

Significance
We show that intracellular ATP at physiological concentrations acts as a signaling molecule to activate the slowly activating K+ channel IKs that regulates heart rate adaptation. ATP binding to the pore-forming α-subunit of IKs, KCNQ1, allows the channel to open. Congenital mutations that reduce ATP binding or subsequent opening of the IKs channel are associated with cardiac arrhythmias in human patients. Electrical abnormalities are often the cause of fatality in cardiovascular diseases, including ischemia and heart failure, in which ATP level is reduced in cardiac cells. Our results open up new possibilities to study and manage these diseases, and the ATP site provides a unique target for therapies.
Keywords: ischemia, heart failure
Abstract
Gating of ion channels by ligands is fundamental to cellular function, and ATP serves as both an energy source and a signaling molecule that modulates ion channel and transporter functions. The slowly activating K+ channel IKs in cardiac myocytes is formed by KCNQ1 and KCNE1 subunits that conduct K+ to repolarize the action potential. Here we show that intracellular ATP activates heterologously coexpressed KCNQ1 and KCNE1 as well as IKs in cardiac myocytes by directly binding to the C terminus of KCNQ1 to allow the pore to open. The channel is most sensitive to ATP near its physiological concentration, and lowering ATP concentration in cardiac myocytes results in IKs reduction and action potential prolongation. Multiple mutations that suppress IKs by decreasing the ATP sensitivity of the channel are associated with the long QT (interval between the Q and T waves in electrocardiogram) syndrome that predisposes afflicted individuals to cardiac arrhythmia and sudden death. A cluster of basic and aromatic residues that may form a unique ATP binding site are identified; ATP activation of the wild-type channel and the effects of the mutations on ATP sensitivity are consistent with an allosteric mechanism. These results demonstrate the activation of an ion channel by intracellular ATP binding, and ATP-dependent gating allows IKs to couple myocyte energy state to its electrophysiology in physiologic and pathologic conditions.
Significant energy is required to sustain both the electrical and contractile events that accompany each heart beat, suggesting that the level of ATP is one key to normal cardiac functions. Not surprisingly, a reduction in ATP concentration ([ATP]) plays a key role in the pathogenesis and progression of ischemic heart diseases, including heart failure. Intracellular ATP is important not only in providing energy (1) and as a substrate for protein kinases (2), but also as signaling molecules to bind and modulate proteins. Only a handful of results have shown that intracellular ATP serves as a signal for membrane channels and transporters (3). The best-studied example is the KATP channel (4, 5). This channel is inhibited in physiologic conditions by [ATP]s of 5–10 mM (6), but when the ATP levels drop to submillimolar concentrations, as in cardiac ischemia, the KATP channels open, shortening the action potential duration and providing metabolic protection against the insult of ischemia (7). However, at normal physiologic conditions, whether and how ATP serves as a signal connecting the energetic state of the cell to membrane excitability is still unknown.
The slowly activating K+ channel IKs plays an important role in controlling the action potential duration (APD) in cardiac myocytes; it opens in response to depolarization to conduct potassium ions out of the cell, which contributes to repolarization of the membrane, terminating the cardiac action potential and thereby the myocyte contraction. The IKs channel consists of pore-forming KCNQ1 subunits and the single-transmembrane auxiliary subunits KCNE1 (8, 9). Loss-of-function mutations in either KCNQ1 or KCNE1 lead to prolongation of ventricular action potentials and long QT syndrome (LQTS) that manifests as QT (interval between the Q and T waves in electrocardiogram) prolongation in the electrocardiogram. LQTS predisposes patients to cardiac arrhythmias that lead to syncope and sudden death (10). Previous studies show that ATP is required for activation of IKs channels (11), but the molecular mechanism and the physiologic function of this ATP modulation is not known. It was the purpose of this study to investigate the mechanism by which ATP regulated IKs. Our results show that ATP directly binds to the KCNQ1 protein to regulate channel function at concentrations ([ATP]) close to the physiologic intracellular [ATP] in cardiac myocytes. Our studies also reveal a unique ATP binding site and mechanism for modulation of ion channel function. Further, we found that several LQT-associated mutations alter IKs function by reducing ATP sensitivity. These results demonstrate that ATP regulation is vital for IKs channel function; a disruption of these regulations predisposes to life-threatening cardiac arrhythmias.
Results
ATP-Dependent Activation of IKs Channels.
We studied heterologously expressed human KCNQ1/KCNE1 currents (hIKs) in inside-out membrane patches from Xenopus oocytes with application of various intracellular ATP concentrations ([ATP]). Upon patch excision, the current ran down in low [ATP] (Fig. 1A), consistent with previous findings that the loss of native ATP in cytosol resulted in reduced channel activity (11). However, the current ran up in high [ATP], suggesting that the open probability of channels (Po) is not maximal with the native cytosolic [ATP] that is insufficient to saturate channel activation. Thus, a reserve of the IKs channels open in higher applied [ATP], resulting in currents larger than those observed at patch excision (Fig. 1A). The steady-state current amplitude increased with [ATP] with the EC50 at 1.7 mM (Fig. 1B), which is close to the physiological [ATP] in cardiac myocytes (6). This result suggests that cardiac IKs could be sensitive to the cellular energetic state, and fluctuations of [ATP] as in ischemia could alter electrical properties via regulating IKs.
