

Abstract
In the present study, we have found that intestinal flora strongly influence peritoneal neutrophilic inflammatory responses to diverse stimuli, including pathogen-derived particles like zymosan and sterile irritant particles like crystals. When germ-free and flora-deficient (antibiotic-treated) mice are challenged with zymosan intraperitoneally, neutrophils are markedly impaired in their ability to extravasate from blood into the peritoneum. In contrast, in these animals, neutrophils can extravasate in response to an intraperitoneal injection of the chemokine, macrophage inflammatory protein 2. Neutrophil recruitment upon inflammatory challenge requires stimulation by microbiota through a myeloid differentiation primary response gene (88) (MyD88) -dependent pathway. MyD88 signalling is crucial during the development of the immune system but depending upon the ligand it may be dispensable at the time of the actual inflammatory challenge. Furthermore, pre-treatment of flora-deficient mice with a purified MyD88-pathway agonist is sufficient to restore neutrophil migration. In summary, this study provides insight into the role of gut microbiota in influencing acute inflammation at sites outside the gastrointestinal tract.
Keywords: Intestinal flora, neutrophils, inflammation
Introduction
The large intestinal tract of humans and other vertebrates is inhabited by numerous and diverse bacterial populations. The extent of microbial colonization is such that the number of microbial cells outnumbers the total number of cells in the human body 10-fold. The combined microbial gene set similarly exceeds the human gene complement about 150-fold.1,2 The intestinal flora plays a vital role in gut physiology. The mammalian digestive system is limited in its ability to produce all the enzymes that are required to metabolize the vast repertoire of energy substrates that are consumed and the gut flora complements the host's digestive system in maximizing their utilization. The nutritive benefits of gut flora extend to carbohydrate fermentation and absorption, lipid storage and secretion of vitamins and amino acids and absorption of minerals.3 Besides their role in digestion, intestinal flora contributes to intestinal epithelial cell growth and proliferation and development of mucosal immunity.
Mice bred in germ-free conditions are more susceptible to pathogens like Salmonella enterica,4 Listeria monocytogenes,5 Klebsiella pneumonia6 and Candida albicans.7 The role of intestinal flora in preventing enteric infections was initially attributed to its ability to prevent invasion and colonization by opportunist pathogens in the intestinal niche. However, in recent years it has become increasingly apparent that the host microbiota plays a more active role in the development and functioning of the immune system in the gastrointestinal system. Germ-free mice have anatomical defects in the gut-associated lymphoid tissue, including poorly developed Peyer's patches and isolated lymphoid follicles, fewer plasma cells and fewer intraepithelial lymphocytes.8–11 These animals also produce lower levels of antimicrobial peptides and immunoglobulin A in their gastrointestinal tract.10,12 Certain species of the microbiota, namely segmented filamentous bacteria, have been shown to induce the production of T helper type 17 cells in the small intestinal lamina propria.13 Likewise, the gut organism Bacteroides fragilis facilitates the production of inducible regulatory T cells in the gut.14 Hence, commensal microbiota are pivotal for the development of gut-associated immunity.
Recent studies have demonstrated that gut flora have more far-reaching effects on host adaptive systemic immunity. Germ-free mice have a systemic defect in the proliferation of effector CD4+ T cell numbers and exhibit a T helper type 1/type 2 imbalance.15 Mazmanian et al. showed that in the absence of intestinal flora, splenic CD4+ T cells made more interleukin-4 (IL-4) and low levels of interferon-γ, which was characteristic of a T helper type 2 response. There is much less information available as to how the gut flora influences innate immunity at sites outside the gastrointestinal tract, although commensal flora has been shown to influence bone marrow and blood neutrophils in ways that promote their phagocytosis of Streptococcus pneumoniae and Staphylococcus aureus.16 In this study, we sought to determine the contribution of intestinal flora in regulating acute neutrophilic inflammatory responses.
In acute inflammatory responses there is a rapid recruitment of neutrophils from the blood to the affected tissue site. Diverse agents including invading pathogens, injured or dead cells and other irritants like crystals may stimulate this response. These pro-inflammatory agents are sensed by tissue-resident cells like macrophages, dendritic cells and mast cells. The latter, once activated, release inflammatory mediators like histamines, prostaglandins and cytokines like interferon-γ, macrophage inflammatory protein-2 (MIP-2), tumour necrosis factor-α and IL-1. These mediators promote vasodilatation and also activate the endothelium, facilitating the transmigration of leucocytes into the affected tissue.
Neutrophils, once recruited to the site of infection, are powerful phagocytes that ingest and kill microbes by oxidative (e.g. reactive oxygen species) and non-oxidative (e.g. various proteases) mechanisms.17 The importance of neutrophil function is evident in individuals who have defects in neutrophil chemotaxis, phagocytic functions or who have neutropenia.18,19 These individuals are more prone to bacterial infections. On the other hand, microbicidal molecules released from activated and dying neutrophils can cause bystander damage to healthy tissue. The consequent cell injury and death can itself cause or aggravate disease. Accordingly, it is important to elucidate the factors controlling neutrophilic inflammation. In this study we describe the surprising finding that the gut flora influences the ability of animals to mount a systemic acute neutrophilic inflammatory response in the peritoneum and characterize the underlying basis for this observation.
