

Abstract
The glomerular basement membrane (GBM) is the central, non-cellular layer of the glomerular filtration barrier that is situated between the two cellular components – fenestrated endothelial cells and interdigitated podocyte foot processes. The GBM is composed primarily of four extracellular matrix macromolecules – laminin-521, type IV collagen α3α4α5, the heparan sulfate proteoglycan agrin, and nidogen – that produce an interwoven meshwork thought to impart both size- and charge-selective properties. Although the composition and biochemical nature of the GBM have been known for a long time, the functional importance of the GBM vs. podocytes and endothelial cells for establishing the glomerular filtration barrier to albumin is still debated. Together with mouse genetics studies, the discoveries of four human mutations in GBM components in two inherited kidney disorders, Alport syndrome and Pierson syndrome, support essential roles for the GBM in glomerular permselectivity. Here we explain in detail the proposed mechanisms whereby the GBM can serve as the major albumin barrier and discuss possible approaches to circumvent GBM defects associated with loss of permselectivity.
Introduction
The glomerular basement membrane (GBM) is a thin (250 to 400 nm) meshwork of extracellular matrix proteins that is an integral part of the glomerular filtration barrier. Most of the GBM is situated between two cellular layers—glomerular endothelial cells and podocytes—in the peripheral capillary wall (Figure 1A); the remaining GBM segments lie between mesangial cells and podocytes at the bases of the capillary loops.1 The GBM both provides structural support for the glomerular capillaries and harbors ligands for receptors on the surface of the adjacent endothelial cells, podocytes, and mesangial cells.2,3 Importantly, the GBM also contributes to glomerular permselectivity; as the second layer of the capillary wall that is encountered by filtrate, the GBM restricts the passage of plasma proteins across the glomerular filtration barrier. In support of this, among the nine major proteins found in the GBM, mutations in four of them are known to cause human kidney diseases4,5 (Alport syndrome and Pierson syndrome) that include proteinuria, the leakage of valuable plasma protein, most of which is albumin, into the urine.
Figure 1. The components of glomerular basement membrane (GBM) and known alterations in Pierson and Alport syndromes.
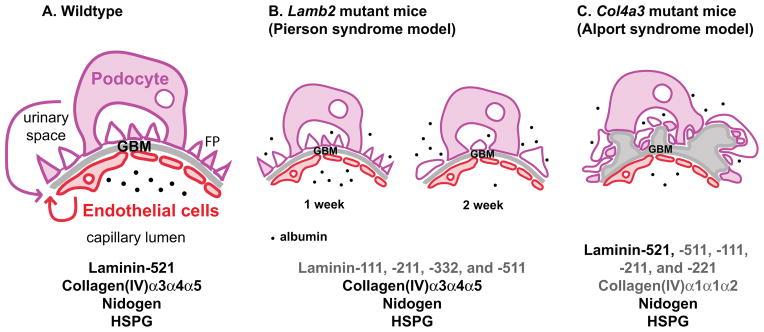
(A) The normal GBM is composed of laminin-521 (α5β2γ1), type IV collagen α3α4α5, nidogen and heparan sulfate proteoglycan (HSPG; primarily agrin). Podocytes and endothelial cells each contribute at least a subset of these components to the GBM (arrows; see text for details). Most of the plasma albumin (black dots) is restricted to the capillary lumen. FP, foot processes. (B and C) Mutations in two GBM components result in albuminuria both in human patients and in mouse models. (B) The GBM of Lamb2 knockout mice lacks laminin-521 and instead contains ectopic laminins (shown in dark gray text), such as laminin-111, -211, -332, and -511. However, the laminin network made of these ectopic laminins is defective, leading to increased passage of plasma protein across the barrier. At 1 week, Lamb2 mutant mice have proteinuria due to the defective GBM, but without podocyte foot process effacement. At 2 weeks, podocyte abnormalities can be detected, followed by increasing proteinuria and widespread effacement. These results indicate that proteinuria precedes podocyte abnormalities in Lamb2 mutant mice, highlighting the importance of the GBM as a barrier to plasma protein. (C) The GBM of Col4a3 mutant mice lacks the collagen α3α4α5(IV) network. Although there is increased deposition of collagen α1α1α2(IV) as a compensatory mechanism, the resulting GBM becomes split and thickened and accumulates multiple ectopic laminins (shown in dark gray text). Eventually there is increased loss of plasma protein across the filtration barrier and proteinuria.
While mutations affecting GBM components are important causes of kidney disease, environmental changes that affect the glomerulus can also lead to alterations in the composition and structure of the GBM. Diabetic nephropathy (DN) is one example in which the GBM is adversely affected by the microenvironment.6 DN is becoming more and more prevalent due to the worldwide increases in obesity and type II diabetes. About forty percent of diabetics develop diabetic nephropathy, which then leads to more patients with chronic kidney disease in need of dialysis.7
Although it is clear that proteinuria and renal failure originate from both genetic and environmental factors, in all but a few cases it is very difficult to clearly define a genetic component. Much research has focused on the cellular components of the glomerulus—the podocytes, endothelial cells, and mesangial cells—because they can actively respond to genetic and environmental changes by producing gene products and cell signaling molecules. However, changes in these cells can also give rise to changes in the GBM, which can secondarily affect the properties and behavior of the neighboring cells through matrix-to-cell (outside-in) signaling events. Similarly, primary changes in the GBM may exert functionally important effects on the neighboring podocytes, endothelial, and mesangial cells and thereby impact glomerular filtration.
