conjugated goat antibodies to rat (Molecular Probes, Eugene, OR). All sections were observed with an Olympus BX51 Microscope (Olympus Corporation, Tokyo, Japan).
Flow cytometry
Mononuclear cell from spleens, lymph nodes, peripheral blood leukocytes or CNS infiltrates were incubated for 4·5 h at 37° with PMA (50 ng/ml; Sigma, St Louis, MO), ionomycin (750 ng/ml; Sigma), and brefeldin A (1·0 μg/ml; eBioscience). Surface markers were stained with the relevant antibodies. For intracellular cytokine staining, cells were then resuspended in fixation/permeabilization solution (Cytofix/Cytoperm kit; BD Pharmingen, San Diego, CA) and intracellular cytokine staining was performed according to the manufacturer's protocol. For Foxp3 staining, cells were not stimulated with PMA and ionomycin; instead, the protocol for the Foxp3 Staining Buffer set was followed (eBioscience). Samples were run with FACSCalibur (BD Biosciences) and analysed using flowjo software (Treestar Inc., Ashland, OR).
Primary CD4 T-cell purification, culture and differentiation
Naive CD4+ CD25− T cells were prepared by magnetic cell sorting from spleens of mice at the age of 6–8 weeks. For in vitro T helper cell differentiation assay, sorted cells were activated with anti-CD3 (2 μg/ml; 145-2C11, soluble; BD Pharmingen) and anti-CD28 (2 μg/ml; 37.51, soluble; BD Pharmingen) and were induced to differentiate into Th1 cells by supplementation with IL-12 (10 ng/ml; Peprotech, Rocky Hill, NJ) plus anti-IL-4; into Th2 cells with IL-4 (40 ng/ml, Peprotech) plus anti-IFN-γ; or into Th17 cells with transforming growth factor-β1 (TGF-β1; 3 ng/ml, Peprotech), IL-6 (30 ng/ml; eBioscience) and anti-IFN-γ. For inducing the conversion of naive CD4+ CD25− cells into Foxp3+ inducible Treg (iTreg) cells, cells were activated by plate-coating anti-CD3 (1 μg/ml) plus soluble anti-CD28 (1 μg/ml) and treated with TGF-β1 (1 or 5 ng/ml) and anti-IFN-γ. Cells stimulated in ‘neutral’ conditions (anti-IL-4 plus anti-IFN-γ but no additional cytokines) were considered to be ‘Th0’ cells. Neutralizing anti-IFN-γ (XMG1.2; BD Pharmingen) was used at a concentration of 5 μg/ml.
Cells were isolated from the spleens of mice at 8–12 weeks old. CD4+ CD25+ cells were purified by depletion of non-T cells with negative selection followed by positive selection of CD25+ T cells. CD4+ CD25− T cells were used as T responder cells. Magnetically sorted CD4+ CD25+ cells were cultured (96 hr, 37°) with CD4+ CD25− (105 cells) T responder cells at different ratios in 96-well round bottom plates together with irradiated T-cell-depleted splenocytes from wild-type mice (5 × 104 cells) as antigen-presenting-cells and anti-CD3 (1 μg/ml; 145-2C11; BD Pharmingen) and pulsed with 1 μCi of [3H]thymidine for the last 16 hr of culture. Cells were then harvested onto a glass-filter using a Tomtec 96-well cell harvester and assessed for thymidine incorporation by liquid scintillation in a Wallac MicroBeta counter (Perkin Elmer, Waltham, MA).
Reverse transcription quantitative real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription of purified RNA was performed using oligo(dT) priming and Moloney murine leukaemia virus reverse transcriptase (Promega). Real-time PCR was conducted in a Light Cycler quantitative PCR apparatus (Stratagene, La Jolla, CA) using the SYBR Green JumpStart Taq ReadyMix kit (Sigma-Aldrich) and primers (Fig. S8).
Western blot
CD4+ CD25− T cells were stimulated under iTreg cell conditions as described. Cells were collected after 24, 36 and 42 hr, and lysed for SDS–PAGE. Protein samples were separated by SDS gel electrophoresis and transferred to a nitrocellulose membrane (GE Healthcare). Membranes were stained with primary antibodies to p-S6Ser235 and p-S6Ser240 (all from Cell Signaling, Beverley, MA) and β-actin. The IRDye800CW-conjugated secondary antibody (Rockland, Gilbertsville, PA) was then added. Data were assessed by the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE).
Statistical analysis
Data are presented as means ± SEM. The statistical significance of the EAE clinical scores between different genotypes was analysed with two-way analysis of variance, followed by Mann–Whitney U-test to assess the significance of difference for individual days. Other analyses, including gene expression, cytokine production and histological analysis, were assessed by Student's t-test. A P-value < 0·05 was considered statistically significant.