Materials and methods
Mouse strains
Specific pathogen free (SPF) C57BL/6 mice and IL-1R−/− mice were purchased from The Jackson Laboratories (Bar Harbor, ME). Germ-free C57BL/6 mice were obtained from The National Gnotobiotic Rodent Resource Center, North Carolina State University Gnotobiotics Unit and Gnotobiotic Research Resource, Medical University of South Carolina. MyD88−/− mice were provided by Dr Shizuo Akira, Osaka University, Osaka, Japan or purchased from The Jackson Laboratories. RIP2−/− mice were provided by Dr Michelle Kelliher and RIG-I−/− and MDA5−/− mice were provided by Dr Kate Fitzgerald (University of Massachusetts Medical School, Worcester, MA). NOD1−/− mice were provided by Dr Grace Chen, University of Michigan, Ann Arbor, MI. For generating the tamoxifen-inducible deletion mutant mice of MyD88, we used a strategy similar to the one described previously.20 MyD88−/− mice were crossed to the whole tissue, tamoxifen-inducible Cre transgenic mice (Rosa26-Cre/ESR+/+) (provided by Dr Roger Davis, University of Massachusetts Medical School, Worcester, MA). The resultant offspring, MyD88+/− Rosa26-Cre/ESR+/− mice were crossed to the MyD88flox/flox mice (provided by Dr Robert Finberg, University of Massachusetts Medical School, Worcester, MA) to generate the MyD88−/flox Rosa26-Cre/ESR+/− (conditional knockout; cKO). Animals were housed and handled according to protocols approved by the University of Massachusetts animal care and use committee.
Peritonitis
Mice were injected intraperitoneally with 0·2 mg zymosan (Sigma-Aldrich, St Louis, MO), 0·5 mg silica crystals (Sigma-Aldrich), 0·5 mg monosodium urate crystals or 5 ng recombinant murine MIP-2 (R&D Systems, Minneapolis, MN) in 0·2 ml PBS. For the thioglycollate injections, 1 ml of 3% thioglycollate (Thermoscientific, Lenexa, KS) was used. The monosodium urate crystals were prepared as described before.21 Mice were killed by exposure to isoflourane 4–16 hr after the injection. The peritoneum was lavaged with 2 ml Dulbecco's modified Eagle's medium with 2% fetal calf serum, 3 mm EDTA and 10 U/ml heparin. The peritoneal cells were stained with antibodies against mouse Ly-6G, CD11b (both from BD Bioscience, San Jose, CA), 7/4, F4/80 (both from eBioscience, San Diego, CA). The percentage and absolute numbers of different cell types were determined by flow cytometric analysis and cell-counting beads (Life Technologies, Grand Island, NY). FACS analysis was performed using a BD Biosciences LSRII Flow cytometer and FlowJo (Tree Star, Ashland, OR) analysis software. In other experiments, cells from blood were analysed and quantified by flow cytometry. Expression of CXCR2, CD62 ligand and CD44 on neutrophils in blood was quantified using antibodies purchased from eBioscience.
Intestinal flora depletion using antibiotics
C57BL/6 and MyD88−/− mice were treated with a cocktail of broad-spectrum antibiotics in their drinking water starting from birth to the time they were used in experiments as described before.22 The antibiotic cocktail consisted of ampicillin 1 g/l, neomycin 1 g/l, metronidazole 1 g/l (Sigma-Aldrich) and vancomycin 0·5 g/l (PhytoTechnology Laboratories, Shawnee Mission, KS). The artificial aspartame sweetener, Equal (Merisant Company, Chicago, IL) was added to the water 5 g/l to make it palatable for the mice to drink. Pups received the antibiotics indirectly via lactating mothers till they were weaned. Drinking water containing the antibiotics was replaced every week.
16S rRNA quantification
DNA was isolated from colonic contents of mice by the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). The quantitative PCR primers used to amplify the bacterial 16S V2 region were sense, 5′-AGYGGCGIACGGGTGAGTAA-3′; and anti-sense, 5′-CYIACTGCTGCCTCCCGTAG-3′. Quantitative PCR primers used to amplify the housekeeping gene GAPDH were sense 5′-TGATGGGTGTGAACCACGAG-3′; and anti-sense 5′-TCAGTGTAGCCCAAGATGCC-3′. Quantitative PCR was performed using the iQ SYBR Green supermix on the CFX96 Touch Bio-Rad machine (Bio-Rad, Hercules, CA). The PCR cycling reaction used was 15 min activation step (95°C); 35 cycles of 30 seconds denaturation (95°C), 30 seconds annealing (60°), and 30 seconds extension (72°C).
Lipopolysaccharide treatment
Lipopolysaccharide (LPS) from Escherichia coli, serotype 026:B6, purified by gel-filtration chromatograph (Sigma Aldrich) was administered in the drinking water of mice at a concentration of 33 mg/l from 3 to 5 weeks of age.
Tamoxifen-induced deletion of MYD88
Tamoxifen (Sigma-Aldrich) solution was prepared in corn oil (Sigma-Aldrich) at 10 mg/ml by incubating at 37°C for 2 hr. To induce deletion of floxed genes in adult mice, tamoxifen (50 mg/kg of body weight) was administered to floxed mice by oral gavage for three alternate days. Mice were used in experiments 7 days after the last administration. For treating pups, lactating mothers were treated intraperitoneally with tamoxifen (200 mg/kg of body weight) from the day of birth for 5 consecutive days.
Quantitative PCR to detect deletion of floxed MYD88 gene
The efficiency of deletion of floxed MyD88 allele was assessed using Taqman PCR using primers and the method described previously.23 The PCR cycling reaction was performed on the C1000 Thermal Cycler (Bio-Rad).
Phagocytosis assay
Efficiency of phagocytosis by peritoneal cells was assessed as described before.24 Briefly, FITC-conjugated zymosan (0·8 mg/ml) was prepared in Dulbecco's modified Eagle's medium + 20% fetal bovine serum. Peritoneal cells plated at 0·5 million cells/well of a 48-well plate were incubated with 500 μl of the FITC-conjugated zymosan solution for 45 min at 37°C. The reaction was terminated by transferring the plate to 0°C. The uningested zymosan was removed by washing wells with Hanks’ balanced salt solution. Cells were scraped off the plate and resuspended in 2 mg/ml trypan blue to quench cell-surface-bound zymosan. In the control group, cells were incubated with zymosan at 0°C throughout the incubation. The efficiency of phagocytosis, ‘phagocytosis index’, was calculated as % of F4/80 cells that were FITC+ × MFI of F4/80 cells.
