

Significance
Glycosyl groups function as essential chemical mediators of molecular interactions in cells and on cellular surfaces. Microbes integrate carbohydrates into secondary metabolism to produce glycosylated natural products (GNPs) that may function in chemical communication and defense. Many glycosylated metabolites are important pharmaceutical agents. Herein, we introduce glycogenomics as a new genome-mining method that links metabolomics and genomics for the rapid identification and characterization of bioactive microbial GNPs. Glycogenomics identifies glycosyl groups in microbial metabolomes by tandem mass spectrometry and links this chemical signature through a glycogenetic code to glycosylation genes in a microbial genome. As a proof of principle, we report the discovery of arenimycin B from a marine actinobacterium as a new antibiotic active against multidrug-resistant Staphylococcus aureus.
Keywords: secondary metabolite, drug discovery, microbial genomics, deoxysugar, polyketide
Abstract
Glycosyl groups are an essential mediator of molecular interactions in cells and on cellular surfaces. There are very few methods that directly relate sugar-containing molecules to their biosynthetic machineries. Here, we introduce glycogenomics as an experiment-guided genome-mining approach for fast characterization of glycosylated natural products (GNPs) and their biosynthetic pathways from genome-sequenced microbes by targeting glycosyl groups in microbial metabolomes. Microbial GNPs consist of aglycone and glycosyl structure groups in which the sugar unit(s) are often critical for the GNP’s bioactivity, e.g., by promoting binding to a target biomolecule. GNPs are a structurally diverse class of molecules with important pharmaceutical and agrochemical applications. Herein, O- and N-glycosyl groups are characterized in their sugar monomers by tandem mass spectrometry (MS) and matched to corresponding glycosylation genes in secondary metabolic pathways by a MS-glycogenetic code. The associated aglycone biosynthetic genes of the GNP genotype then classify the natural product to further guide structure elucidation. We highlight the glycogenomic strategy by the characterization of several bioactive glycosylated molecules and their gene clusters, including the anticancer agent cinerubin B from Streptomyces sp. SPB74 and an antibiotic, arenimycin B, from Salinispora arenicola CNB-527.
Glycosylated natural products (GNPs) produced by microbes comprise many compounds with therapeutic and agrochemical applications, such as the antibiotic erythromycin (1) and the insecticide avermectin (2). A GNP consists of an aglycone and one or multiple glycosyl units (Fig. 1A) (3), which often directly mediate the bioactivity of the compound (4). In microbial genomes, the genes for biosynthesis and attachment of these glycosyl groups are usually coclustered with the biosynthetic genes of the aglycone (5) (Fig. 1B). Here, we report a genome-mining approach for fast characterization of GNP chemotypes from microbial metabolomes by connecting sugar footprints in tandem mass-spectrometric spectra with their corresponding biosynthetic genes in GNP genotypes in genome sequences.
Fig. 1.
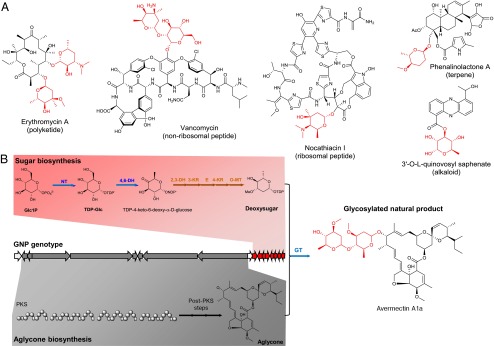
Structure and biosynthesis of GNPs. (A) Selected GNP structures exemplify the diverse biosynthetic origin of the aglycone (black). (B) The simplified genetic organization of the avermectin biosynthetic pathway shows that it can be differentiated into aglycone biosynthetic genes (gray) and sugar biosynthetic genes (red). In a deoxysugar pathway, there are common biosynthetic genes (NT, nucleotidylyltransferase; 4,6-DH, 4,6-dehydratase; GT, glycosyltransferase; blue arrows) and specific biosynthetic genes (here: 2,3-DH, 2,3-dehydratase; 3-KR, 3-ketoreductase; 5-E, 5-epimerase; 4-KR, 4-ketoreductase; O3-MT, O3-methyltransferase; brown arrows). Sugar groups are indicated in red. Abbreviations: Glc1P, D-glucose-1-phosphate; PKS, polyketide synthase; TDP-Glc, 1-TDP-D-glucose.
GNPs are a structurally very diverse class of natural products in terms of the aglycone, i.e., the nonsugar portion of the molecule, and the glycosyl groups. Aglycone diversity is based on the fact that GNPs are found in almost all major biosynthetic classes of natural products (Fig. 1A), e.g., nonribosomal (6) and ribosomal peptides (7), polyketides (8), terpenes (9), and alkaloids (10). Glycosylation diversity arises through sugar monomers and sugar attachment. There are over 100 different sugars found in microbial GNPs, where the majority are 6-deoxysugars (3). These sugar monomers can be attached to the aglycone or to each other through C-, N-, N-O-, O-, and S-glycosidic linkages, with O-glycosidic bonds as the most common. Glycosidic bonds also occur on various sugar ring positions (3). The genes involved in deoxysugar glycosylation of an aglycone can be distinguished into common glycosylation genes, which are found in all deoxysugar pathways, and specific glycosylation genes, which catalyze sugar-specific modifications to yield different sugar monomers (Fig. 1B) (3). The common genes encode a nucleotidylyltransferase (NT), which activates glucose-1-phosphate as TDP-glucose, a 4,6-dehydratase (4,6-DH), that subsequently forms the common deoxysugar intermediate TDP-4-keto-6-deoxy-α-D-glucose, and a glycosyltransferase (GT), which attaches the final sugar monomer to the aglycone or a glycosyl group (Fig. 1B). Specific glycosylation genes such as dehydratases, deoxygenases, dehydrogenases, methyltransferases, aminotransferases, and epimerases modify the common deoxysugar intermediate to yield the large diversity of sugar monomers in microbial GNPs (11, 12). Phylogenetically, the highest GNP sugar diversity is found in actinobacteria (13). Among these bacteria, the families of Micromonosporaceae, Pseudonocardiaceae, Streptomycetaceae, and Thermomonosporaceae have the highest genetic potential to produce GNPs (Dataset S1).
Tandem mass spectrometry (MSn) is a common method to gain structural information of oligosaccharides such as glycans (14). For example, oligosaccharides can be sequenced by MSn based on the cleavage of O-/N-glycosidic bonds in low-energy collision-induced dissociation (CID) (14). The same fragmentation of O-/N-glycosidic bonds has been observed in GNPs such as erythromycin (15), thus enabling a similar fragmentation nomenclature for GNPs (Fig. S1). O-/N-glycosyl groups in GNPs are preferred leaving groups from the parent ion as carbon–heteroatom bonds are generally labile in the gas phase (16). The predictable fragmentation of glycosyl groups in GNPs yields B/C-sugar fragments in the low-m/z region of the MSn spectrum and Y/Z-aglycone fragments in the higher-m/z region (14–18). Both fragmentation footprints correspond to specific sugar losses from the GNP (Fig. S1) and, thus, can reveal these biosynthetic building blocks in MSn experiments.
Genome mining as a natural product discovery strategy is based on the connection of an unknown natural product structure with its biosynthetic genes by applied biosynthetic knowledge. This connection can be done either in the genotype-to-chemotype direction by in silico-guided approaches (19) or in the chemotype-to-genotype direction by experiment-guided approaches (20). Many effective in silico-guided strategies have been developed using genetics (21), substrate labeling (22), and screening for predicted physicochemical properties (23) to characterize new natural products from cryptic and even silent gene clusters in genomes. However, these approaches target only one biosynthetic pathway per experiment, thereby resulting in a slow discovery rate. The experiment-guided approach, such as MS-guided genome mining of peptides, starts with an untargeted analytical step, e.g., MSn analysis of an extract (20), to identify biosynthetic building blocks of an unknown chemotype. This structural information is subsequently used to query the genome sequence of the target organism for corresponding genes associated with the enzymatic assembly of the chemotype based on biosynthetic principles. MS-guided genome mining can target multiple expressed pathways in one experiment and, in combination with automated platforms such as liquid chromatography (LC)-MS, has the potential for automation.