Results
Arrb2−/− mice display exacerbated and sustained EAE
To study the role of β-arrestin 2 in EAE pathogenesis, we immunized Arrb2−/− mice and wild-type littermates with MOG35–55 peptide and monitored disease progression by clinical assessment. Although animals of both groups developed the neurological signs of EAE, the profiles of disease course were significantly altered in Arrb2−/− mice (P < 0·001, Fig. 1a, EAE scoring). As summarized in Fig. 1(d), Arrb2−/− mice showed a slightly delayed disease onset and slower progression; however, they achieved a significantly higher mean maximum of clinical score of 3·76 compared with the mean maximum clinical score of 2·89 in wild-type littermates (Fig. 1d, P < 0·001). Consistently, the percentage of animals that achieved a clinical score of 3 or above was higher in the Arrb2−/− group than that in wild-type controls (89·3% versus 57·7%, P < 0·001). Moreover, that subset of Arrb2−/− mice showed persistence of severe clinical signs (score ≥ 3) for an average of 10·4 days without apparent recovery, whereas wild-type animals showed persistence of severe clinical signs for approximately 2·7 days, followed by disease remission. These features collectively reveal an enhanced EAE severity in Arrb2−/− mice, which was further supported by histological studies on sections of spinal cords, the major anatomical sites where EAE-associated pathological changes (including inflammatory infiltrations and demyelination) occur. Wild-type and Arrb2−/− mice were killed at peak stage (a period from day 22 to day 25 was selected) of disease, and both H&E and Luxol fast blue staining revealed the aggravated neuropathology in Arrb2−/− mice (Fig. 1b,c). Therefore, these data clearly demonstrate that absence of β-arrestin 2 alters the course of EAE development, which is primarily characterized by enhanced and perpetuated disease severity.
Figure 1.

Arrb2−/− mice develop a more severe course of experimental autoimmune encephalomyelitis (EAE). Arrb2+/+ and Arrb2−/− mice on the C57BL/6 background were immunized with MOG35–55 peptide. (a) Clinical scores for Arrb2+/+ (blue circles; n = 26) and Arrb2−/− mice (red triangles; n = 28), 35 days post immunization. Data are expressed as mean EAE score ± SEM. Two-way analyses of variance were applied to compare the entire course of EAE development of both genotypes, followed by Mann–Whitney U-test to assess the significance of difference for individual day. (b) Haematoxylin & eosin (H&E) and Luxol fast blue staining of paraffin sections of spinal cords isolated from naive as well as EAE-affected Arrb2+/+ and Arrb2−/− mice on day 25 after immunization (upper panels, original magnification × 4; lower panels, original magnification × 20, enlarged images of boxed areas outlined in upper panel). (c) Quantitative analyses of the spinal cord leucocyte infiltrates and amount of demyelination presented in (b). Data are represented as means ± SEM. Three animals from each group were killed and five sections of the spinal cord of each animal were analysed. (d) Features of MOG35–55-induced EAE in Arrb2+/+ and Arrb2−/− mice. p.i., post-induction. Data are cumulative results from five independent experiments. *P < 0·05; **P < 0·01, versus wild-type control (two-tailed Student's t-test).
Loss of β-arrestin 2 led to increased CNS accumulation of CD4+ T cells
To further analyse the CNS inflammation in EAE-affected Arrb2−/− mice, immunofluorescence staining of spinal cord frozen sections was performed. Consistent with the H&E staining, marked increases in the number of CD45+ cells (identifying the bone marrow-derived leucocytes) were observed in sections from the Arrb2−/− mice (Fig. 2a,b, upper panel). Notably, signals corresponding to CD4+ T cells were also significantly enhanced in the same sections of Arrb2−/− EAE mice (Fig. 2a,b lower panel). Mononuclear cells were isolated from spinal cords of both animal groups at the peak stage of disease, and flow cytometry measurements showed that CD4+ T cells accounted for an average of 33·9% of the total CD45+ cells in wild-type mice, whereas this value was increased to an average of 51·6% in Arrb2−/− animals (Fig. 2c,d). These data therefore suggest that the loss of β-arrestin 2 leads to increased CD4+ T-cell accumulation in the CNS in response to EAE induction, which is associated with enhanced neuroinflammation.
Figure 2.