Statistical analyses
Data are reported as means ± SEM. Statistical analysis in each independent experiment was performed with an unpaired, two-tailed Student's t-test.
Results
Germ-free and flora-deficient (antibiotic-treated) mice have a defect in peritoneal neutrophilic inflammation
To investigate the role of commensal microbiota in acute inflammation, we examined the recruitment of neutrophils to various inflammatory stimuli in the peritoneal cavity in mice bred in germ-free conditions. We found that germ-free mice showed a dramatic reduction in the number of infiltrating neutrophils compared with SPF mice in the peritoneum after inflammatory stimulation. This defect in acute inflammation was observed in challenge with microbial components like zymosan, a component of yeast cell wall and thioglycollate (Fig. 1a,b), as well as with sterile ligands like silica and monosodium urate crystals (Fig. 1c,d). In subsequent experiments we focused on analysing the responses to peritoneal challenge with zymosan because this agent was easy to administer and gave strong and consistent results. Also, this zymosan-induced neutrophil infiltration is independent of IL-1, which was important for some of the experiments we described below. This phenotype of reduced inflammation observed in germ-free animals was replicated in mice treated with a cocktail of broad-spectrum antibiotics from birth to the time they were used in experiments (Day 0 to Day 45) (see Supplementary material, Fig. S1a); microbial 16S ribosomal RNA was undetectable by PCR in these animals, indicating that they had severely reduced microbial flora, as has been described by others22 (see Supplementary material, Fig. S2). Because of the limited availability of germ-free mice, most subsequent experiments were performed using flora-deficient mice.
Figure 1.
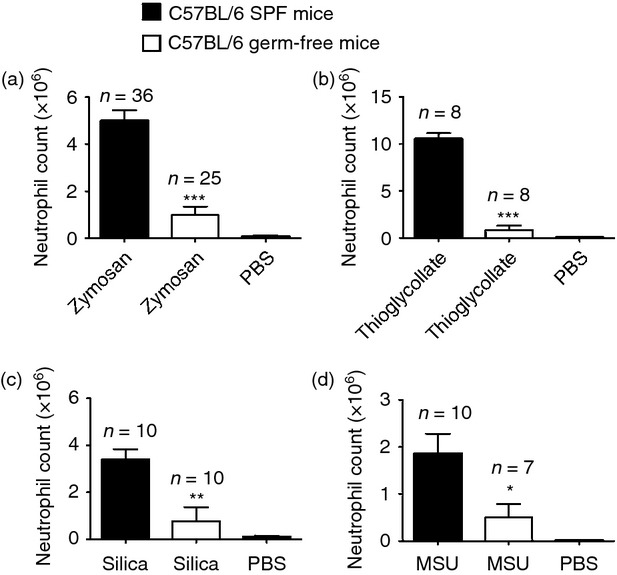
Mice lacking intestinal flora are defective in neutrophil recruitment to diverse inflammatory stimuli. Total neutrophil numbers in the peritoneum of specific pathogen-free (SPF) C57BL/6 and germ-free C57BL/6 mice 4 h after intraperiteonal injection of (a) 0·2 mg zymosan, (b) 1 ml 3% thioglycollate, (c) 0·5 mg silica crystals, and (d) 0·5 mg monosodium urate (MSU) crystals. Numbers of neutrophils were determined by multiplying total cell number by percentage of Ly-6G+ 7/4+. All data are combined results of three or more experiments and represented as mean ± SEM. n = Total number of mice used per datum group. In all panels PBS group represents SPF C57BL/6 mice challenged with PBS and lavaged after 4 hr. *P = 0·04, **P = 0·0025, ***P < 0·0001 versus SPF animals.
The lowered numbers of neutrophils observed in the peritoneum in flora-deficient mice after 4 hr was not the result of delayed migration of neutrophils, because these mice exhibited defective neutrophil migration even 16 hr after inflammatory challenge (see Supplementary material, Fig. S1b). We sought to examine the precise step at which microbiota regulate neutrophil activation and migration. Neutrophils originate and mature in the bone marrow.17 Mature neutrophils subsequently leave the bone marrow and circulate in the bloodstream. Cytokines generated at the site of inflammation stimulate an increase in production of neutrophils in the bone marrow and their release into the bloodstream and chemotactic factors promote their subsequent migration into the inflamed area. We observed that in the absence of an inflammatory challenge, there is no statistically significant reduction in the number of peripheral blood neutrophils in the flora-deficient mice (Fig. 2a). Moreover, when flora-deficient mice were challenged with zymosan, the total blood count of neutrophils was significantly higher than that of their SPF counterparts (Fig. 2c). There was no defect in the maturation of neutrophils in flora-deficient mice before or after an inflammatory stimulus, because we observed similar percentages of mature neutrophils in the periphery as in the SPF animals (Fig. 2b,d). The increased number of peripheral neutrophils in flora-deficient mice after zymosan challenge is presumably the result of a larger pool of marginated cells in the flora-deficient mice compared with control mice, which is then rapidly mobilized upon challenge with zymosan. These data indicated that the defective recruitment of neutrophils in the peritoneum is not the result of lower production of neutrophils in the flora-deficient mice. This suggested a role for intestinal flora in influencing the extravasation of neutrophils from the bloodstream into the inflamed tissue site.
Figure 2.