In this study, we show that sugar substituents of GNPs are identified by MSn and are iteratively connected to the glycosylation genes of the corresponding GNP genotype in a target genome. This concept extends MS-guided genome mining beyond peptide natural products (20) to most biosynthetic classes of natural products that can be glycosylated. We show our approach by characterizing bioactive GNPs from actinobacterial metabolomes.
Results
A MS-Glycogenetic Code Connecting Microbial GNP Chemotypes and Genotypes.
To connect GNP chemotypes by tandem MS with GNP genotypes, a template first had to be established that would link de novo MSn fragmentation data of each sugar with the corresponding biosynthetic genes from characterized microbial GNP pathways. This MS-glycogenetic code comprises 83 microbial sugar monomers, including the most common microbial sugars from the Bacterial Carbohydrate Structure Database (24) and most known deoxysugars, involved in natural product glycosylation (3). For each sugar, calculated masses of an O-/N-glycosidic neutral loss from the parent ion (Y-ion) and of B/C-ions in CID-based tandem MS experiments are listed together with the common and specific biosynthetic genes of the corresponding verified or predicted sugar pathway (Dataset S2).
The MS-glycogenetic code was first tested to determine whether it could connect MSn data of known GNP chemotypes with their corresponding GNP genotypes from GenBank (25). The analyzed GNPs were selected based on the availability of MSn data in the METLIN database (26) (daunomycin, staurosporine, oleandomycin, vancomycin, tylosin, avermectin B1a, nystatin, aclacinomycin A, novobiocin, erythromycin A), the literature [spinosyn A (27), megalomicin (28), chartreusin (29), neocarzinostatin (30), lankamycin (31), Sch40832 (32), lomaiviticin C (33)], or generated for this study (phenalinolactone, amphotericin B, chalcomycin), and the availability of nucleotide sequences associated with biosynthetic gene clusters (Datasets S3 and S4). Eighteen of 20 analyzed GNPs could be connected successfully with their biosynthetic gene cluster by the MS-glycogenetic code (Table 1). Fifteen of the 18 GNPs with observed sugar losses showed sugar-specific B-ions, 15 of 18 showed sugar-specific Y-ion neutral losses, and 1 of 18 showed a sugar-specific Z-ion neutral loss.
Table 1.
Connection of known GNP chemotypes and genotypes by the MS-glycogenetic code
| Compound | MS/MS sugar footprint, m/z | Candidate sugar | Specific glycosylation genes | Common glycosylation genes | Gene cluster, GenBank ID |
| Phenalinolactone | P-128.072 (Y), 129.094 (B) | O-Methyl-L-amicetose | 2,3DH, 3,4DH, 3KR, 4KR, E, O-MT | NT, 4,6DH, GT | DQ230532 |
| 4-O-Methyl-L-rhodinose | |||||
| Daunomycin | 130.085 (B) | L-Daunosamine | 2,3DH, AmT, E, 4KR | NT, 4,6DH, GT | STMDNRLM, STMDNRQ, SPU77891, STMDNRI |
| L-Ristosamine | |||||
| Staurosporine | P-129.080 (Y), 130.086 (B) | L-Daunosamine | 2,3DH, AmT, E, 4KR | NT, 4,6DH, GT (2×) | AB088119 |
| L-Ristosamine | |||||
| Oleandomycin | P-144.084 (Y), 145.086 (B) | L-Oleandrose | 2,3DH, 3KR, E, 4K-R, O-MT | NT, 4,6DH, GT (4×) | AF055579, AJ002638 |
| Olivomose | 2,3DH, 3KR, 4KR, O-MT | ||||
| Spinosyn A | 142.123 (B) | D-Forosamine | 2,3DH, 3KR, 3,4DH, AmT, N,N-MT | GT | AY007564 |
| 188.105 (Y) | 2,3,4-tri-O-Methylrhamnose | — | |||
| Tylosin | P-144.076 (Y), 145.085 (B) | D-Mycarose | 2,3DH, 3KR, C-MT, E, 4KR | NT, 4,6DH, GT (2×) | AF055922, AF147704, SFU08223 |
| L-Oleandrose | 2,3DH, 3KR, O-MT, E, 4KR | ||||
| Olivomose | 2,3DH, 3KR, O-MT, 4KR | ||||
| Vancomycin | P-143.082 (Y), 144.100 (B) | 3-epi-L-Vancosamine | 2,3DH, 3KR, E, 4KR, C-MT | GT (2×) | HE589771 |
| L-Vancosamine | 2,3DH, 3KR, E, 4KR, C-MT | ||||
| Avermectin B1a | P-144.077 (Y) | L-Oleandrose | 2,3DH, 3KR, O-MT, E, 4KR | NT, 4,6DH, GT | AB032523 |
| Olivomose | 2,3DH, 3KR, O-MT, 4-KR | ||||
| Chartreusin | P-160 (Y) | D-Digitalose | 4KR, O-MT | NT, 4,6DH, GT (2×) | AJ786382, AJ786383 |
| 3-O-Methyl-rhamnose | 4KR, E, O-MT | ||||
| 2-O-Methyl-L-rhamnose | 4KR, E, O-MT | ||||
| Aclacinomycin A | P-112.0495 (Y), 113.060 (B) | L-Cinerulose A | 2,3DH, 3,4DH, 3KR, 4KR, E | NT, 4,6DH, GT (3×) | AF264025, AF257324 |
| Novobiocin | P-217.094 (Y), 218.104 (B) | 3-O-Carbamoyl-4-O-methyl-L-noviose | E, 4KR, C-MT, O-MT, CarbT | NT, 4,6DH, GT | AF170880 |
| Neocarzinostatin | 160 (B) | 2′-N-Methyl-D-fucosamine | 2,3DH, 4KR, AmT, N-MT | NT, 4,6DH, GT | AY117439 |
| Erythromycin A | P-158.093 (Y) | L-Cladinose | 2,3DH, 3KR, E, C-MT, 4KR, MT | GT (2×) | AM420293, SEU77459 |
| 158.1168 (B) | D-Desosamine (+5 sugars) | 3,4DH, oxDA, AmT, N,N-MT | |||
| Megalomicin | P-144.08 (Y) | L-Oleandrose | 2,3DH, 3KR, O-MT, E, 4KR | GT (4×) | AF263245 |
| Olivomose | 2,3DH, 3KR, O-MT, 4KR | ||||
| 158.12 (B) | L-Megosamine (+4 sugars) | 2,3DH, 4KR, E, AmT, N,N-MT | |||
| Amphotericin B | P-163.084 (Z) | D-Mycosamine | 3,4IM (CytP450), AmT | 4,6DH, GT | AF357202 |
| Lankamycin | P-200 (Y), 201 (B) | 4-O-Acetyl-L-arcanose | 2,3DH, 3KR, 4KR, E, C-MT, O-MT, AcT | NT, 4,6DH, GT (3×) | AB088224 |
| Chalcomycin | P-144.072 (Y), 145.071 (B) | D-Chalcose | AmT, oxDA, 3KR, O-MT | NT, 4,6DH, GT (2×) | AY509120 |
| Lomaiviticin C | P-144.077 (Y), 145.086 (B) | L-Oleandrose | 2,3DH, 3KR, 4KR, E, O-MT | NT, 4,6DH, GT (2×) | CP000667 |
| Olivomose | 2,3DH, 3KR, 4KR, O-MT |
For detailed MS/MS and gene cluster analysis, see Datasets S3 and S4. For abbreviations, see Dataset S2.