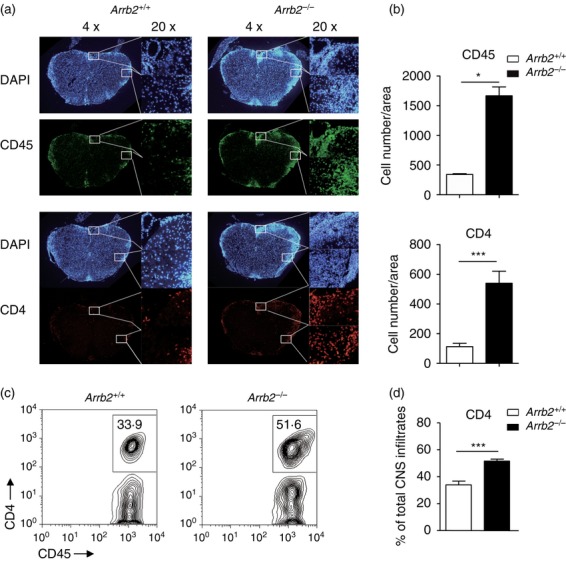
Loss of β-arrestin 2 led to increased central nervous system accumulation of CD4+ T cells (a) Immunofluorescent staining of CD45+ and CD4+ cells in frozen sections of spinal cords isolated from Arrb2+/+ and Arrb2−/− experimental autoimmune encephalomyelitis (EAE) mice on day 25 after immunization (left panels, original magnification × 4; right panels, original magnification × 20, enlarged images of boxed areas outlined in left panel). DAPI was used to stain the nuclei. (b) Quantification of CD45+ and CD4+ staining (indicative a single cell) presented in spinal cord sections shown in (a). Data are shown as means ± SEM. For three mice per group, five serial sections were stained, and representative sections are shown. (c) Mononuclear cell infiltrates were isolated by 37/70% Percoll from Arrb2+/+ and Arrb2−/− EAE mice on day 22 post immunization. Representative flow cytometry graphs show the frequencies of CD4+ T cells among CD45+ leucocytes (d) Quantification of data in (c). Data (means ± SEM) are shown from five mice per genotype *P < 0·05, ***P < 0·001 versus wild-type control (two-tailed Student's t-test).
Decreased numbers of Foxp3+ CD4+ Treg cells in EAE-affected Arrb2−/− mice
In light of the enhanced CNS inflammation (predominantly CD4+ T-cell accumulation) observed in Arrb2−/− EAE mice, we next determined whether in vivo CD4+ T-cell priming and differentiation during EAE development is altered upon depletion of β-arrestin 2. Mononuclear cells were harvested from the spleen (SPL) and draining lymph node (DLN) of the Arrb2+/+ and Arrb2−/− mice at either early stage (day 12–16, throughout this manuscript) or late stage (day 18–25, throughout this manuscript) of EAE. The frequencies of CD11b+ myeloid cells, B220+ B cells, and CD4+ and CD8+ T cells were comparable between the two groups of mice at both disease stages (see Supplementary material, Figs S2, S3). We then focused on the sublineages of effector and regulatory CD4+ T cells (Th1, Th17 and Treg cells). The number of effector CD4+ T cells in peripheral lymphoid organs (SPL and DLN) of Arrb2−/− mice was comparable to that of wild-type controls at both the early and late disease stages. In the CNS, however, CD4+ T cells (and in particular Th1 cells) were less abundant at the early stage, but then rapidly accumulated and even outnumbered those in wild-type controls when disease reached the late stage (see Supplementary material, Fig. S4, S5 and Fig. 3a,b,d). It is therefore likely that effector CD4+ T cells in Arrb2−/− mice entered the CNS more slowly, but were largely unaffected in priming in the periphery. No significant difference in Treg cells was observed at the early stage between the two groups of mice. At the late stage, however, the Treg cells in SPL and DLN of Arrb2−/− EAE mice were significantly reduced both in frequencies among the CD4+ population (Fig. 3a,b) and in the absolute numbers within the entire organ (Fig. 3c). Such a significant reduction of Treg cells was not found in Arrb2−/− CNS lesions, although a remarkably reduced ratio of CNS-resident Treg cells to T effector cells was observed (Fig. 3d–f). Compared with the largely unchanged peripheral effector T-cell repertoire in Arrb2−/− mice throughout the EAE development, the above behaviours of CD4 T-cell lineages suggest a gradual and selective loss of Treg cells in these mice as disease progresses. Since the frequencies of thymus-derived Foxp3+ CD4+ Treg cells were not changed in these mice when unchallenged (see Supplementary material, Fig. S1), the reduction of Treg cells in SPL and DLN of Arrb2−/− EAE mice could be specific to the EAE condition and might be associated with the enhanced and sustained disease symptoms in these animals. We were therefore interested in whether β-arrestin 2 expression in Treg cells is altered during EAE development. At different EAE developmental stages, we sorted GFP+ CD4+ or GFP− CD4+ cells from naive or MOG35–55-immunized Foxp3egfp mice17 and subjected them to gene expression analyses. Foxp3 transcripts were barely detected in GFP− cells, whereas they were abundant in GFP+ cells, and the opposite was found for Il17 transcripts, which was consistent with the T effector and Treg phenotypes of the two groups of cells (Fig. 3g). Notably, the Foxp3 transcript level in those GFP+ cells (i.e. cells in which Foxp3 is or has been actively transcribed) oscillates profoundly during disease development, with a decrease immediately after immunization, sustained low expression throughout the stages of disease priming and progression until the peak of disease is reached, and a significant increase upon disease remission. Interestingly, the level of Arrb2, but not of Arrb1, also increases specifically during the peak-to-remission transition in GFP+ cells, resembling that of Foxp3 (Fig. 3g), implying that β-arrestin 2 might be involved in the regulation of Treg cells during EAE development. Collectively, our data reveal that loss of β-arrestin 2 results in decreased numbers of Treg cells in vivo in response to EAE induction.
Figure 3.