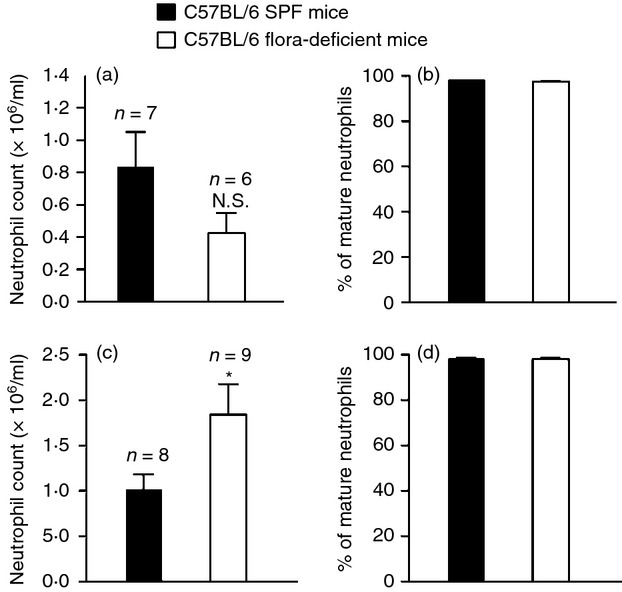
Flora-deficient mice appear to have a specific defect in the recruitment of neutrophils from blood into the inflamed tissue. (a, c) Total number of neutrophils in 1 ml blood in (a) unstimulated specific pathogen-free (SPF) C57BL/6 and flora-deficient C57BL/6 mice and in (c) 1 ml blood of mice injected with 0·2 mg zymosan. (b, d) % of mature neutrophils in SPF C57BL/6 and flora-deficient C57BL/6 mice as assessed by their expression of Ly6G and CD11b by FACS analysis in (b) peripheral blood of unstimulated mice or (d) peripheral blood of mice stimulated with 0·2 mg zymosan. The % of mature neutrophils was calculated as total number of CD11b+ Ly6Ghi cells/total number of CD11b+ Ly6Glo and CD11b+ Ly6Ghi cells × 100. All data are combined results of three or more experiments and are represented as mean ± SEM. n = Total number of mice used per datum group. *P = 0·04, Not significant (N.S.) versus SPF animals.
In the peritoneum, resident macrophages have been shown to sense pro-inflammatory stimuli and produce cytokines that initiate inflammation.25 Therefore, we quantified the numbers of resident macrophages (CD11b+ F4/80+ cells) in the peritoneum of SPF and flora-deficient mice and found that they were similar (see Supplementary material, Fig. S3a). Moreover, peritoneal cells from flora-deficient mice were as efficient as those from SPF mice in their phagocytosis of zymosan (see Supplementary material, Fig. S3b), which was consistent with previous reports.26
Flora-deficient mice do not have a generalized defect in either endothelial or neutrophil function
Neutrophil extravasation through blood vessels into tissues is facilitated by cell adhesion molecules expressed by neutrophils and the endothelium. Neutrophils in the blood of flora-deficient animals showed similar or (higher) percentages and mean fluorescence intensity of expression of cell adhesion molecules like CD44, CD62 ligand, and the chemokine receptor, chemokine (C-X-C motif) receptor 2 (CXCR2) (Fig. 3a–f).
Figure 3.

Neutrophils in flora-deficient mice are not defective in their expression of cell adhesion molecules and can extravasate in response to an intraperitoneal challenge with macrophage inflammatory protein 2 (MIP-2). (a–c) % of neutrophils in blood from zymosan-stimulated, specific pathogen-free (SPF) or flora-deficient C57BL/6 mice expressing (a) CXCR2, (b) CD44, (c) CD62 ligand. (d, e) Mean fluorescence intensity (MFI) of neutrophils in blood from zymosan-stimulated SPF or flora-deficient C57BL/6 mice for their expression of (d) CXCR2, (e) CD44, (f) CD62L. (g) Total number of neutrophils in the peritoneum of SPF C57BL/6 and flora-deficient C57BL/6 mice after intraperitoneal challenge with 5 ng of recombinant murine MIP-2. All data are combined results of three or more experiments and represented as mean ± SEM. n = Total number of mice used per datum group. *P = 0·0143, ***P = 0·0002, Not significant (N.S.) versus SPF animals.
We next examined if flora-deficient mice were able to recruit neutrophils when treated with MIP-2, a chemotactic factor for neutrophils. We injected the mice intraperitoneally with purified recombinant MIP-2 protein. We found that these mice were able to mount a neutrophil response in the peritoneum as well as the SPF mice (Fig. 3g). The response to MIP-2 in flora-deficient mice was intact throughout the dose–response curve and even in limiting amounts. Our data show that normal neutrophil migration to MIP-2 in flora-deficient mice is consistent with the observation that neutrophils from these mice had normal levels of CXCR2, as CXCR2 is the physiological receptor for MIP-2. This indicated that mice lacking microbial flora do not have a generalized defect in the endothelial vasculature and also that neutrophils from these mice are functionally capable of migrating to the inflamed tissue upon receiving the appropriate signals.
Intestinal flora mediate their effects on neutrophilic inflammation through the MyD88-signalling pathway
We hypothesized that the microbiota mediates its effects on inflammatory responses by activating pattern recognition receptor-signalling pathways. To test this hypothesis, we analysed mice deficient in the known pattern recognition receptor pathways and examined their ability to mount a neutrophil response to an intraperitoneal zymosan challenge. Receptor interacting protein-2 (RIP-2) knockout mice, which are defective in nucleotide-binding, oligomerization domain-containing protein-1 (NOD1) or NOD2 signalling, were able to respond normally to zymosan (Fig. 4a). Similar results were obtained in mitochondrial antiviral signalling (MAVS) knockout mice, which were defective in retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) signalling. Also, normal neutrophil recruitment was observed in mice lacking Caspase 1, NOD-like receptor family, pyrin domain containing 3 (NLRP3), or Apoptosis-associated speck-like protein (ASC), which are defective in inflammasome activation (Fig. 4a). However, MyD88 knockout mice showed a markedly reduced recruitment of neutrophils following zymosan stimulation (Fig. 4b), similar to that observed in the flora-deficient mice. Like the flora-deficient animals, MyD88 knockout mice did not have a statistically significant reduction in the number of neutrophils in the blood under basal conditions (see Supplementary material Fig. S4a). Furthermore, on challenge with zymosan, neutrophils in the bloodstream of Myd88−/− mice outnumbered those in wild-type mice (see Supplementary material Fig. S4b), mimicking the phenotype that was observed in mice lacking microbial flora.