Fig. 1.

ATP-dependent activation of IKs channels. (A) KCNQ1 + KCNE1 (hIKs) currents in inside-out patches run down or up in 0 (red), 0.5 (green), and 5 mM (blue) [ATP] from that immediately after patch excision (black, I0). Voltage pulses were +80 mV from a −80-mV holding potential. (Right) It, I0 tail current amplitudes. (B) ATP dose–response of WT (black) and Q357R (red, also scaled to the WT currents at 20 mM ATP) IKs, and fits to the Hill equation (solid curves) with Hill coefficient 1 and 1.1, and EC50 1.66 and 9.60 mM, respectively. (C) Currents of WT (Left) and Q357R (Center) hIKs recorded from inside-out patches and G–V relations after patch excision in solutions containing various [ATP]. Solid curves are fits of Boltzmann equation (Right; Materials and Methods) with V1/2 (mV) at 0.5, 5, and 20 mM [ATP], respectively: WT, 23.7 ± 3.5 (black), 25.6 ± 3.2 (purple), 25.4 ± 5.0 (blue); Q357R, 53.2 ± 2.5 (red), 30.8 ± 2.6 (green), and 26.2 ± 3.8 (cyan). (D) Normalized ATP dose–response of WT hIKs channel activation in control solution [PIP2] = 100 µM (black), 5 µM PIP2 (green), 100 µM Ca2+ (red), and S27D/S92D hIKs in control solution (blue; Materials and Methods). Five and 100 µM [PIP2] are 50% and 100% of saturation for IKs channel activation, respectively (14). Dashed curves in B and C are the fittings of the model in Fig. 7A.
The physiological importance of the ATP sensitivity of IKs is supported by our subsequent finding that a LQT-associated mutation Q357R in KCNQ1 (12) reduces ATP sensitivity, as shown by an increased EC50 of the response to [ATP] and the larger fraction of the current activated by applied high [ATP] (Fig. 1B). Consistent with previous studies (13), we found that Q357R coexpressed with KCNE1 (Q357R hIKs) showed a smaller current amplitude, a slower activation time course, and a shift of the voltage dependence of activation toward more depolarized potentials compared with the WT hIKs measured from whole-cell currents. Each of these changes in channel properties would decrease the ability of IKs to participate in cardiac repolarization, resulting in prolongation of APD. Furthermore, application of a high [ATP] (20 mM) during inside-out patch-clamp recordings of Q357R hIKs restored the WT channel characteristics. Specifically, the current amplitude increased three- to fivefold, sufficient to account for all of the reduction in the whole-cell current (SI Appendix, Fig. S1), and the voltage dependence of channel activation shifted back toward less-depolarized voltages to superimpose on that of the WT hIKs (Fig. 1C). These results suggest that a decrease in ATP sensitivity of the IKs channel due to mutation Q357R can lead to LQT syndrome.
KCNQ1 expressed alone without KCNE1 shows a similar dose–response to [ATP] (Fig. 1D), indicating that the ATP dependence is an intrinsic property of KCNQ1 and not altered by KCNE1 association. IKs channels also require phosphatidylinositol 4,5-bisphosphate (PIP2) for function (11, 14, 15) and are modulated by calmodulin (CaM) (16, 17) and phosphorylation of residues S27 and S92 in KCNQ1 by PKA (18, 19). However, the response of hIKs currents to [ATP] did not change with reduced [PIP2], enhanced [Ca2+], or mutations S27D/S92D that mimic phosphorylation (14, 18) (Fig. 1D), indicating that ATP activates the channel independently from these other intracellular regulatory molecules.
ATP Level Changes Affect Action Potentials in Cardiac Myocytes Through Regulating IKs.
To directly examine the role of ATP sensitivity of IKs in cardiac myocytes, we studied guinea pig ventricular myocytes using whole-cell patch-clamp techniques. IKs currents increased with increasing [ATP] in pipette solutions (0–25 mM); a dose–response curve yielded an EC50 of 1.4 mM (Fig. 2 A and B). These results demonstrate that [ATP] regulates IKs in cardiac myocytes as it does hIKs in oocytes with a similar EC50. To examine the effects of ATP on action potentials (APs), we recorded APs with pipette [ATP] ranging from 2 to 10 mM (Fig. 2 C and D, black). The APD shortened as pipette [ATP] was increased. Application of chromanol 293B, an IKs-specific blocker, lengthened the APD (Fig. 2 C and D, red); the lengthening was greater when pipette [ATP] was higher, from less than 10% at 2-mM pipette [ATP] to more than 40% when pipette [ATP] is 10 mM, consistent with the idea that ATP activates IKs to shorten the action potential. The channel activity and APD are specifically dependent on [ATP] because ATP added as a salt of K+ or Mg2+ had the same effects (Fig. 2). Taken as whole, these results demonstrate that variations in myocyte [ATP] can significantly alter APD by changing IKs magnitude.
Fig. 2.