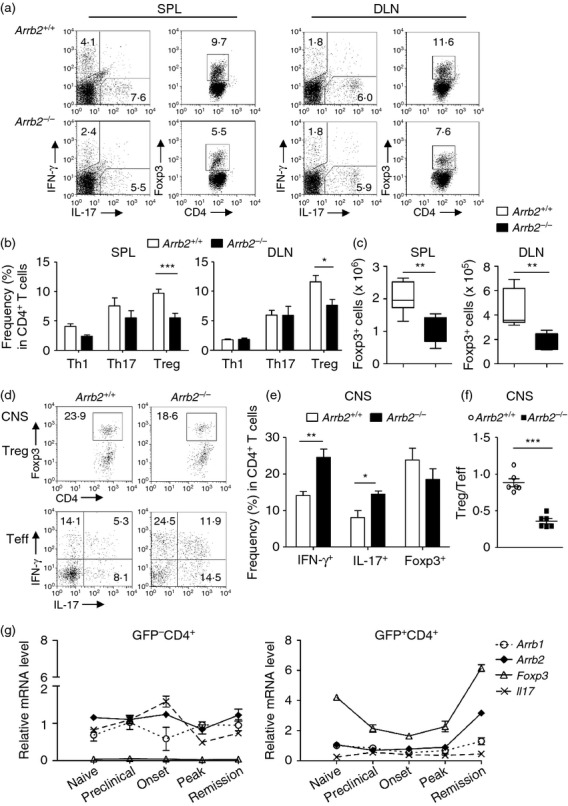
Number of peripheral Foxp3+ CD4+ T cells is decreased in Arrb2−/− experimental autoimmune encephalomyelitis (EAE) mice. (a–c) Mononuclear cells were isolated from the spleens (SPL) and draining lymph nodes (DLN) of Arrb2+/+ and Arrb2−/− EAE mice on day 22 after immunization. (a) Representative flow cytometry graphs show the frequencies of interferon-γ+ (IFN-γ+) (T helper type 1; Th1), interleukin-17+ (IL-17+) (Th17), and Foxp3+ CD4+ (regulatory T; Treg) cells among CD4+ population in SPL (left) and DLN (right), respectively. (b) Quantification of data in (a). (c) Quantification of absolute numbers of Foxp3+ CD4+ Treg cells in SPL or DLN (n = 7). (d–f) Central nervous system mononuclear cell infiltrates were isolated from Arrb2+/+ and Arrb2−/− EAE mice on day 22 post immunization. (d) Representative flow cytometry graphs show the frequencies of Treg cells as well as T effector cells in CD4+ gate, respectively. (e) Quantification of data in (d). (f) Calculation of the ratio of frequencies of Treg cells to T effector cells (a sum of all Th1, Th17 as well as IFN-γ+ IL-17+ cells) in CD4+ cells from the central nervous system lesions of Arrb2+/+ and Arrb2−/− EAE mice (n = 7 for either genotype); (g) GFP+ CD4+ cells (indicative of Foxp3-expressing Treg cells) from either naive or EAE-affected Foxp3egfp mice of distinct disease stages were purified from splenocytes by FACS sorting. Quantitative PCR was performed to analyse gene expression profiles. Gene transcript levels are calculated as the ratio of copy numbers relative to the hprt ‘housekeeping’ gene in the same sample. Data are presented as means ± SEM (n = 5) and are representative of three independent experiments. *P < 0·05, versus wild-type control (two-tailed Student's t-test)
Impaired conversion of naive Arrb2−/− T cells into Foxp3+ CD4+ iTreg cells
We then applied the in vitro T helper cell differentiation assay to investigate the role of β-arrestin 2 in both effector and regulatory CD4+ T-cell lineages. Naive CD4+ CD25− Foxp3− T cells were purified from spleens of Arrb2+/+ and Arrb2−/− littermates, and were cultured under Th1, Th2, Th17, or iTreg cell polarizing conditions. Expression of lineage-specific genes was assessed by intracellular staining. In the presence of exogenous TGF-β, induction of Foxp3 expression was profoundly impaired by β-arrestin 2 deficiency in CD4+ T cells, whereas inductions of signature cytokines corresponding to Th1, Th2 and Th17 lineages were largely unaffected (Fig. 4a,b). By crossing Arrb2−/− and Foxp3egfp reporter mice, we verified the compromised transcriptional up-regulation of Foxp3 in Arrb2−/− T cells. Under iTreg cell conditions, Arrb2−/− GFP− CD4+ cells also showed significantly reduced conversion of GFP+ cells as compared with wild-type controls (Fig. 4c,d), and this was not associated with decreased total cell numbers or compromised cell survival (Fig. 4e and see Supplementary material, Fig. S6). Hence, the absence of β-arrestin 2 can dampen the activation of endogenous Foxp3 transcription. To gain greater insight into how β-arrestin 2 affects this process, we cultured CFSE-labelled CD25− CD4+ naive T cells under