Whether and how the GBM contributes to the establishment and function of the glomerular filtration barrier to protein have been debated for several decades.8 Recent findings gleaned from genetic and physiological studies have provided a better view of how the GBM could function as a barrier. Further understanding the mechanisms in various disease models could help in the design of therapeutics that could prevent or reverse proteinuria by impacting GBM structure and function.
This review focuses mainly on the mechanisms of how the GBM functions to establish and maintain the glomerular filtration barrier. From genetic and biochemical studies in mice and humans, it is evident that the GBM is crucial to prevent the leakage of plasma proteins into the urine. We emphasize the critical role of the GBM as a permselective barrier that can be altered in different ways by genetic defects that cause kidney disease.
The components of GBM and their potential for contributing to the glomerular filtration barrier
In order to understand how the GBM might contribute to permselectivity, it is important to define its composition and to understand the properties of its major components. Like all basement membranes, the GBM is composed of laminin, type IV collagen, heparan sulfate proteoglycan, and nidogen.9 Components of the GBM are synthesized by both podocytes and endothelial cells,10 and during glomerulogenesis there is a fusion of separate podocyte- and endothelium-derived basement membranes to form the immature GBM. These features are together responsible at least in part for the GBM being thicker than most other basement membranes, as both cell layers synthesize ECM components and secrete them into the extracellular space between them. Furthermore, one can infer that changes in either podocytes or endothelial cells can result in altered GBM composition—and vice versa—possibly affecting glomerular filtration barrier function. In this context, podocytes, endothelial cells, and the GBM can be viewed as interconnected; this is evident not only through the obvious direct physical contacts within cell layers and between cells and the GBM, but also across the GBM through both cell-matrix-cell connections and cell-cell communication/signaling via diffusible factors. One excellent example of the latter is the VEGF signaling axis, in which podocyte-derived VEGF is crucial for endothelial cell homeostasis.11
Historically, the GBM was considered by some to be a “crude prefilter”, while slit diaphragms between podocyte foot processes were thought to be responsible for the bulk of glomerular permselectivity.12 However, as proposed by Farquhar and Palade, functional and physiological analyses of the glomerular filtration barrier using various tracers revealed the importance of the GBM as a size- and charge-selective filtration barrier.13–15 When neutral tracers of various sizes were intravenously injected, they found that molecules larger than albumin were restricted to the inside of the glomerular capillary loops and were impaired from traversing the GBM. Likewise, when neutral, anionic and cationic tracers were infused, negatively charged molecules had the most difficulty crossing the GBM.15 From these studies, the function and necessity of the components of the GBM were inferred based on their biochemical properties. However, the most direct evidence as to whether each component of the GBM is essential for establishing the filtration barrier was provided by mouse genetic studies and discovery of human mutations that cause defects in glomerular permselectivity.
Laminins
Laminins are large heterotrimeric glycoproteins composed of three different homologous chains: α, β, and γ. In humans there are five α chains, four β chains, and three γ chains that assemble to form at least fifteen distinct laminin trimers16 that, depending on the α-β-γ chain composition, resemble cruciform, Y-shaped, or rod-shaped structures. Each chain has a laminincoiled-coil domain,17 and interactions among three coiled-coil domains and limited interchain covalent bonding contribute to the formation of the long arm of all laminin trimers.18 The remaining segment of each chain is called a short arm. The short arms of the cruciform trimers contain a laminin N-terminal (LN) domain, which plays an essential role in polymerization of trimers to form a network.19,20 Laminin α chains are unique because they also contain a C-terminal laminin globular (LG) domain, composed of five tandem sub-domains, which is situated distal to the long arm.21
LG domains link laminin trimers in basement membranes to neighboring cells by serving as ligands for two major cellular receptors, integrins and dystroglycan.22,23 Integrins are transmembrane αβ heterodimers that are crucial in many different contexts for cell-to-extracellular matrix signaling and vice versa, as well as for direct cell-cell signaling.24 Integrin α3β1 is the predominant integrin normally present on the basal surface of podocytes, and deletion of α3β1 results in severe kidney glomerular defects during development.25 Integrin α3β1 binds to laminin α5β1γ1 (LM-511) and α5β2γ1 (LM-521) through the α5 chain’s LG domain.26 The binding of α5LG to integrin α3β1 is essential for the formation of the typical glomerular capillary loop structure, likely because the mesangial cells that organize the capillaries also express integrin α3β1.27 Dystroglycan also binds to the LG domain of α chains,28 but the deletion of dystroglycan from most kidney cells does not result in a kidney defect, suggesting that integrin α3β1 is more responsible for extracellular matrix-to-cell adhesion and signaling in the kidney.29
Secretory signal peptides are located at the N-terminus of each laminin chain, which targets them to the endoplasmic reticulum as they are synthesized. Laminin trimers assemble and become glycosylated in the endoplasmic reticulum and are processed further in the Golgi apparatus.30 After secretion into the extracellular space, laminin trimers self-polymerize to form a laminin network by interactions among LN domains.20 This polymerization step is a reversible and calcium-dependent process that requires a critical concentration of laminin trimers to form an initiating complex.19 This initial step is facilitated by the binding of laminin trimers tolaminin receptors via their α chain LG domains,31 which increases the local concentration of laminin trimers and helps trigger laminin polymerization. Interactions that involve the other basement membrane molecules (type IV collagen, nidogen, and sulfated proteoglycan) then enables the assembly of a basement membrane. Interestingly, mice that lack α1 and α2 type IV collagen do assemble basement membranes during early embryonic development, but these basement membranes are unstable, leading to embryonic death by embryonic day 11.32 However, the deletion of laminin γ1 (a component of all early laminins) in mice caused much earlier lethality (embryonic day 5.5) and the total absence of basement membranes.33 These data indicate that laminin is indispensible for the initial formation of basement membranes, whereas type IV collagen is not. And as discussed below, the quantity of efficiently polymerizing laminin in the GBM is important for restricting the passage of plasma macromolecules across the glomerular filter.
