

Significance
Symporters or cotransporters are proteins that use sodium electrochemical potential gradients to drive sugars, amino acids, anions, and neurotransmitters into cells. Progress in structural biology has provided important clues about substrate binding sites, but the identification of Na+ in binding sites has been a challenge owing to the high resolution required. We use a biophysical method to determine the Na+ affinity of electrogenic transporters, and, together with site-directed mutagenesis, we identify the two Na+ binding sites in the human intestinal Na+/glucose cotransporter SGLT1. The results provide insights into the Na+ binding sites of symporters and the mechanism of active transport.
Abstract
Sodium cotransporters from several different gene families belong to the leucine transporter (LeuT) structural family. Although the identification of Na+ in binding sites is beyond the resolution of the structures, two Na+ binding sites (Na1 and Na2) have been proposed in LeuT. Na2 is conserved in the LeuT family but Na1 is not. A biophysical method has been used to measure sodium dissociation constants (Kd) of wild-type and mutant human sodium glucose cotransport (hSGLT1) proteins to identify the Na+ binding sites in hSGLT1. The Na1 site is formed by residues in the sugar binding pocket, and their mutation influences sodium binding to Na1 but not to Na2. For the canonical Na2 site formed by two –OH side chains, S392 and S393, and three backbone carbonyls, mutation of S392 to cysteine increased the sodium Kd by sixfold. This was accompanied by a dramatic reduction in the apparent sugar and phlorizin affinities. We suggest that mutation of S392 in the Na2 site produces a structural rearrangement of the sugar binding pocket to disrupt both the binding of the second Na+ and the binding of sugar. In contrast, the S393 mutations produce no significant changes in sodium, sugar, and phlorizin affinities. We conclude that the Na2 site is conserved in hSGLT1, the side chain of S392 and the backbone carbonyl of S393 are important in the first Na+ binding, and that Na+ binding to Na2 promotes binding to Na1 and also sugar binding.
Ion coupled symporters, or cotransporters, such as hSGLT1 use electrochemical potential gradients to drive solutes into cells. A common finding for these transporters is that external Na+ binds before the substrate. Na+ binding induces a conformational change of the protein, resulting in the substrate vestibule becoming open to the external membrane surface. After substrate binding, the two ligands are transported across the membrane and are released into the cytoplasm. The atomic structures of several sodium dependent transporters have been solved [leucine transporter (LeuT), vibrio parahaemolyticus sodium glucose (vSGLT), sodium hydantoin (Mhp1), and sodium betaine (BetP)] (1–6). They share a common structural fold with a five-helix inverted repeat, the “LeuT fold”. The substrate binding sites are located in the middle of the protein, isolated from the external and membrane surfaces by hydrophobic gates, and putative Na+ sites have been identified. Testing these binding sites is a problem due to the fact that phenomenological kinetic constants for ligand transport (K0.5, half-saturation values) are interdependent on each other (7). Here we have developed a method to estimate the intrinsic Na+ dissociation constants (Kd) for human sodium glucose cotransport (hSGLT1) that may be broadly applicable to other symporters. We then use this to investigate the importance of hSGLT1 residues predicted to be at or near the two Na+ binding sites, Na1 and Na2.
The method is based on the fact that voltage-dependent membrane proteins exhibit transient charge movements (capacitive transients) in response to step changes in membrane potential (8, 9). Voltage-dependent transport proteins, including Na+ cotransporters and pumps, exhibit these capacitive currents (7, 10–12). Transient charge movements may be fit with a Boltzmann relation to estimate the voltage at half-maximal charge transfer (V0.5), where the protein is equally distributed between the two extreme states, the apparent valence of the moveable charge (z) and the number of transporters in the plasma membrane (Qmax) (10, 13). The hSGLT1 Boltzmann parameters are sensitive to the external Na+ concentration [Na+]o: As [Na+]o is lowered there is a reduction in Qmax and z and a shift of V0.5 to more negative voltages. The V0.5 shift is used to determine the Na+ affinity (Kd) for hSGLT1. Basically, it is a thermodynamic method where we determine the voltage where the transporter is equally distributed between two extreme states (outward-facing Na+-bound C2Na2 and inward-facing ligand-free C6), as a function of [Na+]o (Fig. 1). A similar approach has been used to measure the kinetics of Na+/Na+ exchange by the Na+/K+ pump (14, 15). Here we use the method with hSGLT1 mutants to locate the two Na+ binding sites. The results show that Na1 is part of the sugar binding pocket and Na2 is at the same location as other members of the LeuT structural family.
Fig. 1.

Cartoon representing three- and four-state models for Na+ binding to SGLT1. For clarity of presentation, we assume the number (n) of Na+ ions binding to the empty transporter is 2. The ligand-free protein carries a net negative charge and transient capacitive SGLT1 currents are associated with reorientation of the empty transporter between external and internal membrane surfaces (C1 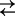 C6) and Na+ binding/dissociation (C1 + 2Na+
C6) and Na+ binding/dissociation (C1 + 2Na+
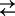 C2Na2 for the three-state and C1+Na+
C2Na2 for the three-state and C1+Na+
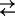 C1Na + Na+
C1Na + Na+
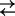 C2Na2 for the four-state model). Kd is the lumped sodium dissociation constant for the three-state model, and KdA and KdB are the sodium dissociation constants for the first and second binding sites for the four-state model (Theory section). Hyperpolarizing voltages drive the transporter to the outward conformation (C6 → C1), as well as promoting the transition C1 → C2Na2.
C2Na2 for the four-state model). Kd is the lumped sodium dissociation constant for the three-state model, and KdA and KdB are the sodium dissociation constants for the first and second binding sites for the four-state model (Theory section). Hyperpolarizing voltages drive the transporter to the outward conformation (C6 → C1), as well as promoting the transition C1 → C2Na2.
Theory
First, consider a three-state model for hSGLT1, where n external Na+ ions bind simultaneously to the empty transporter in the absence of external sugar (Fig. 1). The empty transporter is assumed to be negatively charged and opens to the internal (C6) or external (C1) membrane surfaces, depending on membrane voltage (V). Voltage also affects external Na+ binding and/or the conformational changes associated with it. The transporter is distributed between C6, C1, and C2Nan, depending on voltage and [Na+]o.
We assume the rate constants k12, k21, k16, and k61 depend on voltage according to rate theory with symmetric energy barriers (16, 17)
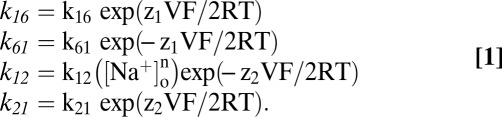 |
k16, k61, k12, and k21 are voltage-independent rate constants. z1 and z2 represent the apparent valence of the moveable charge for the empty transporter (z1) and Na+ binding (z2). F is the Faraday constant, R is the gas constant, and T is absolute temperature.
The occupancy probabilities in C6, C1, and C2Na2 are
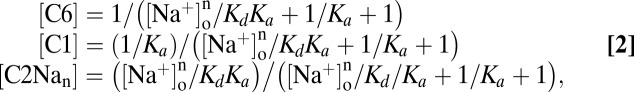 |
where
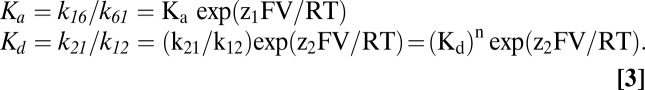 |
Ka (k16/k61) is the equilibrium constant of the distribution of the empty transporter between states C6 and C1 (at 0 mV), and Kd ((k21/k12)1/n) is the intrinsic Na+ affinity at 0 mV. Note that Kd is a lumped parameter for the binding of n Na+ ions.