Figure 4.
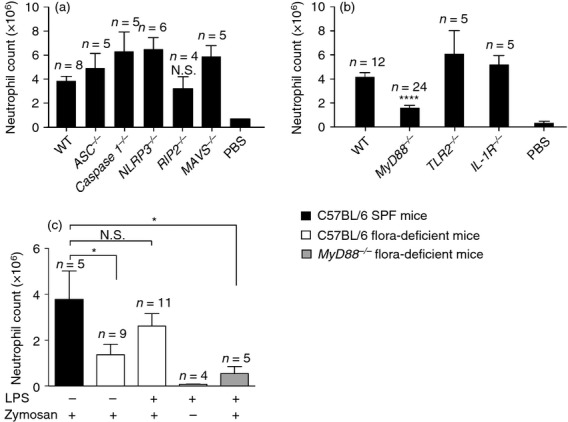
Zymosan-induced neutrophil recruitment requires MyD88 signalling and lipopolysaccharide (LPS) pre-treatment in flora-deficient mice restores neutrophilic inflammation in a process dependent upon MyD88 signalling. (a,b) Total neutrophil numbers in the peritoneum of (a) C57BL/6 (wild-type; WT), ASC−/−, Caspase 1−/−, NLRP3−/−, RIP2−/− and MAVS−/− mice and (b) C57BL/6 (WT), MyD88−/−, TLR2−/− and IL-1R−/− mice. Mice were injected with 0·2 mg zymosan and lavaged after 4 hr. (c) Total neutrophil numbers after zymosan injection in the peritoneum of specific pathogen-free (SPF) C57BL/6, flora-deficient C57BL/6 mice, and flora-deficient MyD88−/− mice in the presence or absence of LPS pre-stimulation. LPS from Escherichia coli was administered in the drinking water of mice at a concentration of 33 mg/l from 3 to 5 weeks of age. At 7 weeks of age, mice injected with 0·2 mg zymosan and lavaged after 4 hr. In one group, flora-deficient mice were pre-treated with LPS in the drinking water but did not receive zymosan injection. Numbers of neutrophils were determined by multiplying total cell number by percentage of Ly-6G+ 7/4+ cells. All data are combined results of two or more experiments and represented as mean ± SEM. n = Total number of mice used per datum group. The PBS group in (a, b) represents WT mice challenged with PBS and lavaged after 4 hr. *P = 0·03, ****P < 0·0001, Not significant (N.S.) versus WT animals.
MyD88 is an adaptor protein for most Toll-like receptors (TLR) and some cytokine receptors, most notably the IL-1 receptor (IL-1R). IL-1 is a pro-inflammatory cytokine that plays a key role in recruiting neutrophils to sites of inflammation in response to some inflammatory stimuli. However, we had previously shown that the neutrophilic inflammatory response to zymosan does not require the IL-1R and we confirmed again that this was the case (Fig. 4b). TLR2 has been reported to be one of the receptors for zymosan and it was possible that this was why MyD88 was required for the inflammatory response to this agent.27 However, we found that TLR2-deficient mice had a normal zymosan-induced infiltration of neutrophils in the peritoneum (Fig. 4b). This is not surprising because the major receptor for zymosan is thought to be Dectin-1, which does not signal though MyD88.28 Therefore the requirement for Myd88 in the response to zymosan was not as an adaptor protein for TLR2 or IL-1R signalling.
LPS treatment working through MyD88 restores zymosan-induced peritoneal recruitment of neutrophils in flora-deficient mice
We hypothesized that microbial flora was functioning in our system as a source of pathogen-associated molecular patterns (PAMPs) that stimulated the TLR–MyD88 pathway in ways that made the host responsive to the pro-inflammatory stimuli. This argument was supported by our observation that when mice were treated with antibiotics starting from birth for 45 days, they had lowered neutrophil migration, but 6-week-old mice treated with antibiotics for the same duration (45 days) did not show a similar defect in neutrophil migration (data not shown). This finding suggested that initial exposure to microbes or microbial ligands might be sufficient to prime neutrophil responses. To test this hypothesis, we sought to determine if MyD88 activation by a purified microbial ligand is sufficient to restore neutrophilic inflammation to zymosan in flora-deficient mice. We added pure LPS from E. coli into the drinking water of mice from 3 to 5 weeks of age in addition to the antibiotic cocktail. We found that flora-deficient mice, which received LPS for 2 weeks, were able to respond to zymosan as well as their SPF counterparts (Fig. 4c). On the other hand, flora-deficient MyD88 knockout mice did not show this restoration in inflammation on LPS administration (Fig. 4c). This shows that MyD88 is required for the downstream signalling initiated by LPS, which enables acute inflammation.