ATP activates IKs and shortens APs in cardiac myocytes. (A) Voltage protocol and current traces at different [ATP]s in the patch pipette in the absence (red) and presence (black) of chromanol 293B (10 µM). (B) IKs vs. [ATP]. n = 5, error bars are SD. Smooth curve: fitting to the data from 1 to 25 mM [ATP] with Hill equation; Hill coefficient = 1 and EC50 = 1.4 mM. The 0 [ATP] data were not included in the curve fitting because 0 [ATP] inside the cell could not be achieved in live myocytes; the predicted value of [ATP] for that magnitude of IKs (gray open circle) is 0.64 mM. Potassium ATP at a concentration of 3 mM and 10 mM (red open circles) were also used in the pipette solution to substitute for MgATP. (C) AP traces in the absence and presence of chromanol 293B, with intracellular infusion of ATP (2–10 mM). (D) Summary of APD90 at various [ATP]. (Upper) The APD for each cell is plotted in the absence and presence of chromanol 293B. (Lower) Average percentage increase in APD induced by chromanol 293B. Note a significantly larger APD prolongation induced by chromanol 293B at 5 or 10 mM ATP compared with that at 2 or 3 mM ATP (P < 0.01, individual t tests). Potassium ATP at two different concentrations (2 mM and 10 mM, in gray) were used to substitute for MgATP in the pipette solution.
ATP Binding in KCNQ1.
ATP could activate the IKs channel by serving as the substrate for phosphorylation, binding to an associated protein or directly binding to the channel proteins. To distinguish these mechanisms, we first measured the ability of various nucleotides to prevent hIKs current run-down due to washout of the native ATP after inside-out membrane patch excision (11). GTP and a nonhydrolyzable ATP analog, 5′-adenylyl-β−γ-imidodiphosphate (AMP–PNP), in the intracellular solution can sustain channel function similarly as ATP, and, furthermore, the dose–response curves of channel function on GTP and AMP–PNP are superimposed with that on ATP (SI Appendix, Fig. S2), whereas the rundown of hIKs currents became progressively faster when ADP and AMP were applied (Fig. 3A). Thus, ATP is not unique in activating the channel and phosphorylation is not required. Furthermore, an ATP analog biotin photoprobe, 2-azidoadenosine 5′-triphosphate 2′,3′-biotin-long chain-hydrazone (AB11) (20) can prevent channel run-down (SI Appendix, Fig. S3) and be photo–cross-linked to the KCNQ1 protein (Fig. 3B), indicating that the nucleotide directly binds to KCNQ1 to modify hIKs channel function. We found that the channels formed by coexpression of KCNQ2 and KCNQ3 do not require ATP for function (Fig. 3 B and C and SI Appendix, Fig. S4), and, correspondingly, AB11 cannot be photo–cross-linked to the KCNQ2 or KCNQ3 proteins (Fig. 3B).
Fig. 3.

ATP binding to KCNQ1. (A) hIKs tail current amplitude elicited by voltage pulses from −80 to +80 mV in intracellular solutions containing 1.5 mM of various nucleotides: ATP (blue), GTP (green), AMP–PNP (pink), ADP (red), AMP (purple), and no nucleotide control (black). (B) Western blot of channel proteins pulled down with avidin beads after UV light photo–cross-linking of the biotin-containing AB11 (Top) or after biotin treatment of intact cells to detect expression in the membrane (Middle). Gbeta is a cytoplasmic protein. Antibodies against KCNQ1 (for lanes of uninjected, hIKs, and KCNQ1), KCNQ2 (KCNQ2), KCNQ3 (KCNQ3), and Gbeta (low bands) were used. (C) Whole-cell currents of WT and chimeric KCNQ1 and KCNQ2/KCNQ3 channels (Upper) and time dependence of current amplitude after inside-out patch excision without application of intracellular ATP (Lower). WT hIKs (black), WT KCNQ2/KCNQ3 (blue), and Q2ctQ1/Q3ctQ1 (red). (D) Mutation scan of all cytosolic basic residues in KCNQ1. (Inset) Experimental design for ATP sensitivity screen. ATP dose–response curves of WT (red) and a hypothetical mutant hIKs with reduced ATP sensitivity (green). Three concentrations of ATP are used: 0.5, 1.5, and 20 mM.
To locate the ATP binding site in KCNQ1, we first studied chimeric channels by transplanting the cytosolic C terminus to KCNQ2 and KCNQ3 to form Q2ctQ1 and Q3ctQ1 (SI Appendix, Fig. S5). Similar to hIKs, channels formed by the coexpression of Q2ctQ1/Q3ctQ1 ran down after inside-out membrane patch excision (Fig. 3C), but intracellularly applied ATP slowed the run-down (SI Appendix, Fig. S6), suggesting that the chimeras acquire ATP sensitivity and the C terminus of KCNQ1 contains the ATP binding site. Because the potency of nucleotides in activating hIKs correlates with the number of phosphates (Fig. 3A), the channel may associate with ATP through electrostatic interactions between basic residues and the negatively charged phosphates of ATP. Interestingly, neutralization mutations of several of the cytosolic basic residues are associated with LQTS. We performed a mutational scan to neutralize each of the basic residues to Ala, LQT-associated mutation, or deletion in the N terminus and the C terminus of KCNQ1 to examine which of these residues affected ATP sensitivity (Fig. 3D). We used a simplified ATP dose–response assay to examine ATP sensitivity of the mutant channels. We chose three ATP concentrations, 0.5, 1.5, and 20 mM, at which the WT IKs channels are activated to 20, 50, and 100% of saturation, respectively (Fig. 3D, Inset). If a mutation reduces ATP sensitivity, the ATP dose–response curve is expected to shift to higher ATP concentrations such that the fractional increments of channel activation at 0.5, 1.5, and 20 mM differ from those of the WT IKs. A more complete dose–response curve would be measured once the mutation is identified as positive for confirmation and further characterization.