Exemplifying the MS-glycogenetic analysis, phenalinolactone A, a glycosylated terpene from genome-sequenced Streptomyces sp. Tu6071 (34), shows a neutral loss of 128.072 Da from the parent ion (738.345 m/z, [M+Na]+) and a complementary 129.094-Da B-ion in its MS2 spectrum (Fig. S2). This putative Y-ion mass shift and B-ion correspond to isomeric O-methyl-L-amicetose or 4-O-methyl-L-rhodinose as MSn candidate sugars (Dataset S2). BLAST analysis of the phenalinolactone gene cluster predicted three common glycosylation genes encoding a nucleotidylyltransferase, a 4,6-dehydratase and a glycosyltransferase, and six specific glycosylation genes, i.e., a 2,3-dehydratase, a 3,4-dehydratase, a 3-ketoreductase, a 4-ketoreductase, an epimerase, and an O-methyltransferase. In the MS-glycogenetic code, these specific genes match to the biosynthetic pathway of the two MSn candidate sugars, O-methyl-L-amicetose and 4-O-methyl-L-rhodinose, thus connecting MSn data of phenalinolactone A with its gene cluster (Fig. S2). The first negative GNP result, nystatin (35), showed no expected sugar fragmentation in the METLIN database MSn spectrum (26) and, thus, could not be matched to its gene cluster. The second negative GNP result, glycosylated thiopeptide Sch40832 (32), showed dideoxysugar-specific Y-ion neutral losses. However, the putative thiopeptide gene cluster in the Micromonospora aurantiaca ATCC 27029 genome contained a glycosyltransferase but not the specific genes involved in dideoxysugar biosynthesis (Datasets S3 and S4) (36). Thus, Sch40832 could not be connected with the gene cluster using a MS-glycogenetic approach as glycogenomics relies on the sugar biosynthetic genes to be coclustered with the remainder of the pathway genes.
MS-Guided Genome Mining of Cinerubin B from Streptomyces sp. SPB74.
The MS-glycogenetic code was integrated into a workflow of MS-guided genome mining of microbial GNPs (Fig. 2). This glycogenomic strategy starts with the LC-MSn analysis of a metabolic extract of a genome-sequenced bacterium (Fig. 2A). Candidate GNP fractions can be identified in the chromatogram by peaks in extracted ion chromatograms (EICs) of sugar-specific B/C-ion masses or Y/Z-ion neutral losses (Dataset S2 and Fig. 2B). Candidate GNPs are then characterized in their putative glycosyl groups by neutral losses and B/C-ions in the corresponding MSn spectra (Fig. 2C). In the next step, the secondary metabolic gene cluster that contains biosynthetic genes to produce the MSn candidate sugars is characterized (Fig. 2D). First, all gene clusters with glycosylation genes are characterized in the genome. Then, the GNP gene cluster with biosynthetic genes corresponding to any MSn candidate sugars is identified. Finally, the analysis of the aglycone genes enables the classification of the GNP chemotype (Fig. 2E). The connection of GNP structure and genes is verified by iterative analysis of MSn and genetic data. When the molecule becomes of sufficient interest, NMR structure elucidation is used to fully establish the GNP chemotype.
Fig. 2.

The glycogenomic workflow for characterization of GNPs from genome-sequenced microbes. (A) Tandem mass-spectrometric analysis of microbial metabolic samples can reveal biosynthetic building blocks such as amino acids and sugar monomers of natural products via tandem MS fragment ions. (B) Identification of putative GNPs in a LC-MSn analysis as peaks in EICs of known sugar fragment masses. (C) Verification of putative GNPs by characterization of candidate sugar monomers by sugar neutral losses and corresponding sugar fragment ions in tandem MS spectra. (D) Connection of putative GNP chemotype with corresponding GNP genotype in target microbial genome by genome mining of GNP pathway with glycosylation genes matching observed sugar fragments. (E) Characterization of GNP chemotype by analysis of aglycone biosynthetic genes of candidate GNP pathway and further structure elucidation.
As a proof-of-concept experiment of the glycogenomic approach, we discovered the glycosylated anthracycline cinerubin B and its gene cluster from a previously unknown producer, Streptomyces sp. SPB74 (37) (Fig. 3 and Fig. S3). An organic extract of this genome-sequenced actinobacterium was analyzed by LC-MSn to give a putative GNP with a parent mass of 825.317 Da (Fig. 3A). Fragmentation of this molecule resulted in two mass shifts and two low-m/z fragment ions that corresponded to MSn candidate sugars. The observed mass shifts of 110.035 and 130.064 Da matched L-aculose and a collection of six dideoxyhexose isomers, respectively, whereas the putative B-ion 158.120 m/z suggested the additional presence of one of eight aminodeoxysugars (Fig. 3B). We next interrogated the genome sequence of Streptomyces sp. SPB74 for the biosynthesis of a natural product adorned with at least three sugar monomers. Of the 16 secondary metabolic gene clusters we identified by antiSMASH analysis (38), just two harbored glycosylation genes and only one contained specific glycosylation genes (Fig. 3C, Fig. S3). Among these specific genes were six associated with the biosynthesis of L-aculose (Dataset S2), a derivative of the trideoxysugar L-rhodinose found in the polyketide antibiotic aclacinomycin Y (39). Additional genes associated with deoxysugar biosynthesis were consistent with the predicted pathways for the four candidate aminodeoxysugars—megosamine, nogalamine, rhodosamine, and angolosamine—and all candidate dideoxysugars, excluding the biosynthetically uncharacterized esperamicin A1 sugar (40). Coclustered with the deoxysugar biosynthesis gene locus were genes predicted for aglycone biosynthesis comprising an aromatic type II polyketide synthase (PKS). Further analysis of the gene set revealed its similarity to the aclacinomycin gene cluster from Streptomyces galileus (41, 42). This enabled the classification of the target compound as a glycosylated anthracycline polyketide. Structure elucidation of the purified compound by NMR identified cinerubin B (calculated mass = 825.32079 Da; Δm = 4.6 ppm), a highly bioactive polyketide first characterized from Streptomyces antibioticus (Fig. S4 and Table S1) (43). The fast characterization of cinerubin B as a glycosylated anthracycline polyketide from a standard LC-MSn run of a crude microbial extract from a genome-sequenced microbe showed the feasibility of the glycogenomic approach in connecting a GNP chemotype with its genotype.
Fig. 3.

Glycogenomic characterization of anthracycline polyketide cinerubin B from Streptomyces sp. SPB74. (A) LC-MSn analysis of an metabolic extract yielded a putative GNP fraction via a product ion corresponding to an aminodeoxysugar (EIC, 158.12 m/z; red). (B) The MSn analysis of the candidate GNP yielded sugar mass shifts for three different groups of candidate MSn sugars, including aminodeoxysugars (red B-ion). (C) Genome mining of Streptomyces sp. SPB74 characterized a candidate pathway for target GNP with the biosynthetic genes corresponding to, e.g., the candidate MSn aminodeoxysugars (red) and biosynthetic genes of a type II PKS aglycone (gray). (D) Chemotype prediction of a glycosylated anthracycline polyketide from tandem MS and genetic data. The target GNP was further characterized as cinerubin B with the aminodeoxysugar L-rhodosamine (red) by NMR. Abbreviations: BPC, base peak chromatogram; EIC, extracted ion chromatogram. For 2,3DH, 3,4DH, 3KR, 4KR, E, AmT, N,N-MT, and OxRed, see Dataset S2.
Glycogenomic Characterization of an Arenimycin Chemotype and Genotype from Salinispora arenicola CNB-527.