MyD88 is essential for pre-conditioning of the inflammatory response before zymosan challenge
We next sought to determine whether MyD88 was needed during the elicitation of the inflammatory response or was needed earlier to somehow condition the innate immune response so as to be responsive to the pro-inflammatory stimulus. We observed that intestinal flora influences acute inflammation during the initial development of the mouse immune system because adult 6-week-old mice treated with antibiotics did not show a defect in neutrophil migration (data not shown), unlike animals treated with antibiotics right from birth. Hence, we hypothesized that the expression of MyD88 in tissues is essential during immune development for commensal flora-induced priming but the presence of MyD88 is dispensable during the actual inflammatory challenge. To test this hypothesis, we used the MyD88 flox/− ROSA26-Cre/ESR+/− (cKO) mice20 to conditionally eliminate MyD88 just before challenge with zymosan. In these mice, one allele of the gene had been deleted from the germline while the other could be inducibly deleted globally by the administration of tamoxifen. Mice were treated with tamoxifen for three alternate days and challenged with zymosan a week after the last tamoxifen injection. Therefore, in these mice MyD88 was reduced at the time of zymosan injection, but present during the maturation of the immune system. Upon administration of tamoxifen, MyD88 was deleted as assessed by quantitative PCR, as described previously23 (see Supplementary material, Table S1). The cKO mice treated with tamoxifen (1 week before zymosan challenge) responded as well as untreated mice to zymosan-induced peritonitis (Fig. 5a). These results showed that the presence of MyD88 is not essential for the signalling initiated by zymosan. While the deletion of MyD88 was partial in these animals, they showed reduced neutrophil recruitment to LPS, confirming the role of the TLR4–MyD88 pathway in detecting LPS and also validating that the deletion was sufficient to impair responses (Fig. 5b). In contrast, tamoxifen treatment of wild-type mice did not impair responses (data not shown). On the other hand, when cKO mice when treated with tamoxifen from Day 0 of birth, these mice exhibited reduced neutrophil recruitment to zymosan as compared with untreated mice (Fig. 5c). These results supported our hypothesis that for inflammatory ligands like zymosan, MyD88 is required during the pre-challenge phase for activation of immune cells but is dispensable during the actual inflammatory challenge.
Figure 5.
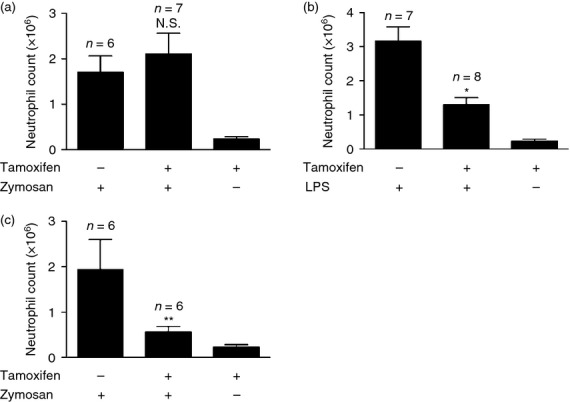
MyD88 is required during the conditioning of the immune system for a subsequent zymosan challenge rather than during the actual zymosan challenge. (a,b) Total neutrophil numbers in the peritoneum of MyD88−/flox Rosa26-Cre/ESR+/− (conditional knockout; cKO) which have been treated with tamoxifen or corn oil (vehicle control) 1 week before challenge with (a) 0·2 mg zymosan and (b) 100 ng lipopolysaccharide (LPS). (c) Total neutrophil numbers in the peritoneum of MyD88−/flox Rosa26-Cre/ESR+/− (cKO) which have been treated with tamoxifen or corn oil (vehicle control) starting from birth and challenged with 0·2 mg zymosan at 7 weeks of age. Mice were lavaged at 4 hr after injection with zymosan or LPS and the numbers of neutrophils in the peritoneum was quantified by flow cytometry. All data are combined results of two experiments and represented as mean ± SEM. n = Total number of mice used per datum group. The PBS group represents cKO mice treated with tamoxifen and challenged with PBS and lavaged after 4 hr. *P = 0·04, **P = 0·004, Not significant (N.S.) versus cKO animals that were treated with vehicle control and injected with zymosan or LPS.
Discussion
One of the major findings of this study is that for neutrophil-mediated acute inflammation to several pro-inflammatory agents, the immune system needs to be previously stimulated by intestinal flora in a MyD88-dependent fashion. This stimulation enables the host to mount a neutrophil response to future inflammatory insults. We have shown that germ-free and flora-deficient mice are defective in neutrophil migration to a number of different microbial and sterile inflammatory ligands. This defect can be corrected by supplementing the drinking water with LPS, a TLR4–MyD88 agonist, before challenge with the inflammatory agent. Furthermore, pre-treatment of flora-deficient MyD88 knockout mice with LPS failed to restore neutrophilic infiltration, showing that LPS specifically acts through MyD88 to prime the immune system. Presumably other PAMPs that stimulate MyD88–TLRs would have similar effects, although this has not yet been tested. There is some evidence that PAMPs derived from intestinal flora are present systemically in the mammalian body under physiological conditions.29,30 These ligands presumably translocate into the circulation via the intestinal epithelium. In a similar fashion, we hypothesize that ligands derived from gut flora, such as LPS (TLR4–MyD88), bacterial DNA (TLR9–MyD88), peptidoglycan (TLR2–MyD88) as well as others, activate MyD88 signalling that then enables systemic neutrophilic inflammatory responses.
A previous report published by our laboratory had shown that MyD88 knockout mice do not show a defect in zymosan-induced neutrophil migration.31 The basis for this discrepancy is unclear. It is possible that this difference was the result of the extent of backcrossing of the MyD88-deficient mice; the mice in the present study were fully backcrossed onto the B6 background whereas those in the earlier study were not. Alternatively, it is possible that in the earlier study the mice had been exposed to some microbial products that were able to condition the animals for responses through a non-MyD88-dependent pathway.