The results revealed three mutations, R380S, K393M, and R397W, that are all associated with LQT and reduced the expression of macroscopic hIKs currents and ATP sensitivity of the channel (Figs. 3D and 4A and B; SI Appendix, Fig. S7). Residue R380 is located in helix A downstream from the S6 gate of the channel, whereas K393 and R397 are located in the linker between helix A and helix B (Fig. 4B and SI Appendix, Fig. S8). Though each of these mutations reduced ATP sensitivity, a combined mutation R380S/R397W eliminated ionic current altogether, although channel expression in the plasma membrane could still be detected (Fig. 4 C and D). Furthermore, R380S/R397W also eliminated photo–cross-linking of the ATP analog AB11 (Fig. 4D). These results suggest that these three residues are part of the ATP binding site; whereas each individual mutation reduces ATP binding, the combined mutations disrupt ATP binding, resulting in the loss of channel function. Likewise, mutating each of these residues to negative charges, which could repel ATP, also eliminated hIKs and AB11 photo–cross-linking (Fig. 4 C and D). The known ATP binding sites in other proteins also contain aromatic residues to coordinate the adenine group (21). A mutational scan of each aromatic residue in helices A and B and the A–B linker (SI Appendix, Figs. S8 and S9) identified one mutation, W379S, that eliminated ionic current and AB11 photo–cross-linking of hIKs (Fig. 4 C and D), suggesting that W379 also participates in ATP binding. Similar to R380S, K393M, and R397W, W379S is also associated with LQT syndrome (22–25), further revealing the physiological importance of ATP modulation of IKs.
Fig. 4.

ATP binds in the C terminus of KCNQ1. (A) ATP dose–response curve of WT and key mutant IKs channels. (B) The KCNQ1 motifs important for ATP interaction (Upper) and EC50 of ATP dose–response of mutant hIKs channels (Lower). Due to insolubility of ATP above 20 mM, dose–response for some mutants did not reach saturation, leading to underestimated EC50. Asterisk indicates no current expression. (C) Whole-cell currents of mutant hIKs. (D) Western blot to detect AB11 labeling of mutant IKs. (Upper) AB11 labeling of mutant IKs and Gβ in the whole-cell lysate to indicate similar inputs. (Lower) Western blot probing for biotinylated mutant IKs and Gβ in the membrane. Gβ is a cytoplasmic protein.
The Mechanism of ATP Regulation of IKs Channels.
As a member of the Kv channel family, KCNQ1 is comprised of a voltage-sensing domain (VSD) and a pore-gate domain (PGD; SI Appendix, Fig. S10). The IKs channel contains four KCNQ1 subunits with the VSDs surrounding a central pore across the membrane; in response to membrane depolarization, voltage sensors move to trigger pore opening (26). Does ATP binding affect voltage sensor movements, pore opening, or the coupling between the two processes? We used voltage clamp fluorometry (VCF) to answer this question.
Fluorescence signals from a fluorophore (Alexa 488 C5 Maleimide) attached to the VSD (Materials and Methods; SI Appendix, Fig. S10) were recorded to monitor VSD movements, and ionic currents were simultaneously measured to show pore opening (15, 26) (Fig. 5A). The mutations that disrupt ATP binding, W379S and R380S/R397W (Fig. 4D), eliminated ionic currents of KCNQ1 but did not abolish ΔF/F signals; moreover, the fluorescence–voltage (F–V) relationship and fluorescence signal are superimposed with that of the WT KCNQ1 (Fig. 5 B and C), indicating that ATP binding is not required for VSD movements. Next we tested if ATP is required for the coupling between the VSD movements and pore opening by measuring if opening the pore by the mutation L353K affected VSD activation (15). L353K is located in the S6 gate and the mutation locks the channels constitutively open (15, 27), such that instantaneous currents were observed at every applied voltage, and these currents were not inhibited by disrupting ATP binding (Fig. 6A and SI Appendix, Fig. S11). The opening of the pore facilitated the VSD activation via the VSD–pore coupling, resulting in a leftward shift in the F–V relation by comparing the F–V relation of WT and that of locked open L353K channels (Fig. 6B). Our recent study showed that this VSD–pore coupling is mediated by PIP2 bound to the channel, and depletion of PIP2 abolishes the coupling and thus the L353K-induced F–V shift (15). However, when we depleted ATP binding by mutations, the F–V relationship remained to be left-shifted by L353K, indicating that, unlike PIP2, ATP is not required for the coupling between VSD activation and pore opening. These results (Figs. 5 and 6) lead to a conceptual model for ATP-dependent activation of KCNQ1 and IKs channels (Fig. 7A and SI Appendix, Fig. S12) such that ATP binding to the channel PGD is a prerequisite for the pore to open, but does not affect VSD activation either directly or via the VSD–pore coupling. We were unable to detect fluorescence signals of the mutant channels in the presence of KCNE1, but the model can fit the experimental data of the hIKs - [ATP] dose response (Fig. 1B) and conductance–voltage (G–V) relationship of hIKs at various [ATP]s (Fig. 1C), suggesting that the coexpression of KCNE1 does not alter the fundamental mechanism of ATP-dependent activation. The lack of influence of KCNE1 on ATP-dependent activation is also supported by the result that both KCNQ1 and hIKs channels showed a similar response to [ATP] (Fig. 1D).