To test glycogenomics for discovery of new glycosylated chemotypes and genotypes, we analyzed organic extracts of several genome sequenced strains of the actinobacterial genus Salinispora, which is known for its prolific production of bioactive secondary metabolites (44), including GNPs (33, 45). LC-MSn analysis of an organic extract of marine actinobacterium Salinispora arenicola CNB-527 (46) yielded a compound of mass 808.304 Da that showed a forosamine sugar B-ion EIC (142.12 m/z; Fig. 4A). The MS2 spectrum of the compound also showed a forosamine Y-ion mass shift (141.117 m/z) and another putative methyldeoxysugar mass shift in the B- and Y-ion series (Fig. 4B). The candidate MSn sugars of the methyldeoxysugars were digitalose, O-methylrhamnose, and 6-deoxy-3-C-methylmannose (Fig. 4B). The antiSMASH analysis of the S. arenicola CNB-527 genome revealed four gene clusters with putative glycosylated products—a type II PKS pathway, an indole pathway, an enediyne PKS pathway, and a type I PKS pathway. The type II PKS gene cluster and the indole gene cluster both had the specific glycosylation genes for digitalose or O-methylrhamnose biosynthesis, i.e., a 4-ketoreductase, an epimerase, and an O-methyltransferase (Fig. 4C, blue). However, only the type II PKS cluster had the specific genes for forosamine production, i.e., a 2,3-dehydratase, 3,4-dehydratase, 3-ketoreductase, an aminotransferase, and an N,N-dimethyltransferase (Fig. S5). We thus suspected that the unknown 808.304-Da metabolite was associated with the orphan type II PKS gene cluster (Fig. S5). The presence of type II PKS genes and the characterized glycosylation genes and MSn data indicated an aromatic polyketide product with a disaccharide group. Purification and comprehensive NMR structure elucidation revealed that the compound is an arenimycin derivative with an additional L-forosamine glycosyl group attached at the 4-hydroxyl group of the 2-O-methyl-L-rhamnose unit (Fig. 4D, Fig. S6, and Tables S2 and S3).
Fig. 4.

Glycogenomic characterization of arenimycin B genotype and chemotype from Salinispora arenicola CNB-527. (A) LC-MSn analysis of a metabolic extract yielded a putative GNP fraction via product ions corresponding to a dimethylaminotrideoxysugar (EIC, 142.1 m/z; red). (B) The MSn analysis of a candidate GNP (809 m/z, z = 1) yielded sugar mass shifts for a candidate MSn forosamine sugar (red) and methyldeoxysugars (blue). (C) Genome mining of S. arenicola CNB-527 characterized a candidate GNP pathway with the biosynthetic genes corresponding to the MSn candidate forosamine sugar (red) and O-methyldeoxysugars (blue) and biosynthetic genes of a type II PKS aglycone (gray). (D) Chemotype prediction of a glycosylated aromatic polyketide from MSn and genetic data, which was further characterized by NMR as a GNP, arenimycin B. Coproduced arenimycin A is shown for comparison.
Arenimycin A is a rare benzo[α]naphthacene quinone natural product originally isolated from a different S. arenicola isolate, strain CNR-647 (47), and structurally related to SF2446B1 from Streptomyces sp. SF2446 (48). Arenimycin also exhibits antibacterial activity against rifampin- and methicillin-resistant Staphylococcus aureus (47). The arenimycin derivative from S. arenicola CNB-527, termed arenimycin B, was similarly evaluated in antimicrobial and anticancer screens. Arenimycin B showed slightly lower cytotoxicity against HCT-116 cancer cells than arenimycin A but a twofold or greater increase in activity against clinically relevant, multidrug-resistant strains of Staphylococcus aureus (Table S4). Both arenimycin A and B showed minimal inhibition of Gram-negative bacteria (Table S4). We further analyzed extracts of S. arenicola CNB-527 by LC-MSn analysis (Fig. S7) and confirmed by NMR (Fig. S8, Tables S3 and S5) the coproduction of arenimycin A (47). This result suggests that the corresponding type II PKS pathway (arn) codes for the biosynthesis of arenimycins in general and that the diglycosylated arenimycin B is the ultimate pathway product. The discovery of an arenimycin chemotype and its connection to an orphan genotype validates the glycogenomic approach. Although the arenimycin biosynthetic gene cluster arn had not been identified before this study, a homologous gene cluster was previously assigned to the structurally related aromatic polyketide pradimicin (49). Pradimicin shares the benzo[α]naphthacene quinone core with arenimycins but has different oxidation patterns and different glycosylation sites and groups (50). However, the biosynthetic genes of the pradimicin benzo[α]naphtacene quinone core are conserved in the arn cluster (Fig. S5). In contrast to the pradimicin cluster, the arenimycin pathway comprises a flavin-dependent monooxygenase that has homology to TcmG, a monooxygenase from the tetrenomycin pathway (51), that may rationalize the hydroxylations at positions 6a and 14a (Fig. S5). Uncommon biosynthetic features of the arenimycins are the N-glycosylation of the aglycone and the rare forosamine glycosylation of arenimycin B. Ultimately, the characterization of a bioactive glycosylated compound and its biosynthetic pathway from S. arenicola CNB-527 highlights how targeting glycosylation on small molecules as a genome-mining approach can rapidly lead to the joint discovery of bioactive molecules and their biosynthetic pathways.
Discussion
In this study, we introduced an experiment-guided genome-mining strategy to characterize GNPs with their biosynthetic gene clusters in microbial genome sequences. Our glycogenomic approach is based on a MS-glycogenetic code that connects predictable glycosylation fragments from MSn experiments of GNPs with their glycosylation genes in microbial genomes. Our approach led to the rapid characterization of cinerubin B, a glycosylated anthracycline antibiotic, and its gene cluster from Streptomyces sp. SPB74, and to the discovery of arenimycin A and B, glycosylated aromatic polyketides with significant anti–methicillin-resistant Staphylococcus aureus activity (47), and their biosynthetic gene cluster from S. arenicola CNB-527.
Genome sequences are becoming a standard resource in microbial research (52). In the analysis of microbial secondary metabolism, genome sequences have revealed a large pool of uncharacterized or so-called “cryptic” natural product pathways as a potential source of new therapeutics (5, 53). Harvesting these orphan pathways has been mainly done by in silico-guided approaches in which predictions of biosynthetic genes in these pathways select the experiments to isolate the target cryptic natural products (19). This workflow allows for the characterization of only one pathway per experiment. In light of an exponential growth of genome sequences, a one-by-one connection of an unknown chemotype with its genotype cannot match the pace of sequencing new cryptic pathways. Therefore, new methodologies are urgently needed.
Experiment-guided genome mining, such as our glycogenomic approach, starts at the chemotype level to identify biosynthetic building blocks from an unknown natural product. The connection to the corresponding biosynthetic genes in the genome sequence is based on current biosynthetic knowledge. This chemotype-to-genotype flow of information should enable a characterization of multiple cryptic pathways by initial parallel analyses of unknown secondary metabolites and subsequent genome mining of their pathways (20).