a range of TGF-β concentrations; TGF-β is the key factor for inducing the conversion of Foxp3− conventional T cells into Foxp3+ iTreg cells. At a lower TGF-β concentration (1 ng/ml), a significantly lower degree of conversion to Foxp3+ cells was found in Arrb2−/− CD4+ T cells as early as day 2 (frequency of Foxp3+ cells among CD4+ cells, 8·72% versus 19·1% in Arrb2+/+ iTreg cell culture, Fig. 5a, upper panel). This attenuated conversion of Foxp3+ CD4+ cells in Arrb2−/− iTreg cell cultures was maintained through the end of in vitro culture (11·9% versus 34·6% in Arrb2+/+ iTreg culture, Fig. 5a, lower panel and Fig. 5b), and was irrespective of the number of cell divisions completed (Fig. 5c). Higher TGF-β concentration (5 ng/ml), however, largely compensated for the inability of Arrb2−/− CD4+ cells to produce Foxp3+ cells, as the levels in each genetic background were similar by day 5 (43·3% versus 53·4% in Arrb2+/+ iTreg culture, Fig. 5a, lower panel). Gene expression analyses on iTreg cells cultured in vitro (harvested at day 2 and cultured in the presence of 1 ng/ml TGF-β) also revealed significantly reduced levels of Foxp3 transcript, as well as several genes associated with Foxp3− status, including Ctla4, Gitr and Lag3. This was accompanied by insufficient suppression of genes that are essential for effector T-cell functions (including Il12p40, Ifng, Il17 and Il2) in Arrb2−/− cells (Fig. 5d). These data convincingly demonstrate that loss of β-arrestin 2 leads to remarkable impairment of the ability of Foxp3− CD4+ T cells to transform into Foxp3+ iTreg cells. This is an early event and is associated with a reduced response to exogenous TGF-β while the induction of effector T-cell lineages is largely unaffected, and is consistent with the reduced Treg : T effector cell ratio observed in Arrb2−/− EAE mice.
Figure 4.
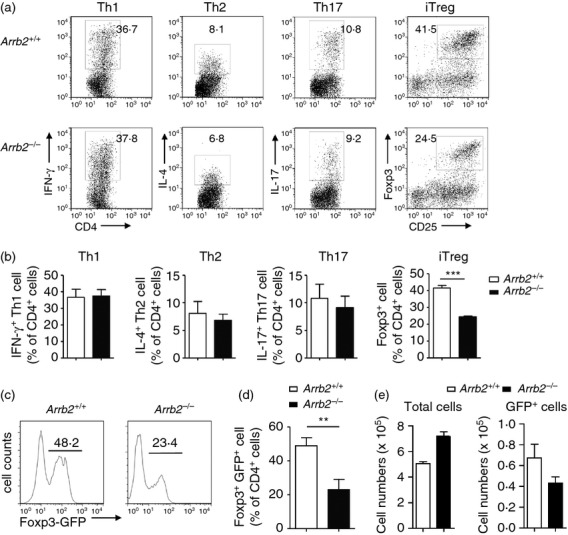
Impaired conversion of naive Arrb2−/− T cells into Foxp3+CD4+ inducible regulatory T (Treg) cells. Naive (CD25− CD4+) T cells purified from spleens of Arrb2−/− or Arrb2+/+ mice were cultured with anti-CD3 and anti-CD28. For T helper type 1 (Th1) differentiation, cells received interleukin-12 (IL-12; 10 ng/ml; PeproTech). For Th2 differentiation, cells received anti-interferon-γ (IFN-γ; 5 mg/ml; XMG1.2; BD Pharmingen) and IL-4 (40 ng/ml; PeproTech). For Th17 differentiation, cells received anti-IFN-γ, transforming growth factor-β1 (TGF-β1) (3 ng/ml; PeproTech), IL-6 (30 ng/ml; eBioscience). For inducible Treg cells, cells received TGF-β1 (1 ng/ml), and anti-IFN-γ. (a) Representative flow cytometry graphs show the generation of IFN-γ+ IL-17+ Foxp3+ cells in Arrb2−/− or Arrb2+/+ cell cultures. Numbers adjacent to outlined areas indicate frequency of cells within the CD4+ gate. (b) Quantification of results in (a). Data were shown as mean ± SEM from three independent experiments. (c) Flow cytometry analysis of GFP expression (indicative of endogenous Foxp3 transcription) by naive T cells purified from either Arrb2−/− Foxp3egfp or Arrb2+/+ Foxp3egfp mice (n = 3 for each group) and cultured under iTreg conditions (TGF-β1; 1 ng/ml) for 5 days. (d) and (e), quantification of results in (c).
Figure 5.