Our focus is on the ability of [Na+]o to alter the distribution of hSGLT1 conformations. The distribution (in steady state) is characterized by the midpoint voltage V0.5, where the protein is equally distributed between C2Nan and C6.
To obtain the relation for any given [Na+]o, consider the charge moved (Q) when membrane voltage (V) is stepped from a very negative (where all transporters are in C2Nan) to a more positive value (17, 18). Q is the sum of the charge transferred from C2Nan to C1 (each transition contributing a net charge z2) and from C2Nan to C6 (contributing a total charge z = z2 + z1). For a membrane with a transporter density NT, the total charge is
and maximal charge Qmax (= NTe(z1 + z2), where e is elementary charge). The Q-V relation is obtained by substituting for [C2Nan], [C1], and [C6] from Eq. 2:
At V = V0.5, Q = Qmax/2, and rearranging (with u = F/RT), we obtain the relation between V0.5 and [Na+]o:
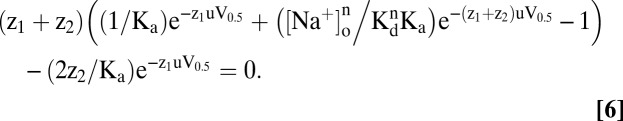 |
Simulation of the V0.5/[Na+]o Relations.
Ka and Kd can be estimated from Eq. 6 (18). In the absence of external Na+, the empty transporter alternates between C6 and C1, depending on voltage. The voltage where C6 and C1 are equally distributed, V0.5, is given by
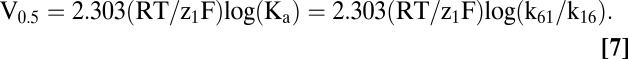 |
At high [Na+]o (>>Kd), the transporter is distributed between C2Nan and C6 as a function of voltage. It can be shown from Eq. 6 (18) that as [Na+]o approaches saturation, V0.5 is linearly related to log [Na+]o with slope given by the ratio of the number (n) of Na+ ions bound and the net charge (z) of the protein:
Eq. 8 is equivalent to the conservation of energy: The energy required to move charge z (z = z1 + z2) through a potential difference (ΔV0.5) is provided by the chemical potential for n Na+ ions.
The theory may be extended to the sequential binding of n Na+ ions. For clarity, we restrict our discussion to n = 2 (Fig. 1) (18), but retain “n” to emphasize the dependence on the number of Na+ ions. The V0.5 vs. [Na+]o relation for sequential binding is given by
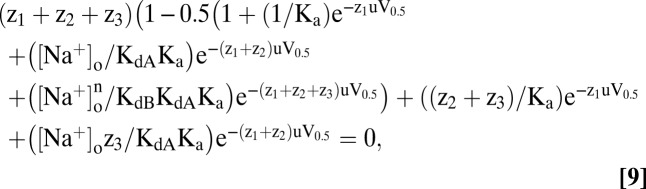 |
where Ka is the equilibrium constant of the empty transporter, and KdA (= kN1/k1N), and KdB (= k2N/kN2) are the Na+ affinities of the first and second sites (at 0 mV), respectively. z1, z2, and z3 are the charge associated with the empty transporter and the first and second Na+ binding steps (Fig. 1). KdA and KdB are related to the lumped Kd by Kd = √ (KdAKdB).
The limiting behaviors of the V0.5 vs. [Na+]o relations are identical to those for the three-state model. In the absence of Na+, V0.5 depends on the apparent valence (z1) of the empty transporter and the ratio of rate constants k16/k61 (Eq. 7); and in saturating [Na+]o, V0.5 is linearly related to log [Na+]o with the slope given by the ratio (n/z) of the number of Na+ ions and the total charge z (Eq. 8). The difference lies in the approach to the limits. For the four-state model, as [Na+]o approaches zero, the V0.5 vs. [Na+]o relations are governed by the affinity (KdA) of the first Na+ site. In contrast, as [Na+]o nears saturation the approach to maximal slope (115.6 mV/decade) is determined by the affinity (KdB) of the second binding site. We anticipate differences in the predictions of the three- and four-state models if there are significant differences between KdA and KdB (see below).
Estimating Parameters.
Ka is estimated from V0.5 in the absence of Na+ (Eq. 7) and k16 and k61 from the ratio and the time constant (τo) [τo = 1/(k16 + k61) and Ka = k61/k16]. Apparent valences z1 and z (= z1 + z2) are obtained from the Q-V curves in the presence and absence of external Na+. For hSGLT1 mutant Q457C, we have shown that n = 2 (19). Na+ affinities (Kd or KdA and KdB) can then be extracted by numerically fitting the V0.5 vs. [Na+]o data to Eqs. 6 and 9 (see Figs. 5 and 6).
Fig. 5.

Dependence of V0.5 on [Na+]o for WT hSGLT1 and mutant W291C. (A) The V0.5 vs. log [Na+]o data are from Fig. 4. The error bars (SE) for V0.5, less than the size of the symbols, are the error of the fit to the experimental data in Fig. 4. The curves are simulations of the three- and four-state models (Eqs. 6 and 9) for the individual oocytes shown in Fig. 4. For WT, Kd = 20 mM for the three-state and KdA = 33 mM, KdB = 10 mM for the four-state model. For W291C, Kd = 55 mM for the three-state and KdA = 30 mM, KdB = 110 mM for the four-state model. k16/k61 = 100 for WT and 1.6 for W291C. For both transporters, z1 = 0.7, z2 = 0.15, and z3 = 0.15. Net charge z (= z1 + z2 + z3) = 1. (B) Replots of the three- and four-state simulations for WT and W291C as the slope of V0.5 vs. log[Na+]o (ΔV0.5/Δlog [Na+]o). This shows the transition between the two limiting slopes for each model and the distribution of the experimental data. The asterisks indicate the [Na+]o where V0.5 could be reliably estimated (A).
Fig. 6.

Dependence of V0.5 on [Na+]o for Y290C, Y290S and Y290F (A) and H83C and S392A (B). Data shown are from individual oocytes, with error of the fit. When V0.5 was more negative than −100 mV (for Y290F, H83C, and S392A at low [Na+]o), the open symbols were obtained from fitting the Q/V curves under the constraint that z = 0.7. The curves are from simulations of the four-state model (Eq. 9): KdA = 30 mM, KdB = 110 mM, and k16/k61 = 1 for Y290S; KdA = 30 mM, KdB = 110 mM, and k16/k61 = 2.4 for Y290C; KdA = 30 mM, KdB = 112 mM, and k16/k61 = 8 for Y290F; KdA = 30 mM, KdB = 480 mM, and k16/k61 = 15 for H83C; and KdA = 50 mM, KdB = 300 mM, and k16/k61 = 10 for S392A. z1 = 0.7, z2= 0.15, z3 = 0.15, z = 1 (z = 1 = z1 + z2 + z3), and n = 2 for all five proteins.
Results
The selection of mutants at putative Na1 and Na2 sites was guided by previous findings on the kinetics of hSGLT1 mutants (20, 21) and the location of the Na1 and Na2 sites in LeuT (6). Fig. 2 shows the location of the glucose binding site and the Na2 site in a homology model of hSGLT1 (20). The glucose binding site mutants that express well in the plasma membrane of oocytes are H83C, Y290C/S/F, W291C, and Q457C. All showed an increase in sugar K0.5 compared with WT, consistent with their role in sugar binding (20), but the K0.5 for Na+ also increased >10-fold for Y290C and W291C (21). The interpretation of reductions in apparent sugar and Na+ affinities is ambiguous because the apparent affinities (K0.5) for sugar and Na+ are interdependent (17, 20, 22).
Fig. 2.