The first steps of glycogenomics rely on tandem mass-spectrometric identification of O- and N-glycosyl groups from microbial GNPs. These sugars can often be characterized as B-ion fragments of CID experiments in the low-m/z region. We implemented this fragmentation behavior in our analysis by creating EICs from LC-MSn data for all 46 B-ion masses of the 71 known sugars involved in natural product glycosylation (Dataset S2). Putative GNP fractions based on sugar EIC peaks can then be verified by identification of corresponding Y-ion neutral losses or B/C-ion fragments of the observed sugar and/or other sugar fragments. The result of the LC-MS analysis is a list of MSn candidate sugars of a putative GNP that are used for finding the GNP genotype by genome mining their corresponding glycosylation genes in a secondary metabolic gene cluster. A limitation of GNP characterization by MSn is variability in ionization, fragmentation, and fragment stability of structurally diverse GNPs. This variability in spectral outcome can be due to compound-inherent properties, e.g., a better ionization of aminosugars versus nonaminosugars, or instrument- and experiment-based differences. For example, more B/C-ions and less Y/Z-ions are observed in experiments with higher CID energies. Different instruments can also yield differences in MSn fragment intensities or fragmentation patterns. However, the general B/Y- and C/Z-fragmentation of O- and N-GNPs applies across different mass spectrometers with CID capabilities (Fig. S1). For our glycogenomic approach, quadrupole time-of-flight (Q-TOF) MS instruments are better suited than ion trap MS instruments because more low-m/z information such as B/C-ions is observed by Q-TOF mass spectrometers. It is also likely the method can be extended to ion traps that are specifically configured with methods such as pulsed-Q dissociation to capture low-m/z ions or even triple-Q–type instruments in precursor ion scanning mode for targeted analysis to capture the low-m/z fragment ions. LC-MS–based identification of NDP-sugar species could be an alternative application of glycogenomics for characterization of GNPs and, in general, glycosylated molecules. Analysis of such activated sugar species via EIC screening of sugar B-ions as [M-H]− species could be integrated in the glycogenomic workflow by ion-pairing LC conditions and MSn analysis in negative ion mode (54). However, a reliable glycogenomic connection of an NDP-sugar MS/MS spectrum to its biosynthetic genes in a microbial genome may only be possible for rare deoxysugars with distinct biosynthetic gene combinations. MSn analysis of GNPs has the advantage of obtaining the retention time of the pathway product, confirmation of sugar B-ions by their Y-ion species, and possible characterization of the aglycone structure.
Genome mining of GNP pathways and connecting observed glycosyl groups with glycosyl genes in these pathways are the next steps in the MS-glycogenomic approach. First, all secondary metabolic gene clusters are predicted from a target genome by antiSMASH (38) and analyzed for the presence of common and specific glycosylation genes. For functional prediction of glycosylation genes by BLAST, only some glycosylation genes enable a reliable sequence-based regioselectivity prediction of their enzymatic products (3), e.g., 3- vs. 4-ketoreductases and 2,3- vs. 3,4-dehydratases, whereas regioselectivity of aminotransferases, epimerases, and methyltransferases was not predicted in our analysis. Among methyltransferases, the methylation site was predicted in terms of its element, i.e., N-, C-, or O-methylation. It is generally difficult to accurately predict a glycosyl group de novo from a set of common and specific glycosylation genes in a GNP gene cluster because these enzymes are often promiscuous in substrates and even catalyzed reactions (55). The bioinformatic assignment of GNP glycosidic bonds, either between sugar units or with the aglycone, also cannot be predicted reliably from specific glycosylation or glycosyltransferase genes. Glycosidic bond types of putative GNPs in glycogenomic analysis are assumed as N-/O-glycosidic based on their major occurrence in GNPs and their efficient MSn fragmentation. Regiospecificity is rather determined experimentally during the LC-MSn analysis or by NMR characterization of the purified GNP. In the case of LC-MSn analyses, glycosidic bond connectivities are characterized by identifying the position of the eliminated glyosidic hydroxyl or amine group in A-/X-fragment ions (14), e.g., from additional MS/MS analysis (MS3) of detected sugar B-ions (MS2).
Cross talk with primary and secondary metabolic pathways involved in carbohydrate biosynthesis, such as cell wall formation, can sometimes lead to lack of important genes in a GNP pathway and, thus, lead to a false or no sugar prediction. For example, the putative gene cluster of glycosylated thiopeptide Sch40832 (26, 27) from Micromonospora carbonacea ATCC 39149 harbors one glycosyltransferase gene but none for the synthesis of the NDP-sugar that are likely encoded in other sugar pathways in the genome (Datasets S3 and S4). In glycogenomic analysis, the glycosylation genes in candidate GNP pathways are used to test several natural product glycosylation hypotheses based on the MSn candidate sugars rather than to do de novo sugar prediction. A match of a putative GNP with its biosynthetic genes is made by reanalysis of MSn data and glycosylation genes, and, ultimately, by genetic deletion or NMR structure elucidation.
Glycogenomic analysis may also accelerate natural product glycodiversification efforts by preliminary LC-MS screening for successful glycosylation of aglycone libraries or specific aglycones via in vivo or in vitro glycoengineering strategies (56). For monitoring glycodiversification with unnatural semisynthetic NDP-sugars (56), the MS-glycogenetic code would need to be extended in its B-/Y-ion mass list. Screening for natural product glycosylation with unknown sugars, e.g., from new or engineered pathways in glycodiversification experiments, would be limited and rely on manual characterization of putative glycosyl groups as B-/Y-ion pairs of unknown mass in MSn spectra and subsequent structure prediction in the genetic context of the unusual sugar’s pathway.
In summary, we introduced a genome-mining approach that can characterize and link unknown GNP chemotypes and their genotypes in microbial genomes by iterative identification of O-/N-glycosyl groups in tandem MS spectra and of their glycosylation genes in secondary metabolic gene clusters. This work extends the concept of experiment-guided genome mining to more natural product classes such as glycosylated polyketides and, therefore, sets another blueprint for future automated characterization of complex secondary metabolomes by a combined application of MSn and genomics. The implementation of the MS-glycogenetic code and glycogenomic workflow in data acquisition and processing programs could lead to a faster characterization of new GNP chemistry, biochemistry, and bioactivity from the increasing microbial genome resources. This advance would also enable accelerated access and understanding of cryptic GNP pathways in microbial communities and as a therapeutic source. Implementation of this concept into new metabolomic (57) and metagenomic (58) approaches, in combination with newer tools that map mass spectrometry-detectable molecular space such as molecular networking or MetaMapp (57, 59, 60) can facilitate studies of more complex microbiome systems where parallel characterization of metabolomes and metagenomes will require connections of expressed chemotypes with present genotypes in a more automated fashion. Glycogenomics could be adapted to study any glycosylated molecule, including human molecules such as heparins, glycosylated virulence factors from pathogens, or even be used to study the composition of cell walls such as lipopolysaccharides.
Materials and Methods
Cultivation and Extraction of Actinobacteria.
A liquid ISP2 starter culture of Streptomyces sp. SPB74 was inoculated from a spore suspension and incubated at 28 °C, 225 rpm for 6 d. All incubations were performed on an Innova 2300 platform shaker (New Brunswick Scientific). A 50-mL ISP2 culture (ISP2 medium: 4 g of yeast extract, 10 g of malt extract, 4 g of D-glucose, and 1,000 mL of Millipore-filtered water) was inoculated with 1% of the starter culture and incubated at 28 °C, 225 rpm for 7 d. The supernatant and cells were extracted with ethyl acetate. The crude extract was dried by rotovaporation and analyzed by LC-MS for presence of GNPs. A liquid A1 starter culture (A1 medium: 4 g of yeast extract, 10 g of soluble starch, 2 g of peptone, 1 g of calcium carbonate, 30 g of InstantOcean mix, and 1,000 mL of Millipore-filtered water) of Salinispora arenicola CNB-527 was inoculated from a spore suspension and incubated at 28 °C, 225 rpm for 6 d. A 50-mL A1 culture was inoculated with 1% of the starter culture and incubated at 28 °C, 225 rpm for 7 d. The supernatant was extracted with ethyl acetate, and the cells were resuspended in methanol and stirred for 30 min. Ethyl acetate and methanol extracts were combined and dried by rotovaporation. The crude extract was analyzed by LC-MS for the presence of GNPs.
MS Analysis of Microbial Metabolic Extracts.