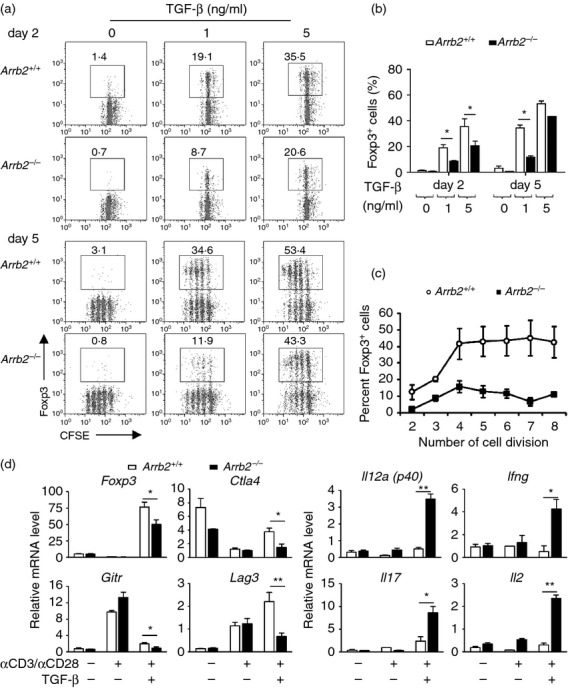
Naive (CD25− CD4+) T cells purified from spleens of Arrb2−/− or Arrb2+/+ mice were labelled with CFSE and cultured with anti-CD3 and anti-CD28, in the presence of increasing concentrations of transforming growth factor-β (TGF-β; 0, 1, 5 ng/ml). Cells were harvested for analyses on day 2 and day 5, respectively. (a) Representative flow cytometry graphs show the generation of Foxp3+ cells in Arrb2−/− or Arrb2+/+ inducible regulatory T (iTreg) cell cultures at the indicated time-points (upper panel, day 2; lower panel, day 5). Numbers adjacent to outlined areas indicate frequency of Foxp3+ cells within the CD4+ gate. (b) and (c) Quantification of results in (a). Data were shown as mean ± SEM from three independent experiments. (d) Quantitative PCR analyses of gene expressions in Arrb2−/− or Arrb2+/+ CD4+ T cells both untreated (naive) or anti-CD3 plus anti-CD28 activated in the presence or absence of 1 ng/ml TGF-β for 2 days. Gene transcript levels are calculated as the ratio of copy numbers relative to the hprt ‘housekeeping’ gene in the same sample. Data for an individual gene expression in different samples are shown as the fold change (means ± SEM, from three independent experiments) of its transcript level assessed in Arrb2+/+ cells activated in the absence of TGF-β. *P < 0·05 (two-tailed Student's t-test).
Discussion
A striking feature of MS is that as many as 80% of patients will initially present with a relatively benign relapsing–remitting disease course, suggesting that most sufferers have the capacity to limit the inflammation. It is well documented that the function of Treg cells is impaired during development of MS and recovers after therapy,18,19 indicating that Treg cells represent one of the host's defence mechanisms against inflammation.20 Consistent with this, our study showed that loss of β-arrestin 2 in mice leads to increased EAE severity with a decreased number of Treg cells in lymphoid tissues, which suggests that expression of β-arrestin 2 might correlate with the severity of EAE. However, our previous study demonstrated that β-arrestin 1 enhanced EAE development by promoting CD4+ T-cell survival, and loss of β-arrestin 1 in CD4+ T cells did not affect in vitro iTreg cell differentiation.21 In our in vitro observations, loss of β-arrestin 2 did not affect the viability of CD4+ T cells. Moreover, the generation of thymus-derived Treg cells in Arrb2−/− and that in wild-type naive mice (even at the early phase of EAE) were comparable. Hence, the impaired ability to convert Arrb2−/− CD4+ Foxp3− cells into Foxp3+ CD4+ Treg cells upon TGF-β stimulation might contribute to the reduced number of Treg cells in Arrb2−/− EAE mice. It is notable that the population of Treg cells in the CNS was relatively scarce because the ratio of Treg to effector T in CNS from Arrb2−/− EAE mice was significantly lower than that in wild-type littermates, although the frequency of Arrb2−/− Treg cells in CNS mononuclear cells did not change. We also noticed that at a specific ratio of Treg to T responder, (1 : 4 ratio of Treg : T responder in Fig. S7, see Supplementary material), partial down-regulation of the suppressive ability of Arrb2−/− Treg cells did occur in vitro. However, this was not observed at other ratios, suggesting that loss of β-arrestin 2 marginally affects the ability of Treg cells to inhibit CD4+ T-cell proliferation upon T-cell receptor stimulation in vitro. However, we cannot exclude the possibility that Arrb2−/− Treg cells might also be defective for inhibition of CD4+ T-cell proliferation or inflammatory cytokine secretion in vivo, particularly in CNS during the relapsing–remitting courses of EAE. Collectively, our results lead us to propose that the reduced number of Treg cells in Arrb2−/− mice might contribute to the increased severity of EAE.
Notably, our in vivo data also show that more β-arrestin 2-deficient CD4+ T cells appeared in the CNS compared with the wild-type controls, and that these Arrb2−/− CD4+ T cells had markedly enhanced secretion of IFN-γ and IL-17 compared with their wild-type counterparts. Though we propose here that impairment of peripherally derived Treg cell generation in Arrb2−/− mice results in sustained accumulation of these pathogenic CD4+ T cells in CNS lesions, it is conceivable that deficiency of β-arrestin 2 might also up-regulate trafficking and infiltration of Th1 and Th17 cells into the CNS, which would in turn cause tissue damage and inflammatory demyelination. Our further in vivo investigations also reveal that the compositions of CD11b+ cells, B220+ B cells, and CD8+ T cells were not altered in β-arrestin 2-deficient mice with EAE. However, the development of EAE is thought to involve an imbalance among multiple immune cells, cytokines and autoantibodies.22 To address whether the exacerbated clinical signs of EAE due to deficiency of β-arrestin 2 were related to the involvement of multiple immune cells, further studies using tissue-specific genetic deletion of β-arrestin 2 are needed.