Fig. 5.

ATP is not required for VSD movement. (A) VCF recordings of the WT and ATP-binding disruptive mutant KCNQ1 channels. Whole-cell current (Upper) and fluorescence signal (Lower) of WT (black), W379S (red), and R380S/R397W (green) in response to increasing voltages. (B) Superimposed fluorescence signal (Left) of WT (black), W379S (red), and R380S/R397W (green) in response to a series of voltage pulses with increasing voltages. The currents activation time constants in response to voltages (Right). (C) Steady-state fluorescence changes vs. voltage (F–V).
Fig. 6.

ATP is not required for VSD–PGD coupling. (A) VCF recordings of WT KCNQ1 (black), W379S (red), and R380S/R397W (green) with and without L353K. (B) Steady-state fluorescence change vs. voltage (F–V). Solid curves are fits of a Boltzmann equation with V1/2. WT (black solid): −53.5 ± 1.1 mV; W379S (red solid): −56.2 ± 0.8 mV; R380S/R397W (green solid): −50.3 ± 0.9 mV; L353K (black open): −72.3 ± 0.8 mV; W379S/L353K (red open): −76.9 ± 0.9 mV; R380S/R397W/L353K (green open): −70.9 ± 1.1 mV.
Fig. 7.

ATP is required for pore opening. (A) The scheme of voltage and ATP-dependent activation of hIKs channels. Voltage sensor movements are simplified as one ATP-independent transition between the resting (V black) and activated (V red) state; the transition of the pore from closed (P black) to the open (P red) state can happen only after voltage sensor activation and ATP binding. K1: 300 M−1, L(V): 4.2 × 10−4exp(0.94VF/RT) (V, voltage; F, Faraday constant; R, gas constant; and T, absolute temperature), and K2: 1,340 and 360 for WT and Q357R IKs, respectively, are obtained from fittings to ATP dose–responses (Fig. 1B) and G–V relations at various [ATP] (Fig. 1C). (B) VCF recordings and F–V relations of Q357R and Q357R/R380S/R397W. (C) Activation (Upper) and deactivation (Lower) kinetics of fluorescence for both WT and Q357R. (D) G–V relations of hIKs WT (black), R380S (green), K393M (blue), and R397W (red). Smooth curves in B–D are Boltzmann fits to the data.
The mutation Q357R also does not alter the fundamental mechanism of ATP-dependent activation, because the disruption of ATP binding in the background of Q357R does not affect the F–V relation (Fig. 7B). Q357 is located just downstream from the S6 gate toward the C terminus in KCNQ1 and away from the amino acid cluster that is important for ATP binding (Fig. 4B), yet it causes a reduction in ATP sensitivity of hIKs activation (Fig. 1B). Perhaps more strikingly, although the WT hIKs activation requires ATP binding, the properties of hIKs activation, including steady-state G–V relation (Fig. 1C) as well as the time course of voltage-dependent activation and deactivation (SI Appendix, Fig. S13), do not change with [ATP], whereas Q357R renders the G–V relation dependent on [ATP] (Fig. 1C). Although Q357R alters the slope of the F–V relation compared with the WT KCNQ1 (Figs. 5C and 7B), the mutation does not change the kinetics of fluorescence signals (Fig. 7C) or properties of the hIKs G–V relation at high [ATP] (Fig. 1C), suggesting that the change in the ATP dependence of G–V relations may not be due to a change in VSD movement. It is also unlikely that all these changes are brought about by a direct influence of the mutation on ATP binding because, unlike Q357R, mutations that directly affect ATP binding do not cause a shift of the G–V relation to different voltages at the native [ATP] (Fig. 7D). Interestingly, a simple change in the equilibrium constant of pore opening in the proposed model (Fig. 7A) can recapitulate the mutation-caused changes in ATP sensitivity (Fig. 1B) and ATP dependence of G–V relations (Fig. 1C). These results suggest that the lack of [ATP] dependence in the WT hIKs activation properties (Fig. 1C and SI Appendix, Fig. S13) is due to a strong mass reaction pulling the VSD-activated and ATP-bound channels to the open state (Fig. 7A), whereas a reduction of the equilibrium constant for this transition by the mutation Q357R unmasks the ATP dependence of activation properties of the channel.