Homology model (side view) of hSGLT1 in the inward-occluded conformation (20) based on the inward-occluded vSGLT structure (Protein Data Bank ID code 3DH4) (2). Glucose coordinates with H83 and N78, Y290 and W291 (CH–π interactions), Q457 (O5 and O6), and E102 (not shown for clarity). The hydroxyl (OH−) of Y290 forms an H bond with N78; Y290 and W291 are involved in a π T-stacking interaction. Na+ (yellow sphere) is shown 10 Å away from the sugar site at the Na2 site with S392 and S393.
The putative Na2 site is 10 Å from the sugar binding site (Fig. 2) and is formed by residues on TM1 and TM8—S393, S392, and the backbone carbonyls of S389, I79 and A76. Mutations of S392 (S392A/C) produced dramatic increases in K0.5 for both sugar and Na+, whereas mutations of S393 (S393A/C) resulted in only modest changes (20). Our aim was to measure the intrinsic Na+ affinity (Kd) for H83C, Y290C/S/F, W291C/F, Q457C, and S392A and other mutants not expected to be involved in ligand binding, i.e., T287C, F101C, F453C, and G507C (20, 23).
WT hSGLT1.
Representative current records for an oocyte expressing wild-type hSGLT1 in 0-, 10-, and 100-mM NaCl buffers are shown in Fig. 3A. The membrane potential was held at −50 mV (Vh) and stepped (ON pulse) to test values (between +50 mV and −150 mV) for 100 ms before returning to Vh (OFF pulse). In response to the voltage jump, total membrane current relaxation consists of an initial capacitive spike (with a time constant τ ∼ 1 ms) due to the membrane lipid bilayer, followed by a slower hSGLT1 transient current that decays to a steady state (τ ∼ 3–30 ms). When membrane voltage was returned to Vh, both the bilayer and the hSGLT1 transient currents were in the opposite direction. hSGLT1 pre–steady-state currents were blocked by phlorizin (a SGLT1-specific inhibitor) and glucose (10, 22, 24). In 10 mM Na+, the currents were smaller and the relaxations were faster. In the absence of Na+, charge movements were clearly observed when membrane voltage was returned from a large hyperpolarizing voltage (e.g., −150 mV) back to the holding potential, but these were less than 1 nC (Fig. 3A). The small charge movements in the absence of Na+ are due, in part, to the fast time constants, <2 ms at −50 mV (see Fig. 7) and the shift of V0.5 to extreme negative values beyond the range of the experimental measurement: Theory predicts that V0.5 approaches −200 mV in the absence of [Na+]o (see Fig. 5A).
Fig. 3.

Current records for oocytes expressing wild-type hSGLT1 and mutant W291C. The experiments were performed with membrane potential held at −50 mV (Vh) and stepped to a series of test voltages (from +50 mV to −150 mV) for 100 ms before returning to Vh. Current records for hSGLT1 (A) and for mutant W291C (B) in 0 mM, 10 mM, and 100 mM. Na+ concentration was varied by equimolar replacement of NaCl with choline chloride.
Fig. 7.

Dependence of relaxation time constants (τ) on membrane voltage (Vm) for WT hSGLT1 and mutant W291C in the absence of Na+. Data were obtained from the experiments in Fig. 3. The solid symbols represent the ON response when Vm was stepped from holding (Vh = −50 mV) to various test values, and the open symbols are from the subsequent OFF response when Vm was returned from test voltage to Vh. For the OFF, τ was independent of test voltage, and data shown are the mean of the 10 voltage pulses. The error bars are SEs of the fit and when absent were smaller than the size of the symbol.
The hSGLT1 transient currents were integrated to obtain the total charge transferred (Q) for each voltage pulse. Q's for the ON and OFF transients were equal and opposite at each voltage. A Boltzmann relation (Eq. 11) was used to fit the charge vs. voltage (Q-V) curves (Fig. 4A) to obtain Qmax, V0.5, and z. In saturating [Na+]o, Qmax provides an index of the number of hSGLT proteins in the plasma membrane. Qmax was 10.6 nC in 100 mM [Na+]o, corresponding to 1 × 1010 transporters in the oocyte plasma membrane (10, 13). V0.5 was −39.5 ± 2 mV; and z was 1.0. Reducing [Na+]o decreased Qmax and z and shifted V0.5 to more negative voltages. At 25 mM Na+ V0.5 shifted from –39.5 ± 1 mV at 100 mM Na+ to −90 ± 2 mV (Fig. 4A). Qmax decreased 30% from 10.6 ± 0.2 nC to 7.6 ± 0.2 nC and z from 1.0 ± 0.1 to 0.8 ± 0.1. Below 25 mM Na+, the Q-V curves did not saturate at the largest hyperpolarizing voltage that could be applied (−150 mV).
Fig. 4.

Characteristics of the charge movement for wild-type hSGLT1 and mutant W291C. The data shown are from individual oocytes. The charge vs. voltage (Q-V) relations are shown for hSGLT1 (A) and W291C (B) when [Na+]o varied from 100 mM to 0 mM. The curves were obtained by fitting the Q/V curves to the Boltzmann relation (Eq. 11) and are aligned at the depolarizing limit. Q/V data for hSGLT1 at 0 mM could not be fitted due to the extremely negative V0.5 (∼ −200 mV, Fig. 5).
W291C.
Representative current records for mutant W291C are shown in Fig. 3B. In 100 mM Na+, the pre–steady-state currents were observed only with depolarizing voltage pulses. In the absence of Na+, the pre–steady-state currents are similar for both hyperpolarizing and depolarizing voltage pulses—the symmetry of the current is particularly clear in the OFF records.
The Q-V relations for mutant W291C as a function of [Na+]o are shown in Fig. 4B. In 100 mM Na+, the Boltzmann parameters Qmax, V0.5, and z were 13 ± 1 nC, 7 ± 2 mV, and 0.9 ± 0.1. As [Na+]o is reduced to zero, Qmax decreased by 20% from 13 nC to 10 nC, z decreased from 0.9 ± 0.05 to 0.6 ± 0.05, and V0.5 shifted from 7 ± 2 mV to −52 ± 5 mV. The values obtained for V0.5 and z in 0 mM and 100 mM Na+ for W291C and all mutants are summarized in Table 1. Note that the apparent valence (z) averaged 1.0 in 100 mM Na+ and 0.7 in the absence of Na+, consistent with the hypothesis that 70% of the charge transfer was associated with the reorientation of the empty transporter (between C1 and C6) and 30% to Na+ binding (C1 to C2Na2), possibly through a Na+-well effect and/or a change in conformation (10, 17).
Table 1.
V0.5 and z at 100 and 0 mM [Na+]o
| V0.5, mV |
Z |
|||
| Mutant | 100 Na+ | 0 Na+ | 100 Na+ | 0 Na+ |
| hSGLT1 | −39 ± 2 | < −120 | 1.0 ± 0.1 | n.m. |
| Near sugar binding | ||||
| H83C | −42 ± 3 | −120 to −140 | 0.9 ± 0.1 | n.m. |
| T287C | −47 ± 1 | < −120 | 1.0 ± 0.1 | n.m. |
| Y290C | 3 ± 4 | −54 ± 4 | 0.9 ± 0.1 | 0.6 ± 0.1 |
| Y290S | 18 ± 1 | −40 ± 6 | 0.9 ± 0.1 | 0.7 ± 0.1 |
| Y290F | −49 ± 13 | < −120 | 0.9 ± 0.1 | n.m. |
| W291C | 12 ± 2 | −53 ± 2 | 0.9 ± 0.1 | 0.6 ± 0.1 |
| W291F | −49 ± 9 | < −120 | 1.0 ± 0.1 | n.m. |
| Q457C | −55 ± 4 | < −120 | 1.0 ± 0.1 | n.m |
| Near Na2 site | ||||
| D204E* | −50 ± 2 | −67 ± 3 | ∼1 | ∼1 |
| S392A/C | −56 ± 3 | −100 to −140 | 0.9 ± 0.1 | n.m. |
| Other | ||||
| F101C | −3 ± 3 | < −120 | 1.0 ± 0.1 | n.m. |
| F453C | −28 ± 4 | < −120 | 1.0 ± 0.1 | n.m. |
| G507C | −35 ± 1 | < −120 | 1.0 ± 0.1 | n.m. |
Kinetics are mean ± SE of three to nine oocytes from at least two donor frogs. For mutants with Q/V curves that did not saturate (within the range −150 mV and +50 mV) at 0 Na+, we were unable to obtain reliable estimates of V0.5 and z. V0.5 was more negative than −120 mV (< −120 mV) and z was designated by nonmeasurable (n.m.). At 0 Na+, z = z1, and at 100 mM Na+, z = z1 + z2 + z3 (Eq. 9).