As a cytosolic signalling mediator, β-arrestin 2 not only causes receptor desensitization and internalization but also serves as a signalling molecule in G protein-coupled receptor signal transduction. Previous studies revealed that β-arrestin 2 regulates the innate immune activation of Toll-like receptor–IL-1R signalling by interacting with tumour necrosis factor receptor-associated factor 6, and also mediates cross-talk between β2-adrenergic receptor and nuclear factor-κB by interacting with IκB.23,24 Here, we showed for the first time that β-arrestin 2 deficiency enhanced pS6 (mTOR1) activation in Treg cells without influencing AKT activation (see Supplementary material, Fig. S9). We therefore hypothesize that a β-arrestin 2-mTOR1-Foxp3 axis, together with its inhibitory effect on Foxp3 expression, might constitute a novel signalling pathway that influences iTreg differentiation.
We noticed that loss of β-arrestin 2 can result in increased airway inflammation, and more severe rheumatoid arthritis,12,25 implying that β-arrestin 2 plays a regulatory role in a broad range of inflammatory diseases. It is also reported that depletion of β-arrestin 2 leads to enhanced pro-inflammatory cytokine secretion by macrophages.23 Consistent with previous findings that Treg cells play an important role in regulating inflammation,26 our current observations underscore the role of β-arrestin 2 in the interplay between Treg cells and neuroinflammation. Our observations are also consistent with a previous finding regarding the anti-inflammatory role of β-arrestin 2 in fibroblast-like synoviocytes in an experimental arthritis model.12
It is also known that β-arrestin 2 can transduce signals from chemokine receptors, including CXCR4.25,27 Although β-arrestin 2 deficiency did not alter the expression of such receptors in splenocytes or DLNs during EAE development (data not shown), it remains possible that β-arrestin 2 may modulate the motility of CD4+ T cells. Taken together, our data show that β-arrestin 2 plays a regulatory role in multiple immune cells, particularly in Treg cells in autoimmune diseases such as MS, and that its effects are the opposite of those of β-arrestin 1. Hence, approaches to up-regulate β-arrestin 2-dependent signalling with specific therapeutic agents may be a strategy for treatment of MS in the future.
Acknowledgments
We are grateful to Robert J. Lefkowitz (Duke University Medical Center) for the Arrb2−/− mice and Dr Honglin Wang (Shanghai Jiaotong University) for the Foxp3egfp mice. We thank all members of the laboratory for sharing reagents and advice. We thank Ru Zhang (Tongji University) and Jian Zhao (Shanghai Institutes for Biological Sciences) for helpful discussions; and Ao Guo, Shichao Huang, Shunmei Xin, Hui Xu and Xianglu Zeng for technical assistance. We also thank all laboratory members for sharing reagents and advice. This research was supported by the Ministry of Science and Technology (XDA01010302), and the Chinese Academy of Sciences (2011CB946102).
Glossary
- APC
allophycocyanin
- Arrb2
β-arrestin 2
- CNS
central nervous system
- DLN
draining lymph node
- EAE
experimental autoimmune encephalomyelitis
- egfp
enhanced green fluorescent protein
- H&E
haematoxylin & eosin
- IFN-γ
interferon-γ
- IL-17
interleukin-17
- IRES
internal ribosomal entry site
- iTreg
inducible regulatory T
- MOG
myelin oligodendrocyte glycoprotein peptide
- MS
multiple sclerosis
- PE
phycoerythrin
- SPL
spleen
- TGF-β
transforming growth factor-β
- Th1
T helper type 1
- Treg cell
regulatory T cell
Disclosures
The authors declare no financial or commercial conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Frequency of Foxp3+ CD4+ natural occurring regulatory T (Treg) cells in naive Arrb2−/− or Arrb2+/+ mice.
Figure S2. Immune cell populations from central nervous system and peripheral immune organs of Arrb2+/+ and Arrb2−/− experimental autoimmune encephalomyelitis (EAE) mice during early stage of EAE (day 12–16).
Figure S3. Immune cell populations from peripheral immune organs of Arrb2+/+ and Arrb2−/− mice during late stage of experimental autoimmune encephalomyelitis (day 18–22).
Figure S4. Frequencies of effector T cells in Arrb2+/+ and Arrb2−/− experimental autoimmune encephalomyelitis (EAE) mice during early stage of EAE (day 12–16).
Figure S5. Frequencies of peripheral blood effector and regulatory T cells in Arrb2+/+ and Arrb2−/− experimental autoimmune encephalomyelitis mice during late disease stage.