Crude microbial extracts were dissolved in methanol and filtered through Acrodisc MS Syringe Filter (polytetrafluoroethylene membrane, 25 mm, 0.2 μm; PALL Life Sciences). The samples were adjusted to a concentration of 200 μg/mL and injected into an Agilent 1260 LC system (injection volume: 5 μL) with an Agilent Extend-C18 RP UPLC column (2.1 × 100 mm, 1.8 μm) connected to an Agilent 6530 Accurate-Mass Q-TOF LC/MS. For analysis of Salinispora arenicola extract, the LC gradient was as follows: 10% (vol/vol) acetonitrile (ACN) (0.1% TFA, 0–3 min), 10–100% (vol/vol) ACN (0.1% TFA)/0.1% TFA (3–23 min), 100% ACN (0.1% TFA, 23–25 min), 10% (vol/vol) ACN (0.1% TFA, 25–30 min). The column compartment temperature was 25 °C. For Streptomyces sp. SPB74 extract analysis, the LC gradient was as follows: 10–100% (vol/vol) ACN (0.1% TFA)/0.1% TFA (0–20 min), 100% (vol/vol) ACN (0.1% TFA, 20–24 min), 10% (vol/vol) ACN (0.1% TFA, 24–30 min). For Salinispora arenicola extract analysis, the Q-TOF settings were as follows: acquisition mode auto-MS2—MS range: 125–1,500 m/z; MS scan rate: 1 spectrum/s; MS/MS scan rate: 2 spectra/s; isolation width: 4 m/z; CID energy: 20 eV; precursor selection static exclusion: 100–500 m/z; electrospray ionization (ESI) source—gas temperature: 300 °C; gas flow: 11 L/min; nebulizer: 45 psig, positive ion polarity; scan source parameters: VCap, 3,000 V; fragmentor, 100 V. For Streptomyces sp. SPB74 extract analysis, the Q-TOF settings were as follows: acquisition mode auto-MS2—MS range: 100–3,000 m/z; MS scan rate: 1 spectrum/s; MS/MS scan rate: 3 spectra/s; isolation width: 4 m/z; CID energy: 30 + 0.1(x[m/z]) eV; ESI source—gas temperature: 350 °C; gas flow: 11 L/min; nebulizer: 45 psig, positive ion polarity; scan source parameters: VCap, 4,000 V; fragmentor, 200 V. LC-MS/MS data were analyzed with Qualitative analysis software of MassHunter software, version B.05.00 (Agilent). LC-MS/MS data were searched for sugar footprints in EICs of B/C-ion fragments of Dataset S2 and/or Y-ion neutral loss chromatograms (NLCs). Peaks in EICs or NLCs were verified or discarded as candidate GNPs by reanalysis of MS/MS spectra for corresponding sugar B/C-ions and Y/Z-ion neutral losses. From a candidate GNP MS/MS spectrum, a list of candidate MS/MS sugars was generated by including all sugars from Dataset S2 that matched observed sugar mass shifts. For Q-TOF MS/MS analysis, vancomycin was injected by an electrospray ionization source into the inlets of the Agilent 6530 Accurate-Mass Q-TOF mass spectrometer or of a Bruker microQ-TOF mass spectrometer. MS/MS data were acquired as described above for the Agilent Q-TOF MS and under the following Q-TOF settings for the Bruker Q-TOF MS: CID: 63.5 eV; radiofrequency, 200 Vpp.
For IonTrap MS/MS analysis, vancomycin was injected by a nanomate-electrospray ionization robot (Advion) for consecutive electrospray into the MS inlet of a LTQ 6.4T Fourier transform–ion cyclon resonance mass spectrometer (Thermo Finnigan). MS/MS data were acquired in FTMS mode (CID: 30 eV; precursor isolation width: 3 m/z) and analyzed using QualBrowser, which is part of the Xcalibur LTQ-FT software package (Thermo Fisher).
Gel filtration and HPLC fractions of Streptomyces sp. SPB74 were analyzed by MALDI-TOF MS. Fractions were mixed 1:1 with a saturated solution of Universal MALDI matrix in 70% (vol/vol) ACN containing 0.1% TFA and spotted on a Bruker MSP 96 anchor plate. The sample was dried and analyzed with a Microflex Bruker Daltonics mass spectrometer equipped with Compass 1.2 software package (Bruker Daltonics). The mass spectrometer was calibrated externally with a standard peptide mixture before each measurement.
Genome Mining of GNPs.
Genome sequences of Streptomyces sp. SPB74 (GenBank files GG770539 and GG770540) and Salinispora arenicola CNB-527 [Department of Energy (DOE) Joint Genome Institute; genome ID 2515154093] were analyzed by antiSMASH (38) for prediction of secondary metabolic gene clusters. Each predicted gene cluster was analyzed for presence of common and specific glycosylation genes (i) based on gene annotation in “Genes and detection info overview” of each cluster and (ii) based on BLAST analysis of putative glycosylation genes. Glycosylation gene functions were assigned based on gene annotation and closest functional BLAST homologs. Specific glycosylation genes were differentiated (if possible) into the following: 2,3DH, 3,4DH, 3KR, 4KR, 3,4IM, E, FuPyIM, AmT, O-MT, N,N-MT, N-MT, C-MT, N-ET, AcT, CarbT, PyT, oxDA, OxRed, Dhg, ThiS, N-Ox (see Dataset S2 for abbreviations). A list of all gene clusters with glycosylation genes was generated.
Each gene cluster was tested if the specific glycosylation genes match any of the observed MS/MS candidate sugars based on Dataset S2, i.e., if the biosynthetic genes of an observed sugar are present in a candidate GNP gene cluster. A putative match was confirmed by matching of additional candidate MS/MS sugars to genes in the candidate gene cluster. Next, the candidate GNP gene cluster was fully analyzed by BLAST analysis of closest functional homologs and a natural product class was assigned based on nonglycosylation biosynthetic genes.
To analyze the distribution of GNP pathways in actinobacterial genomes, 199 strains with complete genomes from the DOE Joint Genome Institute database (October 2012) were first analyzed by antiSMASH. Putative GNP gene clusters were characterized by presence of common glycosylation genes, e.g., a glycosyltransferase, and specific glycosylation genes.
Purification of GNPs.
Cinerubin B was isolated from a 1 L ISP2 medium culture of Streptomyces sp. SPB74, which was inoculated with a 10-mL ISP2 starter culture (6 d, 28 °C, 225 rpm) from spore suspension inoculation and incubated for 7 d at 28 °C and 225 rpm. The liquid culture was extracted with ethyl acetate (three times). The crude extracts were combined and dried completely by rotovaporation. The crude extract was resuspended in methanol and separated by gel filtration chromatography (solid phase: Sephadex LH20; GE Life Sciences; mobile phase: methanol). Gel filtration fractions were analyzed by dried-droplet MALDI-TOF MS for the presence of cinerubin B. Gel filtration fractions with cinerubin B were further purified by semipreparative reverse-phase HPLC [Phenomenex Luna C18, 5u, 250 × 10 mm, 100 Å; 0–5 min—10% (vol/vol) ACN (0.1% TFA)/0.1% TFA; 5–45 min—10–100% (vol/vol) ACN(0.1% TFA)/0.1% TFA]. HPLC fractions were analyzed by dried-droplet MALDI-TOF MS for the presence and purity of cinerubin B for subsequent NMR analysis.
Arenimycin A and B were isolated from a 4 L A1 medium culture of Salinispora arenicola CNB-527, which was inoculated with a 10-mL A1 starter culture (6 d, 28 °C, 225 rpm) from spore suspension inoculation and incubated for 8 d at 28 °C and 225 rpm. On day 8, XAD-7 Amberlite resin (Sigma-Aldrich) was added to the culture (20 g/L) and incubated for another 2 h. The culture was subsequently filtered through cheesecloth. One-half of the cheesecloth with the cells and XAD-7 resin was soaked for 1 h in methanol, whereas the other half was soaked in acetone. The acetone and methanol extracts were decanted and rotovaporated to dryness. The crude extract was concentrated in vacuo, resuspended in methanol (2 mL), and loaded on a reversed-phase C18 silica gel column for flash-column chromatography with a 20–100% (vol/vol) methanol/water gradient in five steps. Chromatography fractions were analyzed for arenimycin A and B by LC-MS or MALDI-TOF MS. Arenimycin A eluted at 80% (vol/vol) methanol and arenimycin B mainly at 100% methanol. Arenimycin A or B fractions were combined, concentrated in vacuo, and further purified by semipreparative reverse HPLC [Phenomenex Luna C18, 5u, 250 × 10 mm, 100 Å; 0–5 min—25% (vol/vol) ACN(0.1% TFA)/0.1% TFA; 5–55 min—25–75% (vol/vol) ACN(0.1% TFA)/0.1% TFA); 55–60 min—75–100% (vol/vol) ACN(0.1% TFA)/0.1% TFA]. HPLC fractions were analyzed by LC-MS for the presence of arenimycin A or B for subsequent NMR analysis.