Type IV Collagen
Type IV collagen is the most abundant protein found in basement membranes, comprising about 50% of total protein mass. There are six genetically distinct collagen IV α chains, α1 through α6, that assemble to form three different heterotrimers referred to as protomers: α1α1α2, α3α4α5, and α5α5α6. Like all collagen chains, the collagen IV chains contain Gly-X-Y amino acid triplet repeats. However, unlike fibril-forming collagens of bone and cartilage, the Gly-X-Y repeat region of collagen IV displays multiple interruptions, imparting flexibility to the collagen IV protomer and to the network that it forms in basement membranes.
Each collagen IV α chain has three domains: the N-terminal 7S domain, the collagenous domain containing the interrupted Gly-X-Y repeats (with X and Y usually being Lys or Pro), and the noncollagenous domain (NC1) at the C-terminus. Collagen IV protomers are assembled inside the ER and secreted into the extracellular space. There, they self-polymerize into a chicken wire-like network through hexameric and dodecameric interactions involving the NC1 and 7S domains, respectively, and become heavily cross-linked through disulfide bonds, sulfilimine bonds, and lysyl oxidase-mediated crosslinks.34,35
In the GBM, there is a transition in the composition of collagen IV chains, from α1α1α2 in the immature GBM to α3α4α5 in the mature GBM.36 This transition occurs coincidentally with the transition of laminin chains in the GBM. The molecular mechanisms controlling the switch of collagen IV and laminin chains in the GBM are unknown. However, the collagen IV transition might be required to accommodate the increased blood pressure in the adult, since α3α4α5 type IV collagen produces a more heavily cross-linked and more protease-resistant network compared to the (α1)2α2 type IV collagen network.37 The importance of the α3α4α5 type IV collagen network in the glomerular capillary wall is illustrated by the fact that its absence causes Alport syndrome, a glomerular disease discussed in detail below.
Nidogen
The nidogens, also known as entactins, are two homologous glycoproteins containing three globular-like (G) domains with two rod-like domains separating the G domains.38 Nidogen-1 and nidogen-2, both found in the GBM, are encoded by two different genes. Nidogen-1 binds to both laminin γ1 and type IV collagen, leading to the hypothesis that nidogen acts as a bridge linking the separate laminin and type IV collagen networks in basement membranes.39 Deletion of either nidogen gene in mice does not cause any significant abnormalites, likely because of overlapping expression and redundant functions.40–43 Nidogen-1 and -2 double knockout mice, however, exhibit perinatal lethality due to lung and heart malformations, but many basement membranes form normally without nidogen, despite nidogen’s ability to link laminin and type IV collagen networks.44 The data suggest that nidogens provide extra stability to basement membranes under unusual stress, but they are not required for their initial formation. There is no definitive evidence that either nidogen alone contributes to the GBM’s barrier properties, so they will not be discussed in that context.
Heparan Sulfate Proteoglycan
Heparan sulfate proteoglycans (HSPGs) have sulfated glycosaminoglycan side chains linked to a protein core. Although perlecan seems to be the prominent HSPG in most basement membranes39 and in the mesangial matrix, agrin is the major heparan sulfate proteoglycan in the GBM.45 Agrin’s sulfated glycosaminoglycan side chains give it a highly negative charge that is reflected by anionic sites within the GBM that can be detected with cationic probes such as polyethyleneimine and cationized ferritin.46,47 The N-terminal domain of agrin (NtA) binds avidly to the LM-521 long arm, and its C-terminus bears domains that can bind to cell surface receptors such as dystroglycan and integrins.48 These properties of agrin suggest that it could have importance in mediating charge selectivity within the glomerular filtration barrier and in linking the GBM to the adjacent cells. In addition, heparan sulfate side chains are important in some contexts for binding and sequestering growth factors; one such growth factor secreted by podocytes that must cross the GBM and could benefit from the presence of such side chains is VEGF.11,49 However, podocyte-specific mutation of agrin in mice did not lead to any structural or functional defects in the glomerulus, although it did greatly reduce the density of negatively charged sites within the GBM and caused a dramatic reduction in the level of full length agrin protein in the GBM. These results suggest that agrin and the negative charge of the GBM do not play critical roles in the filtration barrier.46 Furthermore, deletion of both agrin and the heparan sulfate side chains on perlecan also had little if any effect on permselectivity,50 despite the fact that a previous report had revealed a potential role for perlecan in filtration in the context of protein overload.51 But because no critical role for agrin in the GBM, either as a matrix protein or as an anionic macromolecule, has been proven, agrin will not be discussed further in relation to the glomerular filtration barrier.