Data are from Quick et al. (25), where the data at 0 Na+ were actually obtained in 5 mM Na+.
Dependence of V0.5 on [Na+]o.
The V0.5 vs. log [Na+]o data for WT hSGLT1 are shown in Fig. 5A. Between 25 mM and 100 mM Na+, the regression line through the data yielded a slope of 93 ± 5 mV per 10-fold change in [Na+]o, but above 50 mM the slope approached the theoretical limiting value of 115.6 mV/decade (Eq. 8 at 22 °C) (ref. 10, figure 23E in ref. 22, and figure 3C in ref. 25). The limiting slope data are consistent with two Na+ ions binding to hSGLT1 (Eq. 8 with n = 2 and z =1 for hSGLT1). For all mutants studied the V0.5 vs. log [Na+]o curves also approached the limiting value at high [Na+]o (for example, Figs. 5A and 6). Together with z values of 1 (Table 1), we conclude n = 2 for these mutants.
As the [Na+]o is reduced toward zero, the theory predicts that V0.5 approaches the limiting value determined by Eq. 7 (V0.5 = 2.303 (z1F/RT) log(k61/k16)). For wild-type hSGLT1, V0.5 is predicted to be between −150 mV and −200 mV (Fig. 5), well beyond the experimental working range. However, for W291C, V0.5 was essentially independent of [Na+]o, below 10 mM with a limiting value of −57 mV (Fig. 5A); i.e., there is a charge movement associated with the ligand-free protein. Similar observations were made for Y290C, Y290S, Y290F, H83C, and S392A (Fig. 6) with limiting values between −40 mV and −120 mV. The V0.5 at 0 [Na+]o yields k16/k61 ratios for the reorientation of the empty transporter between the outward and inward facing conformations (Eq. 7), and these range from 100 for WT to 2 for Y290C, Y290S, and W291C (Table 2).
Table 2.
Na+ affinity of hSGLT1 mutants
| Kd, mM | KdA, mM | KdB, mM | k16/k61 | τo, ms | k16, s−1 | k61, s−1 | |
| hSGLT1 | 20 | 33* | 10* | 100 | <2† | 530 | 5 |
| Near sugar binding site | |||||||
| H83C | 120 | — | >150 | 6 | 6 | 170 | 10 |
| T287C | 20 | — | — | 200 | — | — | — |
| Y290C | 57 | 30 | 110 | 2.4 | 9 | 80 | 33 |
| Y290S | 57 | 30 | 110 | 1.4 | 17 | 60 | 25 |
| Y290F | 120 | 30 | >150 | 24 | 10 | 100 | 4 |
| W291C | 55 | 30 | 110 | 2 | 16 | 21 | 43 |
| W291F | 20 | — | — | 100 | 4 | 275 | 3 |
| Q457C | 20 | — | — | 250 | 2 | 530 | 2 |
| Near Na2 site | |||||||
| D204E | >150 | — | — | 2.6 | — | — | — |
| S392A | 122 | 50 | >150 | 10 | 19 | 48 | 5 |
| Other | |||||||
| F101C | 18 | — | — | 35 | — | — | — |
| F453C | 20 | 33* | 10* | 125 | — | — | — |
| G507C | 20 | 33* | 10* | 100 | — | — | — |
Na+ affinities, Kd (= √(k21/k12)) for the three-state and KdA (= kN1/k1N) and KdB (= k2N/kN2) for the four-state model (Fig. 1), were estimated from the V0.5 vs. log [Na+]o data (Figs. 5 and 6). Parameters are those obtained in individual cells, but representative of three to five experiments conducted on different batches of oocytes. For all proteins, the variation in Kd was 10% and, excluding the WT group, the variation in KdA was 10%. τo was obtained from the time constants for relaxation of the transient charge movements in the absence of sodium (e.g., Fig. 7). The values given for KdA and KdB are for those where V0.5 in the absence of Na+ fell within the experimental range (Table 1), reducing the number of unknown parameters in Eq. 9. For all proteins, the number of Na+ ions (n) was 2, and net charge (z) was 1. z1 = 0.7, z2 = 0.3, for the three-state model and z1 = 0.7, z2 = 0.15, z3 = 0.15 for the four-state model. The absence of data for mutants (—) reflects those where V0.5 in the absence of Na+ fell outside the experimental range (Table 1) and those not determined.
The Na+ affinities of the two sites for the WT group are similar, because the three-state model fitted the data, and Kd = √KdAKdB.
Loo et al. (24).
Estimation of Kd.
First we use the three-state model to estimate the lumped Na+ binding constant Kd, by simulation of Eq. 6. The unknowns in Eq. 6 are Ka and Kd given that z1 = 0.7, z = 1 (Table 1), and n =2 (see above): In some cases Ka is known for those mutants where V0.5 falls within the experimental range at [Na+]o = 0 (Table 1). In general, Kd and, if necessary, Ka were varied until the simulated V0.5/log [Na+]o curves fit the experimental data. For WT and for F101C, T287C, W291F, F453C, Q457C, and G507C, the estimated parameters were Kd ∼ 20 mM and k16/k61 ∼ 100 (Table 2). In contrast, the Kd for mutants at the glucose and Na+ binding sites, H83C, Y290C/S/F, W291C, and S392A, was three- to sixfold higher than for WT, 57–122 mM, and the k16/k61 ratio decreased from 100 to less than 25 (Table 2). Replacing the cysteine side chains at W291C with W291F restored the Kd to the wild-type value, but replacing Y290C with Y290F did not.
Next, we use the four-state model to estimate KdA and KdB by numerical simulation of Eq. 9. The limiting conditions, at [Na+]o = 0 and at high saturating [Na+]o, are identical for both the three- and four-state models; therefore, n = 2, z1 = 0.7, and z1 + z2 + z3 = 1. Assuming that z2 = z3 = 0.15, the unknowns in Eq. 9 are reduced to KdA and KdB. For WT hSGLT1 (Fig. 5) and F101C, T287C, W291F, F453C, Q457C, and G507C, the data are equally well simulated by both models with a Kd = 20 mM, KdA = 33 mM, and KdB = 10 mM. Recall that Kd = √ (KdAKdB) = √ (33 × 10) ∼ 20 mM. This indicates that Na+ affinities for the two sites in hSGLT1 are similar. To distinguish between these requires that we extend the experiments to lower [Na+]o but this is impractical as V0.5 moves well outside the experimental range (more negative than −120 mV, Fig. 5).