While there is a clear role for MyD88 in the ability of conventional mice to mount neutrophilic inflammation to zymosan, we found that several other innate immune signalling pathways were not required for this response. Although Clarke et al. have reported that commensal bacteria prime neutrophils via NOD1 signalling in ways that enhance their phagocytic potential to various bacteria,16 we found that RIP2 knockout mice did not show reduced inflammation to zymosan. Since RIP2 is required for NOD1/2 signalling, this finding argued against a role for either NOD1 or NOD2 in mediating a gut flora-induced effect in our system.32 Therefore, NOD1/2 signalling may be important for phagocytosis but is not needed for neutrophilic inflammation to this agent. Similarly, we found no contribution of the inflammasome components (NLRP3/ASC/caspase 1) or the RNA-sensing RIG-I like receptors in mediating zymosan-induced inflammation. Hence, we show that intestinal flora affect the ability of the immune system to mount neutrophilic inflammation via the MyD88 pathway.
To examine when the MyD88 pathway was required, we took advantage of the ROSA26-Cre system, in which the MyD88 gene could be temporally deleted by the addition of tamoxifen. We showed that for zymosan-induced peritonitis, the presence of MyD88 was not required at the time of challenge. This eliminates the possibility that zymosan needs to signal through MyD88 via TLR2 or IL-1R or any other MyD88-dependent receptor. These data therefore, make a strong case for the necessity of priming by intestinal flora-induced MyD88 activation for zymosan-induced neutrophil migration, before the actual zymosan challenge. Hence a significant finding of this study is that although the MyD88 pathway is essential for creating an innate immune system that is poised to respond to inflammatory agent, this pathway is not needed at the elicitation phase of an inflammatory response (unless of course the pro-inflammatory stimulus was using MyD88-dependent receptors such as TLRs).
An implication of our study is that the set point of the naive (i.e. never exposed to microbes) innate immune system may be anti-inflammatory for many stimuli. However, in conventionally reared mice the immune system is perturbed by exposure to microbial flora in ways that alter the cytokines that are made. As part of this process MyD88-dependent pattern recognition receptor signalling by microbial flora appears to alter this set point in ways that promote inflammatory responses.
In summary, we postulate that TLR ligands derived from the intestinal flora constitutively enter the blood and tissues. Here, they prime tissue-resident cells via MyD88 signalling, so that they provide appropriate stimulatory signals that condition the innate immune system to be able to respond to future inflammatory insults in ways that promote neutrophil migration into tissue sites. This study provides insights on how intestinal flora affects systemic immunity and specifically innate responses against microbial and sterile agents.
Acknowledgments
We thank Frederich Cruz, Jeff Colbert, Sharlene Hubbard and Diego Farfan for technical assistance, and Hajime Kono for assistance in designing the experiments. We thank Maureen Bower and Ashley Weaver, Gnotobiotic Core of the Center for Gastrointestinal Biology and Disease, for assistance with experiments using germ-free mice. Support for the Center for Gastrointestinal Biology and Disease is provided by National Institutes of Health (NIH) grant P30 DK034987. This work was supported by grants to K.L.R from the NIH and Diabetes Endocrinology Research Center.
Glossary
- ASC
apoptosis-associated speck-like protein
- CD11b
cluster of differentiation molecule 11b
- cKO
conditional knockout
- CXCR2
chemokine (C-X-C motif) receptor 2
- IL-10
interleukin-10
- IL-1R
interleukin-1 receptor
- LPS
lipopolysaccharide
- MAVS
mitochondrial antiviral signalling
- MDA5
melanoma differentiation-associated gene 5
- MIP-2
macrophage inflammatory protein 2
- NLRP3
NOD-like receptor family, pyrin domain-containing 3
- NOD1
nucleotide-binding, oligomerization domain-containing protein-1
- RIG-I
retinoic acid-inducible gene-I
- RIP-2
receptor interacting protein-2
- SPF
specific pathogen-free
- TLR
Toll-like receptor
Conflicts of interest
The authors declare no financial or commercial conflicts of interests.
Disclosures
The authors disclose no financial or commercial conflicts of interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Flora-deficient mice (antibiotic-treated mice) are defective in neutrophil recruitment to inflammatory stimuli. (a, b) Total neutrophil numbers in the peritoneum of specific pathogen-free (SPF) C57BL/6 and flora-deficient C57BL/6 mice after intraperitoneal injection of 0·2 mg zymosan and lavaged after (a) 4 hr. (b) 16 hr after injection. Flora-deficient mice were given a cocktail of antibiotics (ampicillin, neomycin, vancomycin and metronidazole) from birth through to the day of experimental use. All data are combined results of two or more experiments and represented as mean ± SEM. In both panels PBS group represents SPF C57BL/6 mice challenged with PBS and lavaged after 4 hr or 16 hr. **P = 0·008 versus SPF animals.
Figure S2. Quantitative PCR to determine the efficiency of antibiotic treatment. (a) The bacterial load in the colonic contents of specific pathogen-free versus flora-deficient mice was quantified by determining the number of copies of bacterial 16S ribosomal RNA by quantitative PCR.
Figure S3. Flora-deficient mice do not have a defect in either the numbers of resident peritoneal macrophages or their capacity to phagocytose zymosan. (a) Total number of resident peritoneal macrophages in unstimulated specific pathogen-free (SPF) C57BL/6 and flora-deficient C57BL/6 mice. Numbers of resident macrophages were determined by multiplying total cell number by percentage of CD11b+ F4/80+ cells. (b) Efficiency of phagocytosis of zymosan by resident peritoneal macrophages isolated from unstimulated SPF C57BL/6 and flora-deficient C57BL/6 mice. Peritoneal cells were incubated with FITC-conjugated zymosan solution for 45 min at 37°C. The reaction was terminated by transferring the plate to 4°C. In the control group, cells were incubated with zymosan at 0°C throughout the incubation. After washing unbound zymosan, cells were scraped off the plate and resuspended in trypan blue to quench cell surface-bound zymosan. The efficiency of phagocytosis was termed as ‘phagocytosis index,’ which was calculated as % of macrophages that have taken up zymosan × mean fluorescence intensity (MFI) of macrophages. n = Total number of mice used per datum group. All data are combined results of two or more experiments and are represented as mean ± SEM. Not significant (N.S.) versus SPF animals.