Diseases associated with malfunction of glomerular basement membrane
As mentioned above, there are two known and relatively well understood genetic kidney diseases that target components of the GBM: Pierson syndrome and Alport syndrome. Although both of these diseases involve primary defects in GBM components, they have very different clinical presentations; these differences provide some clues as to the specific roles for laminin and type IV collagen networks in influencing glomerular permselectivity.
Pierson syndrome
In the early 1960s, Pierson et al. described two patients from one family with a microcoria-congenital nephrosis syndrome52. Both patients presented with severe congenital nephrotic syndrome and early onset end-stage renal failure, and they died 2 weeks after birth. Their eyes’ iris dilator muscles showed aplasia and/or atrophy with an abnormal lens. In 2004, Zenker and colleagues reported a congenital nephrotic syndrome with clinical features that included microcoria and mesangial sclerosis, similar to the features described by Pierson, thus the moniker Pierson syndrome.53 From homozygosity mapping of five affected families, autosomal recessive mutations in LAMB2, the gene encoding laminin β2, were identified.53 Follow up of the original cases via screening of healthy living family members revealed two different mutations in LAMB2, suggesting that the original cases were compound heterozygotes.54 These findings highlight the critical role of laminin β2 for normal kidney function, especially glomerular filtration barrier function.
The recognition of Pierson syndrome as a specific disease entity by discovery of LAMB2 mutations spurred more reports of Pierson syndrome cases.5,55 With increasing numbers of clinical and mutation reports, it has become clear that the onset and severity of Pierson syndrome varies greatly among patients in terms of progression of nephrotic syndrome and extra-renal manifestations, such as muscular hypotonia and neurodevelopmental deficits, resulting in a Pierson syndrome “spectrum” with genotype-phenotype correlations. The level of mutated laminin β2 expression and/or function seems critical for determining the severity of Pierson syndrome.56,57 Truncating mutations, which are dispersed throughout the LAMB2 gene, generally show more severe phenotypes than missense mutations do, as truncated proteins often lack the coiled-coil domain that is critical for the assembly of LM-521 trimers.5 In contrast, pathogenic missense mutations in LAMB2 are found primarily in the LN domain, likely resulting in laminin polymerization and/or secretion defects.5,58,59 Thus, for patients in the Pierson syndrome spectrum, the mutations in laminin β2 result in a total absence or reduced level of LM-521 in the GBM or in the formation and secretion of dysfunctional LM-521 trimers.
The straightforward interpretation for the mechanism of proteinuria in Pierson syndrome is that the lack of LM-521 causes a defect in the GBM’s ability to attenuate the passage of albumin from plasma to Bowman’s space. But given the evidence for the involvement of both podocytes and endothelial cells in regulating permselectivity, an alternative hypothesis is that defects in the GBM prevent proper cell/matrix interactions and signaling to cells, resulting in cellular defects leading to proteinuria. Rigorously investigating these hypotheses requires experiments that cannot be performed in humans. Fortunately, mice with a null mutation in Lamb2 have been an excellent model to study Pierson syndrome, as they recapitulate the congenital nephrotic syndrome and die at about three weeks of age with severe proteinuria and neuromuscular defects.60,61 They also show accumulation of ectopic laminin chains in the GBM, including α1, α2, α3, β1, β3, and γ2 (Figure 1B).62 This deposition of ectopic laminins into the GBM may be a compensatory response to the loss of laminin β2/LM-521, but it is not sufficient to establish a fully functional glomerular filtration barrier.62 Interestingly, the glomerular ultrastructure of proteinuric one week old Lamb2 null mice reveals intact podocyte foot processes (Figure 1B) and slit diaphragms, suggesting a role for the GBM as an independent and indispensible filtration barrier to plasma proteins.62 Importantly, these data show that proper GBM laminin composition is not required for what appears to be the elaboration by podocytes of proper foot processes with slit diaphragms in functioning glomeruli, at least at early stages. In mice, and presumably in humans also, foot process effacement follows (Figure 1B), and perhaps is even caused by, the increasing proteinuria that eventually reaches nephrotic range.