For some mutants, for example W291C, the V0.5 at 0 mM [Na+]o falls within the experimental range, 0 mV to −120 mV (Figs. 5 and 6 and Table 1), and Ka is determined experimentally (Eq. 7) (Table 2). For W291C the four-state model gives a better fit to the data with KdA = 30 mM and KdB = 110 mM: This was facilitated by the distribution of the experimental data between the two limiting slopes (Fig. 5B). Similar results were obtained for Y290C/S/F with KdA ∼ 30 mM and KdB > 100 mM (Fig. 6 and Table 2).
The V0.5 vs. log [Na+]o plots for H83C and S392A were intermediate between those for WT and W291C, with V0.5 in the absence of Na+ falling just outside the experimental range, −100 mV to −140 mV. For S392A, with the constraint that z = 0.7 (at 0 Na+), we estimated the KdA = 50 ± 5 mM (4) and KdB > 150 mM. We were unable to obtain reliable estimates of KdA for H83C.
Time Constants for Charge Transfer.
The time constants (τo) for charge transfer in the absence of Na+ at 0 mV are needed to resolve Ka into its components, k61 and k16 (Eq. 7). These are shown in Fig. 7 for hSGLT1 and W291C. τo was <2 ms for WT, F101C, T287C, W291F, F453C, Q457C, and G507C (Table 2). τo was 16 ms for W291C and ranged between 6 ms and 19 ms for H83C, Y290C/S/F, W291C, and S392A (Table 2). Making the simplifying assumption that the transitions between C1 and C6 occur in one step, the k16/k61 values obtained from V0.5 in the absence of Na+ (Eq. 7 and Table 1) and the relationship τo = 1/(k16 + k61) give estimates of k16 and k61 (Table 2). The most striking results were for Y290C, Y290S, and W291C, which show a reduction in k16 from ∼500 s−1 to 20–80 s−1 and an increase in k61 from 5 s−1 to 25–45 s−1. These changes caused by removing the aromatic side chains were partially restored with Y290F and W291F, suggesting aromatic interactions between Y290 and W291 play an important role in the transitions between C1 and C6 (Fig. 1).
Discussion
Na+ plays a key role in the alternating mechanism of Na+ cotransporters. Substituted cysteine accessibility method (SCAM) studies on hSGLT1 show that external Na+ binding triggers a conformational change to open the sugar binding pocket to the external membrane surface (20, 26, 27). Identifying the Na+ binding sites and the coupling between Na+ and glucose transport is crucial to our understanding of the transport mechanism. Locating Na+ binding sites in membrane proteins is a nontrivial task even with the recent advances in structural biology. To distinguish between the densities of sodium and water in crystals it is necessary to resolve structures to greater than 1.2 Å (28). So far, the resolution for the LeuT structural family of transporters is between 1.6 Å and 3.5 Å (2, 4–6).
A structural motif was obtained from a survey of Na+ binding sites in 132 proteins in the Protein Data Bank (29), showing that approximately half are “neutral” and half contain one charged coordinating residue. In both the coordination number is ∼5 with an average Na+ to ligand distance of 2.35 Å. In the highest-resolution structure of cotransporters obtained to date, LeuT at 1.6 Å (6), two Na+ sites have been proposed on the basis of an indirect valence test (30). Na1 has six bonds between Na+ and the protein, including two carbonyl oxygens (A22 and T254), the hydroxyl oxygen from T254, side–chain amide oxygens from N27 and N286, and the charged carboxylate group from the leucine substrate. The Na2 site is neutral with five bonds between Na+ and the protein, including three backbone carbonyl oxygens (G20, V23, and A351) and two side chain –OH groups (T354, S355). The Na2 site is conserved in the three other members of the LeuT structural family, vSGLT, Mhp1, and BetP, and the mammalian SGLTs, but not in the Na+-independent transporters (1, 3). Substrate transport by SGLT1 is driven by two Na+ ions, unlike vSGLT and Mhp1, which are driven by one (20, 21, 25, 31). Thus, there is a second binding site in hSGLT1 (Na1).
We and others have attempted to identify ligand binding sites by mutating residues and measuring the kinetics of Na+ and substrate transport. However, it is not generally appreciated that the K0.5 for Na+ and the K0.5 for substrate are interdependent, and changes in Na+ K0.5 may actually be due to changes in the substrate K0.5 and vice versa (7, 23). New methods are needed to determine the Kd of ligand binding to cotransporters in the absence of the cosubstrate, and we have developed a method to determine the Na+ Kd in electrogenic cotransporters. The underlying principle is that external Na+ binding in the absence of substrate alters the equilibrium distribution of the transporter between two conformational states, C6 and C2Na2 (Fig. 1). We measure the midpoint voltage (V0.5) where the protein is equally distributed between C6 and C2Na2 as a function of [Na+]o. Na+ binding shifts the proteins from C6 to C2Na2, and the Na+ affinities are determined from the relationship between V0.5 and [Na+]o (Figs. 5 and 6).
Overall, the theoretical predictions are borne out experimentally in that there are two limiting cases for both the three- and four-state models in the absence of Na+ and at high [Na+]o, and the Na+ affinity determines the transition between the two limiting conditions. For wild-type hSGLT1 (and mutants F101C, T287C, W291F, F453C, Q457C, and G507C) the experimental data are equally well described by both three- and four-state models (Fig. 5) with Kd = 20 mM, KdA = 33 mM, and KdB = 10 mM (Kd = √ (KdAKdB)). This is in part due to the similarity of the Kds for the two sites and/or the lack of data within the experimental limits (Fig. 5). At low [Na+]o, V0.5 falls outside the experimental range (Fig. 4). However, for some mutants, a better fit is obtained with the four-state model (e.g., W291C in Fig. 5), permitting a robust measure of KdA and KdB (Table 2). How specific are the observed reductions in Na+ affinity? We found that Kd was unaffected by mutation of other residues at (i) the sugar coordinating residue Q457C; (ii) residue T287C, near the sugar binding pocket; (iii) two outer gate residues, F101C and F453C; and (iv) a remote residue, G507C, on the external loop linking TM11 and TM12.
hSGLT1 Na2 Site.
This putative site is conserved between vSGLT (A62, I65, A361, S364, and S365) and hSGLT1 (A76, I79, S389, S392, and S393) (Fig. 8A). We have previously reported that mutation of one (S392) of the two conserved serines (S392, S393) produced significant decreases in the apparent affinity for Na+, sugar, and phlorizin. The increase in sugar K0.5 (from 0.5 mM to >100 mM) and phlorizin Ki for this mutant (20) can be explained by the loss of intrinsic Na+ affinity. The lack of effect of S393 mutations (S393A/C) suggests that it is not the side chain, but perhaps the backbone carbonyl of this residue that serves as a Na2 site. Mutations of T467 and S468 in the Na2 site in BetP produce similar increases in the Kd (32).
Fig. 8.

Predicted Na1 and Na2 sites shown in a homology model of hSGLT1 (Fig. 2). Na+ ions are colored yellow. (A) Na2 site. The Na2 site is 10 Å from the sugar binding site. Na+ binding is predicted to involve H bonds with carbonyl oxygens of A76, I79, and S389 (bond length <4 Å) and the side chain hydroxyl (OH−) from S392 (bond length 5 Å). The Na+ bond with D204 is predicted to occur via a water bridge (35). No Na+ bonding is shown to the side chain of S393 in accordance with the experimental data. (B) Na1 site. A Na+ ion is placed ∼2.5 Å from the carbonyl oxygen of N78 and H83 [corresponding residues are in the LeuT Na1 site (6)]. Also shown are distances between Na+ and Y290 (3.3 Å), W291 (6.3 Å), E102 (7.5 Å), and the O2 of glucose (3.5 Å).