NMR Analysis of GNPs.
Purified cinerubin B, arenimycin A, and arenimycin B were each dissolved in MeOD-d4 and subjected to NMR structure elucidation [1H, double-quantum–filtered correlation spectroscopy (DQF-COSY), 1H-13C heteronuclear multiple-bond correlation spectroscopy (HMBC), 1H-13C heteronuclear single-quantum coherence (HSQC), NOESY]. NMR data were analyzed with Topspin 2.1.6 software (Bruker).
Bioactivity Tests of GNPs.
Minimum inhibitory concentration testing was performed by broth dilution in cation-adjusted Mueller–Hinton broth according to Clinical and Laboratory Standards Institute methods (61). Companion minimum bactericidal concentration was calculated upon sample transfer via 48‐prong Boekel replicator to antibiotic-free Todd–Hewitt agar to detect surviving bacterial colony-forming units.
Supplementary Material
Acknowledgments
We thank P. R. Jensen for providing Salinispora strains and genome sequences, M. Meehan for Bruker Q-TOF MS training, M. Fischbach and P. Cimermancic for bioinformatics discussions, H.-P. Fiedler for the strain Streptomyces sp. Tu6071, M. Crüsemann for help with cultivation and extraction, and J. Busch for performing the cytotoxicity assay. This work was supported by National Institutes of Health Grants GM085770 (to B.S.M.), GM097509 (to B.S.M. and P.C.D.), HL107150 (to V.N.) for University of California, San Diego Programs of Excellence in Glycosciences, and Instrument Grants S10-RR031562 and S10-RR029121.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315492110/-/DCSupplemental.
References
- 1.Staunton J, Weissman KJ. Polyketide biosynthesis: A millennium review. Nat Prod Rep. 2001;18(4):380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda H, Nonomiya T, Usami M, Ohta T, Omura S. Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis. Proc Natl Acad Sci USA. 1999;96(17):9509–9514. doi: 10.1073/pnas.96.17.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thibodeaux CJ, Melançon CE, 3rd, Liu HW. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew Chem Int Ed Engl. 2008;47(51):9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.La Ferla B, et al. Natural glycoconjugates with antitumor activity. Nat Prod Rep. 2011;28(3):630–648. doi: 10.1039/c0np00055h. [DOI] [PubMed] [Google Scholar]
- 5.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep. 2009;26(11):1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hubbard BK, Walsh CT. Vancomycin assembly: Nature’s way. Angew Chem Int Ed Engl. 2003;42(7):730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 7.Ding Y, et al. Moving posttranslational modifications forward to biosynthesize the glycosylated thiopeptide nocathiacin I in Nocardia sp. ATCC202099. Mol Biosyst. 2010;6(7):1180–1185. doi: 10.1039/c005121g. [DOI] [PubMed] [Google Scholar]
- 8.Ahlert J, et al. The calicheamicin gene cluster and its iterative type I enediyne PKS. Science. 2002;297(5584):1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 9.Gebhardt K, et al. Phenalinolactones A-D, terpenoglycoside antibiotics from Streptomyces sp. Tü 6071. J Antibiot (Tokyo) 2011;64(3):229–232. doi: 10.1038/ja.2010.165. [DOI] [PubMed] [Google Scholar]
- 10.Pathirana C, Jensen PR, Dwight R, Fenical W. Rare phenazine L-quinovose esters from a marine actinomycete. J Org Chem. 1992;57:740–742. [Google Scholar]
- 11.Singh S, Phillips GN, Jr, Thorson JS. The structural biology of enzymes involved in natural product glycosylation. Nat Prod Rep. 2012;29(10):1201–1237. doi: 10.1039/c2np20039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohr J. Modifying oxidation and glycosylation events in biosyntheses of natural product anticancer drugs—Challenges for combinatorial biosynthesis. In: Wrigley SK, Thomas R, Bedford CT, Nicholson N, editors. Functional Molecules from Natural Sources. Cambridge, UK: Royal Society of Chemistry Publishing; 2011. pp. 161–183. [Google Scholar]
- 13.Chen F, et al. Distribution of dTDP-glucose-4,6-dehydratase gene and diversity of potential glycosylated natural products in marine sediment-derived bacteria. Appl Microbiol Biotechnol. 2011;90(4):1347–1359. doi: 10.1007/s00253-011-3112-y. [DOI] [PubMed] [Google Scholar]
- 14.An HJ, Lebrilla CB. Structure elucidation of native N- and O-linked glycans by tandem mass spectrometry (tutorial) Mass Spectrom Rev. 2011;30(4):560–578. doi: 10.1002/mas.20283. [DOI] [PubMed] [Google Scholar]
- 15.Gates PJ, Kearney GC, Jones R, Leadlay PF, Staunton J. Structural elucidation studies of erythromycins by electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom. 1999;13(4):242–246. doi: 10.1002/(SICI)1097-0231(19990228)13:4<242::AID-RCM447>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 16.Gräfe U, Heinze S, Schlegel B, Härtl A. Disclosure of new and recurrent microbial metabolites by mass spectrometric methods. J Ind Microbiol Biotechnol. 2001;27(3):136–143. doi: 10.1038/sj.jim.7000023. [DOI] [PubMed] [Google Scholar]
- 17.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5(4):397–409. [Google Scholar]
- 18.Cuyckens F, Claeys M. Mass spectrometry in the structural analysis of flavonoids. J Mass Spectrom. 2004;39(1):1–15. doi: 10.1002/jms.585. [DOI] [PubMed] [Google Scholar]
- 19.Winter JM, Behnken S, Hertweck C. Genomics-inspired discovery of natural products. Curr Opin Chem Biol. 2011;15(1):22–31. doi: 10.1016/j.cbpa.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Kersten RD, et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7(11):794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laureti L, et al. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc Natl Acad Sci USA. 2011;108(15):6258–6263. doi: 10.1073/pnas.1019077108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gross H, et al. The genomisotopic approach: A systematic method to isolate products of orphan biosynthetic gene clusters. Chem Biol. 2007;14(1):53–63. doi: 10.1016/j.chembiol.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Lautru S, Deeth RJ, Bailey LM, Challis GL. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat Chem Biol. 2005;1(5):265–269. doi: 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- 24.Herget S, et al. Statistical analysis of the Bacterial Carbohydrate Structure Data Base (BCSDB): Characteristics and diversity of bacterial carbohydrates in comparison with mammalian glycans. BMC Struct Biol. 2008;8:35. doi: 10.1186/1472-6807-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benson DA, et al. GenBank. Nucleic Acids Res. 2013;41(Database issue):D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith CA, et al. METLIN: A metabolite mass spectral database. Ther Drug Monit. 2005;27(6):747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 27.Ferrer I, García-Reyes JF, Fernandez-Alba A. Identification and quantitation of pesticides in vegetables by liquid chromatography time-of-flight mass spectrometry. Trends Analyt Chem. 2005;24:671–682. [Google Scholar]