In further support of a direct role for the GBM in mediating permselectivity, the use of ferritin as an electron dense tracer to monitor the permeability of the GBM to plasma macromolecules showed that the lack of LM-521 in Lamb2−/− mice was associated with an increase in permeability relative to control littermates; this increase was apparent before widespread foot process effacement and loss of slit pores.62 And similar to previously reported results using albumin and ferritin tracing in situ,13,14,63 there was no accumulation of ferritin below the slit diaphragms, suggesting that they do not act as restrictive pores in the physiological state.62 These results are, however, in conflict with structural studies of the slit diaphragm that show pore sizes similar to or smaller than the size of albumin.64
Alport syndrome
Alport syndrome is a hereditary glomerular, auditory, and ocular disease caused by mutations in the COL4A3, COL4A4, or COL4A5 genes that encode the type IV collagen α3, α4, and α5 chains, respectively. 4,65 Because the GBM’s collagen IV network consists primarily of cross-linked α3α4α5(IV) protomers made by podocytes,66 the lack of any one of the three chains (for example, due to a null mutation) prevents the protomer from forming. Moreover, missense mutations—particularly those that cause glycine substitutions in the collagenous domain—can lead to defects in protomer structure and in the collagen IV network and therefore also cause Alport syndrome.
In the absence of the α3α4α5(IV) network, there is a compensatory increase in the α1α1α2(IV) network, which is normally a minor component in the subendothelial aspect of the mature human GBM and barely detectable in normal mouse GBM. This compensation allows the GBM to form and function properly for several years, but eventually there is hematuria that accompanies a characteristic splitting and thickening of the GBM (Figure 1C). This is followed by the onset of proteinuria that increases and signifies declining GFR. The majority of patients, most of whom are males carrying X-linked COL4A5 mutations, eventually require renal replacement therapy. In contrast, only a minority of female carriers of X-linked Alport syndrome progress to ESRD. The rarer autosomal forms of Alport syndrome affect males and females equally.
That proteinuria appears late in the disease process suggests that collagen IV is not as important as laminin for maintaining glomerular permselectivity. However, studies of a mouse model of Alport syndrome show that Alport GBM is more permeable to ferritin than is normal mouse GBM, indicating that collagen IV is important for the filtration barrier.67 The areas of increased permeability also showed ectopic deposition of laminin chains (laminin α1 plus increased levels of α5),67 suggesting the possibility that secondary changes in the laminin network, together with the collagen IV network defect, might be responsible for increased permeability in Alport GBM. That proteinuria appears late in the course of the disease may be because any early increase in the level of filtered albumin may be masked by increased albumin uptake by proximal tubular cells.
Treatment of Alport mice and humans with ACE inhibitors has been shown to slow the onset of proteinuria and the decline in GFR.68,69 A reduction in blood pressure should put less stress on the defective, imperfectly cross-linked GBM, on the adjacent podocyte, and on the entire glomerulus, which exhibit increased deformability.70 Moreover, biomechanical strain has been proposed to change gene expression in ways that exacerbate glomerular disease in Alport syndrome.71
Concluding thoughts
What are the mechanisms whereby laminin and collagen IV might be directly involved in glomerular permselectivity? According to concepts proffered by Smithies,72 the GBM behaves like a concentrated gel with size-selective properties into which macromolecules such as albumin permeate primarily by diffusion. As the major components of the GBM, the laminin-521 and collagen IV networks, in concert with the other GBM components, likely impart the GBM with its characteristic porosity. By analogy to the polyacrylamide gel, which also has size-selective properties, it is easy to understand how 1) reducing laminin or type IV collagen concentration might be similar to reducing the percentage of acrylamide; or 2) how changing laminin or type IV collagen isoforms might be similar to changing the ratio of acrylamide to bis-acrylamide. In either of these cases, the result could be increased permeability via increased pore sizes. Consistent with this, our hypothesis that Lamb2−/− mice develop nephrotic syndrome due to a paucity of laminin in the GBM, and therefore a defective laminin network in the GBM, was supported by our data from transgenic mice with podocyte-specific overexpression of laminin β1/LM-511. On the Lamb2−/− background, the forced secretion of high levels of LM-511 from podocytes into the GBM prevented the nephrotic syndrome that would have otherwise developed, and the mice lived a long life.73 These data also suggest that in the GBM the quantity of laminin is important, as LM-511 could efficiently substitute for LM-521 if expressed at a high enough level. An exciting conclusion from this study is that upregulation of the unaffected LAMB1 gene in the podocytes of patients with Pierson syndrome should be an effective therapy. Moreover, our recent study showed that secretion of a secretion-defective mutant laminin β2 (C321R-LAMB2) could be improved in vitro by treatment with a chemical chaperone,74 which promotesproper protein folding. This result suggests a possible drug therapy for Pierson syndrome patients who have missense LAMB2 mutations that impair protein folding and/or secretion. Finally, additional studies in mice suggest that the three C-terminal segments of the LG domain of laminin α5 are critical for a proper filtration barrier75 for reasons not yet understood. Future studies concentrating on the mechanism for this could reveal novel approaches for tightening the glomerular barrier to albumin and reducing albuminuria.