Discussion
IKs currents repolarize ventricular action potentials in the heart, and the IKs channel is an important regulator of heart rhythm. Here we show that intracellular ATP at physiologic concentrations (0.5–7.5 mM) acts as a signaling molecule to activate IKs channels and shorten cardiac action potentials (Figs. 1 and 2). We also identify a cluster of residues important for ATP binding located in the C terminus of KCNQ1 (Figs. 3 and 4) and the mechanism by which ATP binding alters channel function (Figs. 5–7). These results indicate that ATP binding to the cytosolic domain promotes pore opening via an allosteric mechanism.
The IKs ATP binding site has a low affinity for ATP (EC50 = 1.7 mM; Fig. 1) and does not select between ATP and GTP (Fig. 3 and SI Appendix, Fig. S2), but in the cell, ATP is present at a much higher concentration than GTP so that in physiologic conditions it can be considered an ATP binding site. Consistent with the low affinity and nonselective features, sensitivity to nucleotides with different phosphates and mutagenesis (Figs. 3 and 4) suggest that ATP is primarily coordinated by a cluster of basic charges, R380, K393, and R397, via electrostatic interaction with phosphates, whereas hydrophobic residues, including W379, stabilize the nucleoside moieties. We made a mutation scanning on aromatic residues in a part of the cytosolic domain flanking the basic residues to identify W397 (SI Appendix, Figs. S8 and S9). Our study cannot rule out the possibility that other residues may also contribute to ATP binding. Our search of the sequence of KCNQ1 did not find the conserved ATP binding motifs Walker A [GXXXXGK(T)] or Walker B (R/KXXXGXXXLhhhhD), where X represents any of the amino acids and h denotes a hydrophobic amino acid (28). KATP channels are formed by the inward rectifier channel subunit Kir 6.2/6.1 and regulatory sulphonylurea receptor subunit (29), and ATP binds to the Kir subunit. Both N and C termini for ATP binding have been identified by mutagenesis. Positively charged residues play an important role interacting with negatively charged phosphate groups of ATP, with K185 and R201 in the C terminus of one subunit and R50 in the N terminus of another directly interacting with ATP (30). In other known ATP-binding ion channels, the cystic fibrosis transmembrane conductance regulator contains Walker motifs for ATP binding (31), whereas P2X receptors bind ATP with extensive hydrophilic interactions from amino acids located in two different structural domains (32). Unlike in Kir6.2 or P2X, the putative ATP interacting residues in KCNQ1 are located in a close cluster (Fig. 4). The 3D structure of the cytosolic domain of the KCNQ1 channel at the putative binding site has not been solved; it is not clear how the ATP binding site relates to the pore or other parts of the channel.
In addition to voltage, IKs channels require both ATP and PIP2 to fully activate (11) (SI Appendix, Fig. S14), but our current and previous studies (14, 15) show that these two signaling molecules activate the channel with distinct molecular mechanisms. First, in this study, 100 µM PIP2, which is saturating for IKs channels (EC50 5 µM) (14), was always present in intracellular solutions during patch-clamp recordings to prevent reduced channel activities due to PIP2 level variations. GTP and nonhydrolysable ATP analog AMP–PNP could activate the channels with the same doses as ATP (Fig. 3A and SI Appendix, Fig. S2), ruling out the possibility of ATP activating the channel via hydrolysis for the synthesis of PIP2. Second, the PIP2 binding site was identified in between the VSD and the PGD (15), whereas the putative ATP binding site is located in the C terminus toward the S6 gate (Fig. 4). The mutations of the residues in the ATP binding site reduce ATP sensitivity but have no effect on the PIP2 sensitivity of IKs activation (SI Appendix, Fig. S15), supporting the distinctness of the two binding sites. Third, PIP2 activates the channels by mediating the coupling between VSD movements and pore opening (15), whereas ATP activates the channels by opening the pore (Figs. 5–7). The last distinctive properties between PIP2 and ATP-dependent activation is that PIP2 sensitivity of the channels is modulated by the association of the KCNE1 subunit, the EC50 of PIP2-dependent activation decreased more than 100-fold with KCNE1 association (14), whereas ATP-dependent activation is not affected by whether KCNE1 is present (Fig. 1).
Our results show that the IKs channel is a voltage- and ligand-activated K+ channel, with voltage sensor and ATP binding independently regulating pore opening. Likewise, HCN (33) and BK (34) channels are also voltage- and ligand-activated channels, both with voltage-sensing domains and intracellular ligand binding sites. A unique property of the IKs channel is that both voltage and ATP are required for the pore to open, whereas BK channels can be activated by either voltage or Ca2+ binding (35), and HCN channels can be activated by voltage alone (33). Nevertheless, it is not known if at extreme voltages the IKs channel can be activated in the absence of ATP. For both BK and HCN channels, ligand binding shifts the voltage dependence of channel opening (33, 35). However, ATP binding only enhances current amplitude without altering the G–V relation of WT IKs channels (Fig. 1).
Heart rate changes with physical activity and emotion. The modulation of the IKs channel by ATP at physiologic concentrations directly links electrical activity and heart rhythm to the energetic state of cardiac myocytes. In normal physiologic conditions, the cellular ATP level in cells is tightly maintained, but submembrane ATP concentration has been shown to fluctuate with Na+-K+ ATPase activity during action potential firings (36). In cardiac myocytes the change of APD and frequency is accompanied by changes in Na+-K+ ATPase and Ca2+ pump activity (37), which may also alter submembrane ATP concentrations. Therefore, ATP may act as a continuous regulator of heart rhythm during normal physiologic events.