Figure S6. Comparable cell viabilities in transforming growth factor-β induced Arrb2+/+ and Arrb2−/− inducible regulatory T cell cultures.
Figure S7. Mixed lymph node reaction.
Figure S8. Primer list.
Figure S9. Enhanced pS6 (mTOR1) activation in Arrb2−/− inducible regulatory T cell cultures.
References
- 1.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 2.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 3.Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol. 2002;20:101–23. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- 4.Buckner JH. Mechanisms of impaired regulation by CD4+ CD25 + FOXP3+ regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10:849–59. doi: 10.1038/nri2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Boehmer H. Mechanisms of suppression by suppressor T cells. Nat Immunol. 2005;6:338–44. doi: 10.1038/ni1180. [DOI] [PubMed] [Google Scholar]
- 6.Kumar V, Sercarz E. Induction or protection from experimental autoimmune encephalomyelitis depends on the cytokine secretion profile of TCR peptide-specific regulatory CD4 T cells. J Immunol. 1998;161:6585–91. [PubMed] [Google Scholar]
- 7.Cabbage SE, Huseby ES, Sather BD, Brabb T, Liggitt D, Goverman J. Regulatory T cells maintain long-term tolerance to myelin basic protein by inducing a novel, dynamic state of T cell tolerance. J Immunol. 2007;178:887–96. doi: 10.4049/jimmunol.178.2.887. [DOI] [PubMed] [Google Scholar]
- 8.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+ CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuchroo VK, Ohashi PS, Sartor RB, Vinuesa CG. Dysregulation of immune homeostasis in autoimmune diseases. Nat Med. 2012;18:42–7. doi: 10.1038/nm.2621. [DOI] [PubMed] [Google Scholar]
- 10.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 11.Shi Y, Feng Y, Kang J, et al. Critical regulation of CD4+ T cell survival and autoimmunity by β-arrestin 1. Nat Immunol. 2007;8:817–24. doi: 10.1038/ni1489. [DOI] [PubMed] [Google Scholar]
- 12.Li P, Cook JA, Gilkeson GS, Luttrell LM, Wang L, Borg KT, Halushka PV, Fan H. Increased expression of β-arrestin 1 and 2 in murine models of rheumatoid arthritis: isoform specific regulation of inflammation. Mol Immunol. 2011;49:64–74. doi: 10.1016/j.molimm.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Z, Huang Y, Liu Y, et al. β-Arrestin 1 modulates functions of autoimmune T cells from primary biliary cirrhosis patients. J Clin Immunol. 2011;31:346–55. doi: 10.1007/s10875-010-9492-4. [DOI] [PubMed] [Google Scholar]
- 14.Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, Lefkowitz RJ. β-Arrestin-2 regulates the development of allergic asthma. J Clin Invest. 2003;112:566–74. doi: 10.1172/JCI17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basher F, Fan H, Zingarelli B, Borg KT, Luttrell LM, Tempel GE, Halushka PV, Cook JA. β-Arrestin 2: a negative regulator of inflammatory responses in polymorphonuclear leukocytes. Int J Clin Exp Med. 2008;1:32–41. [PMC free article] [PubMed] [Google Scholar]
- 16.Yu MC, Su LL, Zou L, et al. An essential function for β-arrestin 2 in the inhibitory signaling of natural killer cells. Nat Immunol. 2008;9:898–907. doi: 10.1038/ni.1635. [DOI] [PubMed] [Google Scholar]
- 17.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178:2961–72. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 18.Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+ CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci U S A. 2005;102:6449–54. doi: 10.1073/pnas.0502187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korporal M, Haas J, Balint B, Fritzsching B, Schwarz A, Moeller S, Fritz B, Suri-Payer E, Wildemann B. Interferon β-induced restoration of regulatory T-cell function in multiple sclerosis is prompted by an increase in newly generated naive regulatory T cells. Arch Neurol. 2008;65:1434–9. doi: 10.1001/archneur.65.11.1434. [DOI] [PubMed] [Google Scholar]
- 20.Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. 2012;248:156–69. doi: 10.1111/j.1600-065X.2012.01130.x. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Wei B, Guo A, Liu C, Huang S, Du F, Fan W, Bao C, Pei G. Deficiency of β-arrestin 1 ameliorates collagen-induced arthritis with impaired TH17 cell differentiation. Proc Natl Acad Sci U S A. 2013;110:7395–400. doi: 10.1073/pnas.1221608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutton JD, Winston R, Rodman TC. Multiple sclerosis: etiological mechanisms and future directions. Exp Biol Med (Maywood) 2004;229:12–20. doi: 10.1177/153537020422900102. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2006;7:139–47. doi: 10.1038/ni1294. [DOI] [PubMed] [Google Scholar]
- 24.Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G. Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol Cell. 2004;14:303–17. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 25.Nichols HL, Saffeddine M, Theriot BS, et al. β-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc Natl Acad Sci U S A. 2012;109:16660–5. doi: 10.1073/pnas.1208881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol Rev. 2006;212:256–71. doi: 10.1111/j.0105-2896.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Cheng Z, Ma L, Pei G. β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–9. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.