Given the well-demonstrated importance of multiple podocyte proteins for proper foot process architecture and for permselectivity (see accompanying article by Brinkkötter et al.), it is important to determine how this can be reconciled with the concept that the GBM is a major contributor to the physical filtration barrier. We propose, based on cell biological concepts put forth previously,76 that the podocyte cytoskeleton and slit diaphragms are so critical for normal filtration because they are connected (either directly or indirectly) to cell surface receptors—primarily integrins—that link to the GBM via LM-521. In turn, these receptors are critical for organizing the arrangement of the GBM’s components and can thereby regulate its architecture, its porosity, and thus its permselectivity.
Key points.
The glomerular basement membrane is the extracellular matrix component of the glomerular filtration barrier. It is flanked by the podocyte and glomerular endothelial cell layers.
The major GBM components are laminin-521, collagen α3α4α5(IV), nidogen, and the heparan sulfate proteoglycan agrin
Mutations in COL4 genes that result in absence of the collagen α3α4α5(IV) network cause Alport syndrome, a hereditary nephritis accompanied by hearing defects
Mutations in laminin β2 (LAMB2) cause Pierson syndrome, a congenital nephrotic syndrome with eye and neurologic abnormalities
Studies using mouse models of Pierson and Alport syndromes have shown the defective GBM to be more permeable to macromolecules, suggesting a role in permselectivity
Acknowledgments
The authors were supported by NIH grants R01DK078314, R21DK095419, and P30DK079333 and by a grant from the Alport Syndrome Foundation. JHS was supported by NIH training grant T32DK007126.
Footnotes
Competing interests
The authors declare no competing interests.
Bibliography
- 1.Miner JH. Organogenesis of the kidney glomerulus: focus on the glomerular basement membrane. Organogenesis. 2011;7:75–82. doi: 10.4161/org.7.2.15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yurchenco PD, Patton BL. Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des. 2009;15:1277–1294. doi: 10.2174/138161209787846766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miner JH. Building the glomerulus: a matricentric view. J Am Soc Nephrol. 2005;16:857–861. doi: 10.1681/ASN.2004121139. [DOI] [PubMed] [Google Scholar]
- 4.Kruegel J, Rubel D, Gross O. Alport syndrome-insights from basic and clinical research. Nat Rev Nephrol. 2012 doi: 10.1038/nrneph.2012.259. [DOI] [PubMed] [Google Scholar]
- 5.Matejas V, et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat. 2010;31:992–1002. doi: 10.1002/humu.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 7.de Boer IH, et al. Temporal trends in the prevalence of diabetic kidney disease in the united states. JAMA: The Journal of the American Medical Association. 2011;305:2532–2539. doi: 10.1001/jama.2011.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farquhar MG. The glomerular basement membrane: not gone, just forgotten. J Clin Invest. 2006;116:2090–2093. doi: 10.1172/JCI29488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miner JH. The glomerular basement membrane. Experimental Cell Research. 2012;318:973–978. doi: 10.1016/j.yexcr.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.St John PL, Abrahamson DR. Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int. 2001;60:1037–1046. doi: 10.1046/j.1523-1755.2001.0600031037.x. [DOI] [PubMed] [Google Scholar]
- 11.Eremina V, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farquhar MG. Editorial: The primary glomerular filtration barrier--basement membrane or epithelial slits? Kidney Int. 1975;8:197–211. doi: 10.1038/ki.1975.103. [DOI] [PubMed] [Google Scholar]
- 13.Farquhar MG, Palade GE. Glomerular permeability. II Ferritin transfer across the glomerular capillary wall in nephrotic rats. J Exp Med. 1961;114:699–716. doi: 10.1084/jem.114.5.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farquhar MG, Wissig SL, Palade GE. Glomerular permeability. I Ferritin transfer across the normal glomerular capillary wall. J Exp Med. 1961;113:47–66. doi: 10.1084/jem.113.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenner BM, Hostetter TH, Humes HD. Molecular basis of proteinuria of glomerular origin. N Engl J Med. 1978;298:826–833. doi: 10.1056/NEJM197804132981507. [DOI] [PubMed] [Google Scholar]
- 16.Miner JH, Yurchenco PD. Laminin functions in tissue morphogenesis. Annu Rev Cell Dev Biol. 2004;20:255–284. doi: 10.1146/annurev.cellbio.20.010403.094555. [DOI] [PubMed] [Google Scholar]
- 17.Paulsson M. Basement membrane proteins: structure, assembly, and cellular interactions. Crit Rev Biochem Molec Biol. 1992;27:93–127. doi: 10.3109/10409239209082560. [DOI] [PubMed] [Google Scholar]
- 18.Ekblom P, Timpl R. Cell-to-cell contact and extracellular matrix. A multifaceted approach emerging. Curr Opin Cell Biol. 1996;8:599–601. doi: 10.1016/s0955-0674(96)80099-8. [DOI] [PubMed] [Google Scholar]
- 19.Yurchenco PD, Cheng YS. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J Biol Chem. 1993;268:17286–17299. [PubMed] [Google Scholar]
- 20.Cheng YS, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem. 1997;272:31525–31532. doi: 10.1074/jbc.272.50.31525. [DOI] [PubMed] [Google Scholar]