- 28.Useglio M, et al. TDP-L-megosamine biosynthesis pathway elucidation and megalomicin a production in Escherichia coli. Appl Environ Microbiol. 2010;76(12):3869–3877. doi: 10.1128/AEM.03083-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Z, Jakobi K, Welzel K, Hertweck C. Biosynthesis of the antitumor agent chartreusin involves the oxidative rearrangement of an anthracyclic polyketide. Chem Biol. 2005;12(5):579–588. doi: 10.1016/j.chembiol.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 30.Edo K, et al. The structure of neocarzinostatin chromophore possessing a novel bicyclo-[7,3,0]dodecadiyne system. Tetrahedron Lett. 1985;26:331–334. [Google Scholar]
- 31.Martin JR, et al. 3′-de-o-methyl-2′,3′-anhydro-lankamycin, a new macrolide antibiotic from Streptomyces violaceoniger. Helv Chim Acta. 1976;59(5):1886–1894. doi: 10.1002/hlca.19760590545. [DOI] [PubMed] [Google Scholar]
- 32.Puar MS, et al. Sch 40832: A novel thiostrepton from Micromonospora carbonacea. J Antibiot (Tokyo) 1998;51(2):221–224. doi: 10.7164/antibiotics.51.221. [DOI] [PubMed] [Google Scholar]
- 33.Kersten RD, et al. Bioactivity-guided genome mining reveals the lomaiviticin biosynthetic gene cluster in Salinispora tropica. ChemBioChem. 2013;14(8):955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dürr C, et al. Biosynthesis of the terpene phenalinolactone in Streptomyces sp. Tü6071: Analysis of the gene cluster and generation of derivatives. Chem Biol. 2006;13(4):365–377. doi: 10.1016/j.chembiol.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Brautaset T, et al. Biosynthesis of the polyene antifungal antibiotic nystatin in Streptomyces noursei ATCC 11455: Analysis of the gene cluster and deduction of the biosynthetic pathway. Chem Biol. 2000;7(6):395–403. doi: 10.1016/s1074-5521(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 36.Li J, et al. ThioFinder: A Web-based tool for the identification of thiopeptide gene clusters in DNA sequences. PLoS One. 2012;7(9):e45878. doi: 10.1371/journal.pone.0045878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott JJ, et al. Bacterial protection of beetle-fungus mutualism. Science. 2008;322(5898):63. doi: 10.1126/science.1160423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blin K, et al. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41(Web Server issue):W204–W212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexeev I, Sultana A, Mäntsälä P, Niemi J, Schneider G. Aclacinomycin oxidoreductase (AknOx) from the biosynthetic pathway of the antibiotic aclacinomycin is an unusual flavoenzyme with a dual active site. Proc Natl Acad Sci USA. 2007;104(15):6170–6175. doi: 10.1073/pnas.0700579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konishi M, et al. Esperamicins, a novel class of potent antitumor antibiotics. I. Physico-chemical data and partial structure. J Antibiot (Tokyo) 1985;38(11):1605–1609. doi: 10.7164/antibiotics.38.1605. [DOI] [PubMed] [Google Scholar]
- 41.Räty K, et al. Cloning and characterization of Streptomyces galilaeus aclacinomycins polyketide synthase (PKS) cluster. Gene. 2002;293(1-2):115–122. doi: 10.1016/s0378-1119(02)00699-6. [DOI] [PubMed] [Google Scholar]
- 42.Räty K, Kunnari T, Hakala J, Mäntsälä P, Ylihonko K. A gene cluster from Streptomyces galilaeus involved in glycosylation of aclarubicin. Mol Gen Genet. 2000;264(1-2):164–172. doi: 10.1007/s004380000306. [DOI] [PubMed] [Google Scholar]
- 43.Ettlinger L, et al. Stoffwechselprodukte von Actinomyceten, XVI. Cinerubine. Chem Ber. 1959;92:1867–1879. [Google Scholar]
- 44.Udwary DW, et al. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA. 2007;104(25):10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lane AL, et al. Structures and comparative characterization of biosynthetic gene clusters for cyanosporasides, enediyne-derived natural products from marine actinomycetes. J Am Chem Soc. 2013;135(11):4171–4174. doi: 10.1021/ja311065v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jensen PR, Mafnas C. Biogeography of the marine actinomycete Salinispora. Environ Microbiol. 2006;8(11):1881–1888. doi: 10.1111/j.1462-2920.2006.01093.x. [DOI] [PubMed] [Google Scholar]
- 47.Asolkar RN, Kirkland TN, Jensen PR, Fenical W. Arenimycin, an antibiotic effective against rifampin- and methicillin-resistant Staphylococcus aureus from the marine actinomycete Salinispora arenicola. J Antibiot (Tokyo) 2010;63(1):37–39. doi: 10.1038/ja.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomi S, Sasaki T, Itoh J, Sezaki M. SF2446, new benzo[a]naphthacene quinone antibiotics. II. The structural elucidation. J Antibiot (Tokyo) 1988;41(4):425–432. doi: 10.7164/antibiotics.41.425. [DOI] [PubMed] [Google Scholar]
- 49.Kim BC, Lee JM, Ahn JS, Kim BS. Cloning, sequencing, and characterization of the pradimicin biosynthetic gene cluster of Actinomadura hibisca P157-2. J Microbiol Biotechnol. 2007;17(5):830–839. [PubMed] [Google Scholar]
- 50.Tsunakawa M, et al. The structure of pradimicins A, B and C: A novel family of antifungal antibiotics. J Org Chem. 1989;54:2532–2536. [Google Scholar]
- 51.Rafanan ER, Jr, Hutchinson CR, Shen B. Triple hydroxylation of tetracenomycin A2 to tetracenomycin C involving two molecules of O2 and one molecule of H2O. Org Lett. 2000;2(20):3225–3227. doi: 10.1021/ol0002267. [DOI] [PubMed] [Google Scholar]
- 52.Pagani I, et al. The Genomes OnLine Database (GOLD) v.4: Status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40(Database issue):D571–D579. doi: 10.1093/nar/gkr1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corre C, Challis GL. New natural product biosynthetic chemistry discovered by genome mining. Nat Prod Rep. 2009;26(8):977–986. doi: 10.1039/b713024b. [DOI] [PubMed] [Google Scholar]
- 54.Dürr C, et al. The glycosyltransferase UrdGT2 catalyzes both C- and O-glycosidic sugar transfers. Angew Chem Int Ed Engl. 2004;43(22):2962–2965. doi: 10.1002/anie.200453758. [DOI] [PubMed] [Google Scholar]
- 55.Rodríguez E, Peirú S, Carney JR, Gramajo H. In vivo characterization of the dTDP-D-desosamine pathway of the megalomicin gene cluster from Micromonospora megalomicea. Microbiology. 2006;152(Pt 3):667–673. doi: 10.1099/mic.0.28680-0. [DOI] [PubMed] [Google Scholar]
- 56.Gantt RW, Peltier-Pain P, Thorson JS. Enzymatic methods for glyco(diversification/randomization) of drugs and small molecules. Nat Prod Rep. 2011;28(11):1811–1853. doi: 10.1039/c1np00045d. [DOI] [PubMed] [Google Scholar]
- 57.Watrous J, et al. Mass spectral molecular networking of living microbial colonies. Proc Natl Acad Sci USA. 2012;109(26):E1743–E1752. doi: 10.1073/pnas.1203689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen EE, Banfield JF. Community genomics in microbial ecology and evolution. Nat Rev Microbiol. 2005;3(6):489–498. doi: 10.1038/nrmicro1157. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen DD, et al. MS/MS networking guided analysis of molecule and gene cluster families. Proc Natl Acad Sci USA. 2013;110(28):E2611–E2620. doi: 10.1073/pnas.1303471110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barupal DK, et al. MetaMapp: Mapping and visualizing metabolomic data by integrating information from biochemical pathways and chemical and mass spectral similarity. BMC Bioinformatics. 2012;13:99. doi: 10.1186/1471-2105-13-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clinical and Laboratory Standards Institute . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. Wayne, PA: Clinical and Laboratory Standards Institute; 2006. Document M7–A7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.