In the failing human heart, ATP concentration is decreased by 35–40% (38). Complex ventricular arrhythmias are presented in about half of heart failure patients, and sudden death is common (39). Studies showed prolongation of the ventricular action potential both in animal models and in the human heart failure (40, 41). Many ion currents underlie these changes in APD, and among them IKs was found to be decreased in a canine model of heart failure, whereas IKr remained unchanged (40). However, the mechanism of this IKs reduction has not been determined. Our results provide a possible mechanism. In addition, whereas during severe ischemia, ATP concentration falls to submillimolar levels and combined with the increase in ADP levels open KATP to reduce APD and prevent excessive depolarization, thus maintaining excitability, in acute ischemia the ATP level is reduced by 50% (41) but still well above the range where activation of the KATP channel would be expected to occur (42), and a prolonged QT interval is observed (43). This phenomenon, which suggests the involvement of repolarization currents, could be explained by a reduction of the IKs channel activity. The increase of APD in acute ischemia would allow L-type Ca2+ channels to open for a longer time to compensate for the reduced Ca2+ channel activities (44) and maintain contraction. Finally, the activation of IKs by ATP also presents a unique therapeutic opportunity such that at physiologic [ATP] a reservoir of IKs remains unopened (Fig. 1); inducing these remaining closed channels to open would provide additional repolarizing currents that should shorten the APD in all circumstances in which a congenital or acquired LQT syndrome exists, independent of its origins.
Materials and Methods
All mutations were generated using overlap-extension PCR (14) and verified by sequencing. Xenopus laevis oocytes were injected with 0.05–20 ng of cRNA per oocyte, and macroscopic currents were recorded from whole oocytes or inside-out patches in 2–4 d (14). Voltage-clamp fluorometry and Western blot experiments were as previously described (15). Guinea pig single left ventricular myocyte were isolated and recorded for APs and IKs currents as described (45).
The relative conductance was determined by measuring tail current amplitudes at indicated voltages. The G–V and F–V relationships were fitted with the Boltzmann equation: 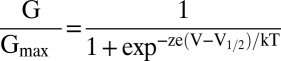 , where z is the number of the equivalent charges, V1/2 is the voltage at which the channel is 50% activated, e is the elementary charge, k is Boltzmann’s constant, and T is the absolute temperature.
, where z is the number of the equivalent charges, V1/2 is the voltage at which the channel is 50% activated, e is the elementary charge, k is Boltzmann’s constant, and T is the absolute temperature.
More detailed experimental procedures and data acquisition and analyses can be found in SI Appendix, Materials and Methods.
Supplementary Material
Acknowledgments
We thank Mark Zaydman for technical help in the VCF recording. This study was funded by National Institutes of Health Grants R01-HL70393 and R01-NS060706 (to J.C.), and R01-HL094410 and HL111401 (to I.S.C.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315649110/-/DCSupplemental.
References
- 1.Dhar-Chowdhury P, Malester B, Rajacic P, Coetzee WA. The regulation of ion channels and transporters by glycolytically derived ATP. Cell Mol Life Sci. 2007;64(23):3069–3083. doi: 10.1007/s00018-007-7332-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krebs EG. Historical perspectives on protein phosphorylation and a classification system for protein kinases. Philos Trans R Soc Lond B Biol Sci. 1983;302(1108):3–11. doi: 10.1098/rstb.1983.0033. [DOI] [PubMed] [Google Scholar]
- 3.Hilgemann DW. Cytoplasmic ATP-dependent regulation of ion transporters and channels: Mechanisms and messengers. Annu Rev Physiol. 1997;59:193–220. doi: 10.1146/annurev.physiol.59.1.193. [DOI] [PubMed] [Google Scholar]
- 4.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 5.Ashcroft SJ, Ashcroft FM. Properties and functions of ATP-sensitive K-channels. Cell Signal. 1990;2(3):197–214. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- 6.Elliott AC, Smith GL, Eisner DA, Allen DG. Metabolic changes during ischaemia and their role in contractile failure in isolated ferret hearts. J Physiol. 1992;454:467–490. doi: 10.1113/jphysiol.1992.sp019274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005;38(6):937–943. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barhanin J, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 9.Sanguinetti MC, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 10.Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358(2):169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 11.Loussouarn G, et al. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: A functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003;22(20):5412–5421. doi: 10.1093/emboj/cdg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S, et al. KCNQ1 mutations in patients with a family history of lethal cardiac arrhythmias and sudden death. Clin Genet. 2003;63(4):273–282. doi: 10.1034/j.1399-0004.2003.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boulet IR, Raes AL, Ottschytsch N, Snyders DJ. Functional effects of a KCNQ1 mutation associated with the long QT syndrome. Cardiovasc Res. 2006;70(3):466–474. doi: 10.1016/j.cardiores.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, et al. KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc Natl Acad Sci USA. 2011;108(22):9095–9100. doi: 10.1073/pnas.1100872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaydman MA, et al. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc Natl Acad Sci USA. 2013;110(32):13180–13185. doi: 10.1073/pnas.1305167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh S, Nunziato DA, Pitt GS. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ Res. 2006;98(8):1048–1054. doi: 10.1161/01.RES.0000218863.44140.f2. [DOI] [PubMed] [Google Scholar]