In our study, there was a fivefold increase in Kd for S392A from 20 mM to 122 mM (Table 2), and this was due to an increase in both KdA from 30 mM to 50 mM and KdB from 10 mM to >150 mM. Because KdB is a measure of Na+ binding to Na1 (see below), we infer that mutation of S392 produces a direct reduction in Na+ binding to Na2 and a long-range effect on Na+ binding to Na1 and indirect effects on the sugar and inhibitor binding pocket.
D204 has a low Na+ affinity (Table 2), consistent with molecular dynamics (MD) studies on vSGLT showing that Na+ leaving the Na2 site interacts with the conserved aspartate (33, 34). Na+ may coordinate with D204 through a water bridge as predicted from the bond distance of 6.4 Å between Na+ and the equivalent D189 in vSGLT (35) (Fig. 8A).
hSGLT1 Na1 Site.
The starting point in our search was the finding that Na1 is close to the substrate binding site in LeuT (6). Residues predicted to be involved in coordinating glucose in hSGLT1 include H83, N78, E102, Y290, W291, K321, and Q457 (20). There were dramatic increases in the lumped Kd for H83C, Y290C, and W291C, but not for Q457C or another residue, T287C, in the sugar binding pocket. The 3- to 6-fold increases in Kd are due to a >10-fold increase in KdB with no change in KdA (Table 2). This suggested that H83, Y290, and W291 are involved in sodium binding to the Na1 site (Fig. 8B) and that KdB is the affinity for this site. Interestingly, the apparent affinity for glucose decreased by >100-fold for these mutants (20) and indicates that Na+ and glucose share binding to H83, Y290, and W291. How is it compatible that sugar and Na+ share binding sites? MD simulations on vSGLT have shown that substrate and Na+ binding is not static, but dynamic: e.g., galactose at the crystal binding sites rapidly exchanges bonds with water and other adjacent residues (34, 36, 37).
Residues W291, Y290, and W289 form an aromatic triad conserved in glucose and inositol transporters, and W291 and Y290 are involved in a T-stacking π–π interaction (21). We have previously found the loss of aromaticity at residue 291 (with mutant W291C) resulted in sugar (αMDG) K0.5 > 100 mM and restoring an aromatic side chain (W291F) largely restored sugar affinity (K0.5 = 4 mM compared with 0.6 mM for WT hSGLT1) (21). The present study shows that Na+ binding to Na1 is also recovered when the aromatic side chain is restored (W291C to W291F) (Table 2), indicating the aromatic side chain at W291 is important in Na+ binding. We also found the Na+ binding affinity to Na1 was lost with the Y290C mutation, but this was not recovered by replacing cysteine with either serine or phenylalanine (Table 2). We conclude that both the aromatic and −OH groups at Y290 are needed for Na+ binding. Sugar binding to Y290F was partially restored relative to Y290C (21), and we suggest that an H bond between Y290 and N78, which increases the electrostatic potential of the aromatic system (38), is important for the strength of the interaction between Y290 and glucose (21). We further suggest that this H bond is crucial for Na+ binding.
N78 corresponds to A22 of LeuT, proposed to bind Na+ (via carbonyl oxygen) to the LeuT Na1 site. We have searched for suitable Na1 coordinating residues within ∼3 Å from the carbonyl oxygen of N78, and a possibility is shown in Fig. 8B. Without modification of our homology model, the distance (in angstroms) between Na+ and N78 (carbonyl oxygen) is 2.6, that for Y290 is 3.3, that for W291 (aromatic plane) is 6.3, and that for H83 is 2.0. H83 corresponds to N27 in the Na1 site of LeuT (6). In LeuT the carboxylate (COO−) of the substrate (leucine) is predicted to coordinate with Na+, but in hSGLT1, E102 in the sugar binding site may substitute for this bond. When sugar is bound, Na+ is 3.5 Å away from the O2 group of glucose, suggesting a direct interaction (39, 40). The distance between the Na+ and aromatics Y290 and W291, 4–7 Å, indicates that cation–π interactions might be involved in Na+ binding.
An intriguing question that has yet to be addressed is cation selectivity; i.e., How can Na+ sites in some transporters recognize H+ and/or Li+ but not Cs+ and Rb+? For example, H+ and Li+ can drive sugar transport by hSGLT1 with exquisite sensitivity for H+ (K0.5 of 7 μM for H+, 12 mM for Li+, and 4 mM for Na+) (22).
Transitions Between C1 and C6.
The data on V0.5, z, and τ0 for 13 mutants in the absence of [Na+]o provide preliminary information on the transitions of hSGLT1 between C1 and C6 (Table 2). The most compelling results are those with the Y290 and W291 mutants, where the k16/k61 ratio was reduced 25- to 50-fold for the cysteine mutants. Replacing the W291C cysteine chain with phenylalanine restored the ratio to that for wild type, but for Y290C there was only partial recovery for Y290F. These changes in k16/k61 were largely due to a decrease in k16 and a smaller increase in k61. We conclude that π–π interactions between Y290 and W291 play an important role in the transitions between C1 and C6, as well as in the formation of the Na1 and sugar binding sites and coupling between Na+ and sugar cotransport (21).
Mechanism of Na+ and Sugar Cotransport.
Voltage sensitivity.
The results provide an insight into the voltage dependence of hSGLT1 in that the ligand-free transporter has an apparent valence of 0.7, and the binding of extracellular Na+ ions is associated with a total charge of 0.3. Originally, we assumed that the valence of the ligand-free protein was 2, as two Na+ bind in the absence of glucose (17, 19, 23). This led to unaccounted discrepancies between models and the observed shift of V0.5 with varying [Na+]o and we had attributed this to a simplifying assumption about simultaneous binding of two external Na+ ions (22, 23). Given the structure of the Na1 and Na2 binding sites and the data presented here, it is no longer necessary to assume that the valence of SGLT1 is 2, especially as a valence of 1 now accounts for the charge movements as a function of [Na+]o. However, because two Na+ ions are coupled to each glucose molecule transported by WT hSGLT1 (20, 21, 26, 27, 31), this raises questions about what other step of the overall transport cycle is voltage dependent (41). Clues may come from closer inspection of the kinetics of Y290F, W291F, and hSGLT2, where the coupling ratio is 1/1 (21, 42).
Na+ binding order.
For hSGLT1, two Na+ ions bind to the empty transporter before glucose. The order of binding to the two Na+ sites (Na1, Na2) is not known, but the available evidence tends to favor Na+ binding first to the Na2 site: First, the Na2 site is conserved in vSGLT and hSGLT2, and these proteins couple only one Na+ to sugar transport (42–44); second, MD studies on LeuT have led to the predictions that Na+ binds first to the Na2 site and then to the Na1 site even in the absence of substrate (45); third, Na+ has also been proposed to bind first to the Na2 site in BetP (32); and fourth, Na+ is proposed to bind first to the Na2 site on the human GABA transporter GAT-1, followed by cooperative binding of the second Na+ to the Na1 site (46). In all cases Na+ binding stabilizes the transporter in an outward-open conformation to facilitate substrate binding, but it is not yet clear whether substrate binding increases the Na1 affinity for Na+.
The coupling ratio between Na+ and sugar transport is 1:1 Na+:sugar for Y290F and W291F, even though the Hill coefficients for Na+ activation are 1.5 and 2.2, respectively (21). Thus, two Na+ ions bind to the mutants to allow sugar binding to hSGLT1, but for these mutants only one Na+ is released to the cytoplasm along with a sugar molecule. We hypothesize the Na+ ion at the Na1 site may be released from these mutants back into the external solution.
Molecular dynamic simulations on vSGLT also predict that water plays an important role in the exit of sugar into the cytoplasm. After Na+ release from the Na2 site (Fig. 8A), water enters the sugar binding site and causes large fluctuations in the position of galactose, in part due to hydration of the sugar and formation of new H bonds before exiting to the cytoplasm (34, 36, 37). Presumably water also causes dissociation of Na+ from the Na1 site and this second Na+ follows sugar exit to account for the coupling of two Na+ ions to sugar transport across the protein.