- 21.Timpl R, et al. Structure and function of laminin LG modules. Matrix Biol. 2000;19:309–317. doi: 10.1016/s0945-053x(00)00072-x. [DOI] [PubMed] [Google Scholar]
- 22.Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 23.Henry MD, Campbell KP. Dystroglycan inside and out. Curr Opin Cell Biol. 1999;11:602–607. doi: 10.1016/s0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 24.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 25.Kreidberg JA, et al. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- 26.Kikkawa Y, Sanzen N, Sekiguchi K. Isolation and characterization of laminin-10/11 secreted by human lung carcinoma cells. laminin-10/11 mediates cell adhesion through integrin alpha3 beta1. J Biol Chem. 1998;273:15854–15859. doi: 10.1074/jbc.273.25.15854. [DOI] [PubMed] [Google Scholar]
- 27.Kikkawa Y, Virtanen I, Miner JH. Mesangial cells organize the glomerular capillaries by adhering to the G domain of laminin alpha5 in the glomerular basement membrane. J Cell Biol. 2003;161:187–196. doi: 10.1083/jcb.200211121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wizemann H, et al. Distinct requirements for heparin and alpha-dystroglycan binding revealed by structure-based mutagenesis of the laminin alpha2 LG4-LG5 domain pair. J Mol Biol. 2003;332:635–642. doi: 10.1016/s0022-2836(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 29.Jarad G, Pippin JW, Shankland SJ, Kreidberg JA, Miner JH. Dystroglycan does not contribute significantly to kidney development or function, in health or after injury. Am J Physiol Renal Physiol. 2011;300:F811–820. doi: 10.1152/ajprenal.00725.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YM, Miner JH. Translational Research. 2012. Glomerular basement membrane and related glomerular disease; pp. 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colognato H, Winkelmann DA, Yurchenco PD. Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol. 1999;145:619–631. doi: 10.1083/jcb.145.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poschl E, et al. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 33.Smyth N, et al. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–160. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanacore R, et al. A sulfilimine bond identified in collagen IV. Science. 2009;325:1230–1234. doi: 10.1126/science.1176811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hudson BG. The molecular basis of Goodpasture and Alport syndromes: beacons for the discovery of the collagen IV family. J Am Soc Nephrol. 2004;15:2514–2527. doi: 10.1097/01.ASN.0000141462.00630.76. [DOI] [PubMed] [Google Scholar]
- 36.Miner JH. Developmental biology of glomerular basement membrane components. Curr Opin Nephrol Hypertens. 1998;7:13–19. doi: 10.1097/00041552-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Gunwar S, et al. Glomerular basement membrane. Identification of a novel disulfide-cross- linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem. 1998;273:8767–8775. doi: 10.1074/jbc.273.15.8767. [DOI] [PubMed] [Google Scholar]
- 38.Kohfeldt E, Sasaki T, Gohring W, Timpl R. Nidogen-2: A new basement membrane protein with diverse binding properties. J Mol Biol. 1998;282:99–109. doi: 10.1006/jmbi.1998.2004. [DOI] [PubMed] [Google Scholar]
- 39.Timpl R. Structure and biological activity of basement membrane proteins. Eur J Biochem. 1989;180:487–502. doi: 10.1111/j.1432-1033.1989.tb14673.x. [DOI] [PubMed] [Google Scholar]
- 40.Miosge N, Sasaki T, Timpl R. Evidence of nidogen-2 compensation for nidogen-1 deficiency in transgenic mice. Matrix Biol. 2002;21:611–621. doi: 10.1016/s0945-053x(02)00070-7. [DOI] [PubMed] [Google Scholar]
- 41.Miosge N, et al. Ultrastructural colocalization of nidogen-1 and nidogen-2 with laminin-1 in murine kidney basement membranes. Histochem Cell Biol. 2000;113:115–124. doi: 10.1007/s004180050014. [DOI] [PubMed] [Google Scholar]
- 42.Schymeinsky J, et al. Gene structure and functional analysis of the mouse nidogen-2 gene: nidogen-2 is not essential for basement membrane formation in mice. Mol Cell Biol. 2002;22:6820–6830. doi: 10.1128/MCB.22.19.6820-6830.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murshed M, et al. The absence of nidogen 1 does not affect murine basement membrane formation. Mol Cell Biol. 2000;20:7007–7012. doi: 10.1128/mcb.20.18.7007-7012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bader BL, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol. 2005;25:6846–6856. doi: 10.1128/MCB.25.15.6846-6856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groffen AJ, et al. Agrin is a major heparan sulfate proteoglycan in the human glomerular basement membrane. J Histochem Cytochem. 1998;46:19–27. doi: 10.1177/002215549804600104. [DOI] [PubMed] [Google Scholar]