- 17.Shamgar L, et al. Calmodulin is essential for cardiac IKS channel gating and assembly: Impaired function in long-QT mutations. Circ Res. 2006;98(8):1055–1063. doi: 10.1161/01.RES.0000218979.40770.69. [DOI] [PubMed] [Google Scholar]
- 18.Lopes CM, et al. Protein kinase A modulates PLC-dependent regulation and PIP2-sensitivity of K+ channels. Channels (Austin) 2007;1(2):124–134. doi: 10.4161/chan.4322. [DOI] [PubMed] [Google Scholar]
- 19.Marx SO, et al. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295(5554):496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 20.Conner SD, Schmid SL. CVAK104 is a novel poly-L-lysine-stimulated kinase that targets the beta2-subunit of AP2. J Biol Chem. 2005;280(22):21539–21544. doi: 10.1074/jbc.M502462200. [DOI] [PubMed] [Google Scholar]
- 21.Roberts JA, Evans RJ. ATP binding at human P2X1 receptors. Contribution of aromatic and basic amino acids revealed using mutagenesis and partial agonists. J Biol Chem. 2004;279(10):9043–9055. doi: 10.1074/jbc.M308964200. [DOI] [PubMed] [Google Scholar]
- 22.Van Langen IM, et al. The use of genotype-phenotype correlations in mutation analysis for the long QT syndrome. J Med Genet. 2003;40(2):141–145. doi: 10.1136/jmg.40.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2(5):507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 24.Napolitano C, et al. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005;294(23):2975–2980. doi: 10.1001/jama.294.23.2975. [DOI] [PubMed] [Google Scholar]
- 25.Kapplinger JD, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6(9):1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osteen JD, et al. KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc Natl Acad Sci USA. 2010;107(52):22710–22715. doi: 10.1073/pnas.1016300108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boulet IR, Labro AJ, Raes AL, Snyders DJ. Role of the S6 C-terminus in KCNQ1 channel gating. J Physiol. 2007;585(Pt 2):325–337. doi: 10.1113/jphysiol.2007.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- 30.Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 2005;24(2):229–239. doi: 10.1038/sj.emboj.7600487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikuma M, Welsh MJ. Regulation of CFTR Cl- channel gating by ATP binding and hydrolysis. Proc Natl Acad Sci USA. 2000;97(15):8675–8680. doi: 10.1073/pnas.140220597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hattori M, Gouaux E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature. 2012;485(7397):207–212. doi: 10.1038/nature11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biel M, Schneider A, Wahl C. Cardiac HCN channels: Structure, function, and modulation. Trends Cardiovasc Med. 2002;12(5):206–212. doi: 10.1016/s1050-1738(02)00162-7. [DOI] [PubMed] [Google Scholar]
- 34.Cui J, Cox DH, Aldrich RW. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J Gen Physiol. 1997;109(5):647–673. doi: 10.1085/jgp.109.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66(5):852–875. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanner GR, Lutas A, Martínez-François JR, Yellen G. Single K ATP channel opening in response to action potential firing in mouse dentate granule neurons. J Neurosci. 2011;31(23):8689–8696. doi: 10.1523/JNEUROSCI.5951-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circ Res. 1994;74(6):1097–1113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 38.Beer M, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40(7):1267–1274. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- 39.Parmley WW. Factors causing arrhythmias in chronic congestive heart failure. Am Heart J. 1987;114(5):1267–1272. doi: 10.1016/0002-8703(87)90215-8. [DOI] [PubMed] [Google Scholar]
- 40.Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283(3):H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 41.Carmeliet E. Cardiac ionic currents and acute ischemia: From channels to arrhythmias. Physiol Rev. 1999;79(3):917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- 42.Elliott AC, Smith GL, Allen DG. Simultaneous measurements of action potential duration and intracellular ATP in isolated ferret hearts exposed to cyanide. Circ Res. 1989;64(3):583–591. doi: 10.1161/01.res.64.3.583. [DOI] [PubMed] [Google Scholar]
- 43.Jiménez Candil J, Martín Luengo C. QT interval and acute myocardial ischemia: Past promises, new evidences. Rev Esp Cardiol. 2008;61(6):561–563. doi: 10.1157/13123059. [DOI] [PubMed] [Google Scholar]
- 44.Irisawa H, Sato R. Intra- and extracellular actions of proton on the calcium current of isolated guinea pig ventricular cells. Circ Res. 1986;59(3):348–355. doi: 10.1161/01.res.59.3.348. [DOI] [PubMed] [Google Scholar]
- 45.Lu Z, et al. Suppression of phosphoinositide 3-kinase signaling and alteration of multiple ion currents in drug-induced long QT syndrome. Sci Transl Med. 2012;4(131):131ra150. doi: 10.1126/scitranslmed.3003623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.