Summary
A biophysical assay has been used to determine multiple sodium equilibrium binding constants (Kd) for electrogenic cotransporters. In hSGLT1 where binding of two sodium ions drive sugar transport, we could resolve the Kd for each site, KdA and KdB. Combined with studies on hSGLT1 mutants, the assay has been used to identify the binding sites.
The Na1 site has been constructed from several residues in the sugar binding pocket, as individually changing sugar crystal contacts at H83, Y290, and W291 to cysteine decreased the Na+ affinity—KdB increased more than 10-fold with no change in KdA. For W291C, but not Y290C, restoring the aromatic side chain with phenylalanine restored sodium affinity to the wild-type value. Mutation of these binding residues also dramatically reduced the apparent sugar and phlorizin affinities.
The canonical Na2 binding site is conserved in hSGLT1, with two –OH side chains, S392 and S393, and three backbone carbonyls. Mutation of S392 to cysteine reduced the lumped sodium affinity by sixfold (due to increases in both KdA and KdB), resulting in a dramatic decrease in the apparent sugar and phlorizin affinities. We conclude that mutation of S392 in the Na2 site produces a structural rearrangement of the sugar binding pocket that disrupts both the binding of the second Na+ and that of sugar. In contrast, the S393 mutations produce no significant changes in sodium, sugar, and phlorizin affinities. We conclude that the Na2 site is conserved in hSGLT1, the side chain of S392 and the backbone carbonyl of S393 are important for the binding of the first Na+, and it subsequently promotes the binding of Na1 in the sugar binding pocket.
Methods
Molecular Biology.
Mutants used (H83C, F101C, D204E,Y290C/S/F, W291C/F, S392A/C, F453C, Q457C, and G507C) were described previously (20, 21, 23, 26). The same procedures were used to generate a new mutant, T287C.
Functional Expression of hSGLT1 in Oocytes.
Protocols were approved by the University of California Chancellor’s Committee on Animal Research. Stage V–VI oocytes were harvested from mature female Xenopus laevis frogs, selected, and maintained at 18 °C in modified Barth’s solution (20, 21). Oocytes were injected with 50 ng of cRNA coding for WT and mutant hSGLT1 and incubated at 18 °C for 4–7 d. Noninjected oocytes served as controls. Experiments were conducted on oocytes bathed in a NaCl buffer containing 100 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 10 mM Hepes, pH 7.5, and the Na+ concentration was varied by isomolar replacement of NaCl with CholineCl. The internal Na+ concentration was assumed to be constant (5–10 meq/L) throughout each oocyte experiment and was supported by no observed change in the electrical parameters between the beginning and the end of the experiment. Data were collected on single oocytes, where we randomly varied the external NaCl concentration from 100 mM to 0 mM.
Electrophysiological Experiments.
These were performed using a two-electrode voltage clamp (19, 20, 23, 26). A standard pulse protocol was applied, where membrane potential was held at −50 mV (Vh) and stepped to various test values (Vt from +50 mV to −150 mV in 20-mV decrements) for 100 ms before returning to Vh. The current records were the averages of three sweeps and were filtered at 500 Hz. All experiments were performed at room temperature (20–23 °C).
Total membrane current (Itot) in response to a voltage pulse is composed of the oocyte membrane bilayer capacitive transient (Icm), pre–steady-state SGLT1 currents (Ipss), and steady-state currents (Iss). Itot was fitted to
where Iss is the steady-state current, Icm exp(−t/τcm) is the bilayer capacitance current with initial value Icm and time constant τcm, and Ipss exp(−t/τ) is the SGLT1 pre–steady-state current (23). SGLT1 pre–steady-state currents (Ipss exp(−t/τ)) were isolated by subtraction of the bilayer capacitive and steady-state components from total current (Ipss exp(−t/τ) = Itot(t) − Icm exp(−t/τcm) − Iss).
At each membrane voltage, SGLT1 charge movement (Q) was obtained from the integral of the pre–steady-state currents. The charge vs. voltage (Q-V) relations were fit to a single Boltzmann function (10, 24, 47),
where Qmax = Qdep − Qhyp, Qdep and Qhyp are the Q (absolute value) at depolarizing and hyperpolarizing limits, V is membrane potential, F is the Faraday constant, R is gas constant, T is absolute temperature, V0.5 is midpoint voltage (membrane potential at 50% Qmax), and the apparent valence, z, is the maximum steepness factor for the dependence of Q on voltage.
For some transporters, the determination of Qmax, z, and V0.5 was problematic, due to (i) practical limits on the range of voltage jumps, generally to −150 mV and +50 mV, because dielectric breakdown and/or activation of ion channels may occur at larger voltages, and (ii) V0.5 falling outside the experimental range; e.g., lowering the external [Na+]o shifts the V0.5 to more negative than −100 mV, leading to errors in estimation of the Boltzmann parameters (Figs. 4–6).
Fits of data to Eqs. 10 and 11 were performed using either Sigmaplot 10 (SPSS) or Clampfit 8.1 (Axon Instruments). On data obtained on a single oocyte, the statistics are given by the estimates and SE of the fit. For a population, the statistics are given by the means and SEMs. Although data may be shown for representative experiments, all experiments were performed on at least three oocytes from different batches. The Na+ affinities (Kd) and the ratio k16/k61 were estimated by numerically solving the equation relating V0.5 and [Na+]o (Eqs. 6 and 9), using Berkeley Madonna (www.berkeleymadonna.com), and the procedure was kindly provided by Ian Forster of the University of Zurich.
Acknowledgments
We acknowledge Dr. Monica Sala-Rabanal who was involved in the preliminary studies leading to this study and Dr. I. C. Forster for stimulating discussions. This work was supported by the National Institute of Diabetes and Digestive Disease Grant DK19567.
Footnotes
The authors declare no conflict of interest.