- 46.Harvey SJ, et al. Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol. 2007;171:139–152. doi: 10.2353/ajpath.2007.061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rennke HG, Cotran RS, Venkatachalam MA. Role of molecular charge in glomerular permeability. Tracer studies with cationized ferritins. J Cell Biol. 1975;67:638–646. doi: 10.1083/jcb.67.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- 49.Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–1326. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldberg S, Harvey SJ, Cunningham J, Tryggvason K, Miner JH. Glomerular filtration is normal in the absence of both agrin and perlecan-heparan sulfate from the glomerular basement membrane. Nephrol Dial Transplant. 2009;24:2044–2051. doi: 10.1093/ndt/gfn758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morita H, et al. Heparan sulfate of perlecan is involved in glomerular filtration. J Am Soc Nephrol. 2005;16:1703–1710. doi: 10.1681/ASN.2004050387. [DOI] [PubMed] [Google Scholar]
- 52.Pierson M, Cordier J, Hervouuet F, Rauber G. An Unusual Congenital and Familial Congenital Malformative Combination Involving the Eye and Kidney. J Genet Hum. 1963;12:184–213. [PubMed] [Google Scholar]
- 53.Zenker M, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 54.Zenker M, Pierson M, Jonveaux P, Reis A. Demonstration of two novel LAMB2 mutations in the original Pierson syndrome family reported 42 years ago. Am J Med Genet A. 2005;138:73–74. doi: 10.1002/ajmg.a.30894. [DOI] [PubMed] [Google Scholar]
- 55.Lehnhardt A, et al. Pierson syndrome in an adolescent girl with nephrotic range proteinuria but a normal GFR. Pediatr Nephrol. 2012;27:865–868. doi: 10.1007/s00467-011-2088-2. [DOI] [PubMed] [Google Scholar]
- 56.Kagan M, Cohen AH, Matejas V, Vlangos C, Zenker M. A milder variant of Pierson syndrome. Pediatr Nephrol. 2008;23:323–327. doi: 10.1007/s00467-007-0624-x. [DOI] [PubMed] [Google Scholar]
- 57.Hasselbacher K, et al. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int. 2006;70:1008–1012. doi: 10.1038/sj.ki.5001679. [DOI] [PubMed] [Google Scholar]
- 58.Chen YM, Kikkawa Y, Miner JH. A missense LAMB2 mutation causes congenital nephrotic syndrome by impairing laminin secretion. J Am Soc Nephrol. 2011;22:849–858. doi: 10.1681/ASN.2010060632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purvis A, Hohenester E. Laminin network formation studied by reconstitution of ternary nodes in solution. J Biol Chem. 2012;287:44270–44277. doi: 10.1074/jbc.M112.418426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noakes PG, et al. The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 61.Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature. 1995;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- 62.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities inLamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest. 2006;116:2272–2279. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryan GB, Karnovsky MJ. Distribution of endogenous albumin in the rat glomerulus: role of hemodynamic factors in glomerular barrier function. Kidney Int. 1976;9:36–45. doi: 10.1038/ki.1976.5. [DOI] [PubMed] [Google Scholar]
- 64.Wartiovaara J, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest. 2004;114:1475–1483. doi: 10.1172/JCI22562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Noone D, Licht C. An update on the pathomechanisms and future therapies of Alport syndrome. Pediatr Nephrol. 2012 doi: 10.1007/s00467-012-2272-z. in press. [DOI] [PubMed] [Google Scholar]
- 66.Abrahamson DR, Hudson BG, Stroganova L, Borza DB, St John PL. Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol. 2009;20:1471–1479. doi: 10.1681/ASN.2008101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abrahamson DR, et al. Laminin compensation in collagen alpha3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol. 2007;18:2465–2472. doi: 10.1681/ASN.2007030328. [DOI] [PubMed] [Google Scholar]
- 68.Gross O, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63:438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 69.Gross O, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81:494–501. doi: 10.1038/ki.2011.407. [DOI] [PubMed] [Google Scholar]
- 70.Wyss HM, et al. Biophysical properties of normal and diseased renal glomeruli. Am J Physiol Cell Physiol. 2011;300:C397–405. doi: 10.1152/ajpcell.00438.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meehan DT, et al. Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int. 2009;76:968–976. doi: 10.1038/ki.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smithies O. Why the kidney glomerulus does not clog: A gel permeation/diffusion hypothesis of renal function. Proc Natl Acad Sci U S A. 2003;100:4108–4113. doi: 10.1073/pnas.0730776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suh JH, Jarad G, Vandevoorde RG, Miner JH. Forced expression of laminin {beta}1 in podocytes prevents nephrotic syndrome in mice lacking laminin {beta}2, a model for Pierson syndrome. Proc Natl Acad Sci USA. 2011;108:15348–15353. doi: 10.1073/pnas.1108269108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen YM, et al. Podocyte endoplasmic reticulum stress in mice expressing a LAMB2 mutant associated with human nephrotic syndrome. J Amer Soc Nephrol. 2013 in press. [Google Scholar]
- 75.Kikkawa Y, Miner JH. Molecular dissection of laminin alpha 5 in vivo reveals separable domain-specific roles in embryonic development and kidney function. Dev Biol. 2006;296:265–277. doi: 10.1016/j.ydbio.2006.04.463. [DOI] [PubMed] [Google Scholar]
- 76.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]