Figure S4. Neutrophil numbers in zymosan-injected MyD88−/− or control mice. (a, b) Total number of neutrophils in 1 ml blood (a) in unstimulated wild-type (WT) and MyD88−/− mice and (b) in mice after 4 hr of zymosan challenge. All data are combined results of two or more experiments and are represented as mean ± SEM. n = total number of mice used per datum group. Not significant (N.S.) versus SPF animals.
Table S1. Quantitative PCR to determine the amount of residual floxed MyD88 after treatment with tamoxifen. DNA was isolated from cells obtained from the blood and peritoneum after zymosan challenge. ΔΔCt is the difference of the normalized threshold-cycle number (Ct) between cell sample obtained from tamoxifen-treated Myd88flox/− Rosa26-Cre/ESR+/− mice and that of corn oil-treated (vehicle control) Myd88flox/− Rosa26-Cre/ESR+/− mice. The amount of flox allele of each sample was normalized to β-actin. Residual MyD88flox allele was quantified as 2−ΔΔCt. % of deletion was calculated as (1–2−ΔΔCt) × 100.
References
- 1.Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–19. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 2.Qin J, Li R, Raes J, Arumugam M, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooper LV, Midtvedt T, Gordon JI. How host–microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 4.Collins FM, Carter PB. Growth of salmonellae in orally infected germfree mice. Infect Immun. 1978;21:41–7. doi: 10.1128/iai.21.1.41-47.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inagaki H, Suzuki T, Nomoto K, Yoshikai Y. Increased susceptibility to primary infection with Listeria monocytogenes in germfree mice may be due to lack of accumulation of l-selectin+ CD44+ T cells in sites of inflammation. Infect Immun. 1996;64:3280–7. doi: 10.1128/iai.64.8.3280-3287.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fagundes CT, Amaral FA, Vieira AT, et al. Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. J Immunol. 2012;188:1411–20. doi: 10.4049/jimmunol.1101682. [DOI] [PubMed] [Google Scholar]
- 7.Phillips AW, Balish E. Growth and invasiveness of Candida albicans in the germ-free and conventional mouse after oral challenge. Appl Microbiol. 1966;14:737–41. doi: 10.1128/am.14.5.737-741.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–10. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 10.Glaister JR. Factors affecting the lymphoid cells in the small intestinal epithelium of the mouse. Int Arch Allergy Appl Immunol. 1973;45:719–30. doi: 10.1159/000231071. [DOI] [PubMed] [Google Scholar]
- 11.Gordon HA. Morphological and physiological characterization of germfree life. Ann N Y Acad Sci. 1959;78:208–20. doi: 10.1111/j.1749-6632.1959.tb53104.x. [DOI] [PubMed] [Google Scholar]
- 12.Moreau MC, Ducluzeau R, Guy-Grand D, Muller MC. Increase in the population of duodenal immunoglobulin A plasmocytes in axenic mice associated with different living or dead bacterial strains of intestinal origin. Infect Immun. 1978;21:532–9. doi: 10.1128/iai.21.2.532-539.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107:12204–9. doi: 10.1073/pnas.0909122107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–18. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16:228–31. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–89. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 18.Frendeus B, Godaly G, Hang L, Karpman D, Lundstedt AC, Svanborg C. Interleukin 8 receptor deficiency confers susceptibility to acute experimental pyelonephritis and may have a human counterpart. J Exp Med. 2000;192:881–90. doi: 10.1084/jem.192.6.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews T, Sullivan KE. Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev. 2003;16:597–621. doi: 10.1128/CMR.16.4.597-621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahman AH, Zhang R, Blosser CD, Hou B, Defranco AL, Maltzman JS, Wherry EJ, Turka LA. Antiviral memory CD8 T-cell differentiation, maintenance, and secondary expansion occur independently of MyD88. Blood. 2011;117:3123–30. doi: 10.1182/blood-2010-11-318485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–71. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–82. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ragsdale RL, Grasso RJ. An improved spectrofluorometric assay for quantitating yeast phagocytosis in cultures of murine peritoneal macrophages. J Immunol Methods. 1989;123:259–67. doi: 10.1016/0022-1759(89)90230-5. [DOI] [PubMed] [Google Scholar]
- 25.Kono H, Karmarkar D, Iwakura Y, Rock KL. Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol. 2010;184:4470–8. doi: 10.4049/jimmunol.0902485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morland B, Smievoll AI, Midtvedt T. Comparison of peritoneal macrophages from germfree and conventional mice. Infect Immun. 1979;26:1129–36. doi: 10.1128/iai.26.3.1129-1136.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–15. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 28.Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36–7. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 29.Krueger JM, Karnovsky ML, Martin SA, Pappenheimer JR, Walter J, Biemann K. Peptidoglycans as promoters of slow-wave sleep. II. Somnogenic and pyrogenic activities of some naturally occurring muramyl peptides; correlations with mass spectrometric structure determination. J Biol Chem. 1984;259:12659–62. [PubMed] [Google Scholar]
- 30.Sartor RB, Bond TM, Schwab JH. Systemic uptake and intestinal inflammatory effects of luminal bacterial cell wall polymers in rats with acute colonic injury. Infect Immun. 1988;56:2101–8. doi: 10.1128/iai.56.8.2101-2108.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, Medzhitov R, Flavell RA. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–9. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.