References
- 1.Abramson J, Wright EM. Structure and function of Na+-symporters with inverted repeats. Curr Opin Struct Biol. 2009;19(4):425–432. doi: 10.1016/j.sbi.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faham S, et al. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science. 2008;321(5890):810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krishnamurthy H, Piscitelli CL, Gouaux E. Unlocking the molecular secrets of sodium-coupled transporters. Nature. 2009;459(7245):347–355. doi: 10.1038/nature08143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature. 2009;458(7234):47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 5.Weyand S, et al. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008;322(5902):709–713. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437(7056):215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 7.Parent L, Supplisson S, Loo DD, Wright EM. Electrogenic properties of the cloned Na+/glucose cotransporter: I. Voltage-clamp studies. J Membr Biol. 1992;125(1):49–62. doi: 10.1007/BF00235797. [DOI] [PubMed] [Google Scholar]
- 8.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117(4):500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9(4):323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 10.Loo DD, Hazama A, Supplisson S, Turk E, Wright EM. Relaxation kinetics of the Na+/glucose cotransporter. Proc Natl Acad Sci USA. 1993;90(12):5767–5771. doi: 10.1073/pnas.90.12.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forster IC, Hernando N, Biber J, Murer H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr Top Membr. 2012;70:313–356. doi: 10.1016/B978-0-12-394316-3.00010-7. [DOI] [PubMed] [Google Scholar]
- 12.Nakao M, Gadsby DC. Voltage dependence of Na translocation by the Na/K pump. Nature. 1986;323(6089):628–630. doi: 10.1038/323628a0. [DOI] [PubMed] [Google Scholar]
- 13.Zampighi GA, et al. A method for determining the unitary functional capacity of cloned channels and transporters expressed in Xenopus laevis oocytes. J Membr Biol. 1995;148(1):65–78. doi: 10.1007/BF00234157. [DOI] [PubMed] [Google Scholar]
- 14.Holmgren M, et al. Three distinct and sequential steps in the release of sodium ions by the Na+/K+-ATPase. Nature. 2000;403(6772):898–901. doi: 10.1038/35002599. [DOI] [PubMed] [Google Scholar]
- 15.Holmgren M, Rakowski RF. Charge translocation by the Na+/K+ pump under Na+/Na+ exchange conditions: Intracellular Na+ dependence. Biophys J. 2006;90(5):1607–1616. doi: 10.1529/biophysj.105.072942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Läuger P, Jauch P. Microscopic description of voltage effects on ion-driven cotransport systems. J Membr Biol. 1986;91(3):275–284. doi: 10.1007/BF01868820. [DOI] [PubMed] [Google Scholar]
- 17.Parent L, Supplisson S, Loo DD, Wright EM. Electrogenic properties of the cloned Na+/glucose cotransporter: II. A transport model under nonrapid equilibrium conditions. J Membr Biol. 1992;125(1):63–79. doi: 10.1007/BF00235798. [DOI] [PubMed] [Google Scholar]
- 18.Andrini O, Meinild AK, Ghezzi C, Murer H, Forster IC. Lithium interactions with Na+-coupled inorganic phosphate cotransporters: Insights into the mechanism of sequential cation binding. Am J Physiol Cell Physiol. 2012;302(3):C539–C554. doi: 10.1152/ajpcell.00364.2011. [DOI] [PubMed] [Google Scholar]
- 19.Meinild AK, Hirayama BA, Wright EM, Loo DD. Fluorescence studies of ligand-induced conformational changes of the Na+/glucose cotransporter. Biochemistry. 2002;41(4):1250–1258. doi: 10.1021/bi011661r. [DOI] [PubMed] [Google Scholar]
- 20.Sala-Rabanal M, et al. Bridging the gap between structure and kinetics of human SGLT1. Am J Physiol Cell Physiol. 2012;302(9):C1293–C1305. doi: 10.1152/ajpcell.00397.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X, Loo DD, Hirayama BA, Wright EM. The importance of being aromatic: π interactions in sodium symporters. Biochemistry. 2012;51(47):9480–9487. doi: 10.1021/bi301329w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91(2):733–794. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 23.Loo DDF, Hirayama BA, Karakossian MH, Meinild AK, Wright EM. Conformational dynamics of hSGLT1 during Na+/glucose cotransport. J Gen Physiol. 2006;128(6):701–720. doi: 10.1085/jgp.200609643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loo DD, Hirayama BA, Cha A, Bezanilla F, Wright EM. Perturbation analysis of the voltage-sensitive conformational changes of the Na+/glucose cotransporter. J Gen Physiol. 2005;125(1):13–36. doi: 10.1085/jgp.200409150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quick M, Loo DD, Wright EM. Neutralization of a conserved amino acid residue in the human Na+/glucose transporter (hSGLT1) generates a glucose-gated H+ channel. J Biol Chem. 2001;276(3):1728–1734. doi: 10.1074/jbc.M005521200. [DOI] [PubMed] [Google Scholar]
- 26.Loo DD, et al. Conformational changes couple Na+ and glucose transport. Proc Natl Acad Sci USA. 1998;95(13):7789–7794. doi: 10.1073/pnas.95.13.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirayama BA, et al. Sodium-dependent reorganization of the sugar-binding site of SGLT1. Biochemistry. 2007;46(46):13391–13406. doi: 10.1021/bi701562k. [DOI] [PubMed] [Google Scholar]
- 28.Page MJ, Carrell CJ, Di Cera E. Engineering protein allostery: 1.05 A resolution structure and enzymatic properties of a Na+-activated trypsin. J Mol Biol. 2008;378(3):666–672. doi: 10.1016/j.jmb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noskov SY, Roux B. Control of ion selectivity in LeuT: Two Na+ binding sites with two different mechanisms. J Mol Biol. 2008;377(3):804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nayal M, Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J Mol Biol. 1996;256(2):228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 31.Mackenzie B, Loo DD, Wright EM. Relationships between Na+/glucose cotransporter (SGLT1) currents and fluxes. J Membr Biol. 1998;162(2):101–106. doi: 10.1007/s002329900347. [DOI] [PubMed] [Google Scholar]
- 32.Khafizov K, et al. Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proc Natl Acad Sci USA. 2012;109(44):E3035–E3044. doi: 10.1073/pnas.1209039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Tajkhorshid E. Ion-releasing state of a secondary membrane transporter. Biophys J. 2009;97(11):L29–L31. doi: 10.1016/j.bpj.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zomot E, Bahar I. The sodium/galactose symporter crystal structure is a dynamic, not so occluded state. Mol Biosyst. 2010;6(6):1040–1046. doi: 10.1039/b927492h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bisha I, Laio A, Magistrato A, Giorgetti A, Sgrignani J. A candidate ion-retaining state in the inward-facing conformation of sodium/galactose symporter: clues from atomistic simulations. J Chem Theory Comput. 2013;9(2):1240–1246. doi: 10.1021/ct3008233. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Tajkhorshid E. A gate-free pathway for substrate release from the inward-facing state of the Na+-galactose transporter. Biochim Biophys Acta. 2012;1818(2):263–271. doi: 10.1016/j.bbamem.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe AC, et al. The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature. 2010;468(7326):988–991. doi: 10.1038/nature09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mecozzi S, West AP, Jr, Dougherty DA. Cation-pi interactions in aromatics of biological and medicinal interest: Electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci USA. 1996;93(20):10566–10571. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heaton AL, Armentrout PB. Experimental and theoretical studies of sodium cation interactions with D-arabinose, xylose, glucose, and galactose. J Phys Chem A. 2008;112(41):10156–10167. doi: 10.1021/jp804113q. [DOI] [PubMed] [Google Scholar]
- 40.Wincel H. Thermochemistry of microhydration of sodiated and potassiated monosaccharides. J Am Soc Mass Spectrom. 2011;22(9):1570–1576. doi: 10.1007/s13361-011-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longpré JP, Sasseville LJ, Lapointe JY. Simulated annealing reveals the kinetic activity of SGLT1, a member of the LeuT structural family. J Gen Physiol. 2012;140(4):361–374. doi: 10.1085/jgp.201210822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hummel CS, et al. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol. 2011;300(1):C14–C21. doi: 10.1152/ajpcell.00388.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veenstra M, Lanza S, Hirayama BA, Turk E, Wright EM. Local conformational changes in the Vibrio Na+/galactose cotransporter. Biochemistry. 2004;43(12):3620–3627. doi: 10.1021/bi0357210. [DOI] [PubMed] [Google Scholar]
- 44.Turk E, et al. Molecular characterization of Vibrio parahaemolyticus vSGLT: A model for sodium-coupled sugar cotransporters. J Biol Chem. 2000;275(33):25711–25716. doi: 10.1074/jbc.M003127200. [DOI] [PubMed] [Google Scholar]
- 45.Zhao C, et al. Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophys J. 2012;103(5):878–888. doi: 10.1016/j.bpj.2012.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meinild AK, Forster IC. Using lithium to probe sequential cation interactions with GAT1. Am J Physiol Cell Physiol. 2012;302(11):C1661–C1675. doi: 10.1152/ajpcell.00446.2011. [DOI] [PubMed] [Google Scholar]
- 47.Hazama A, Loo DD, Wright EM. Presteady-state currents of the rabbit Na+/glucose cotransporter (SGLT1) J Membr Biol. 1997;155(2):175–186. doi: 10.1007/s002329900169. [DOI] [PubMed] [Google Scholar]