

Significance
Cytochrome c oxidase is an important proton pump that utilizes the chemical energy released by oxygen reduction to generate the transmembrane proton concentration gradient. A conserved glutamate residue has been proposed to play a key role in proton pumping, although factors that control the timing and destination of proton transfers by this residue remain poorly understood. By integrating results from multiple computational methodologies, we propose a mechanism in which changes in local hydration and electrostatic interactions regulate the proton affinity of this key residue and, as a result, proton transfer activities. The results highlight the functional significance of local protein motions and hydration state of internal cavities.
Keywords: proton pumping, pKa
Abstract
Cytochrome c oxidase contributes to the transmembrane proton gradient by removing two protons from the high-pH side of the membrane each time the binuclear center active site is reduced. One proton goes to the binuclear center, whereas the other is pumped to the low-pH periplasmic space. Glutamate 286 (Glu286) has been proposed to serve as a transiently deprotonated proton donor. Using unrestrained atomistic molecular dynamics simulations, we show that the size of and water distribution in the hydrophobic cavity that holds Glu286 is controlled by the protonation state of the propionic acid of heme a3, a group on the proton outlet pathway. Protonation of the propionate disrupts hydrogen bonding to two side chains, allowing a loop to swing open. Continuum electrostatics and atomistic free-energy perturbation calculations show that the resultant changes in hydration and electrostatic interactions lower the Glu proton affinity by at least 5 kcal/mol. These changes in the internal hydration level occur in the absence of major conformational transitions and serve to stabilize needed transient intermediates in proton transport. The trigger is not the protonation of the Glu of interest, but rather the protonation of a residue ∼10 Å away. Thus, unlike local water penetration to stabilize a new charge, this finding represents a specific role for water molecules in the protein interior, mediating proton transfers and facilitating ion transport.
Water is essential to the structure, dynamics, and function of biomolecules (1, 2), and its role in protein folding, association (3), and dynamics (4, 5) has been well documented. The highly polar and polarizable water molecules play diverse roles in protein interiors. Water can aid catalysis in enzyme active sites (6–8). Water or water chains are often observed in proteins that are (9, 10) proton or ion transporters or pumps (11–14). Internal cavities holding functional water molecules are believed to have a fairly constant level of hydration throughout the protein reaction cycle, unless significant conformational changes occur (15). Water penetration in response to the ionization or reduction of internal groups has been extensively discussed (16, 17), although it is usually described as part of protein's local dielectric response.
Cytochrome c oxidase (CcO) adds to the transmembrane proton gradient through proton transport coupled to electron transfer reactions (12, 18, 19). In the overall reaction, electrons from four cytochromes c are transferred to oxygen to make two water molecules at the binuclear center (BNC). The four protons needed for chemistry are bound only from the high-pH, N side of the membrane. Coupled to the process, four more protons are transferred across the membrane from the high- to low-pH (P) side of the membrane. Thus, eight charges are transferred across the membrane as each O2 is reduced.
Glu286 is a required, conserved residue that is expected to transfer protons from the D channel either to the BNC or the proton-loading site (PLS) each time CcO is reduced (Fig. 1). Experiments assign a functional pKa to Glu286 near 9.4 (20). Thus, at higher pH, proton binding to the Glu becomes rate-limiting for steady-state turnover. The current understanding of the reaction cycle shows that protons are pumped in each of the four distinct BNC redox states (12, 18, 19). The reaction mechanism needs Glu286 to be deprotonated twice to pass a proton to the PLS and to the BNC in each CcO reduction step. Previous continuum electrostatics (21–24) and semimacroscopic (25, 26) calculations obtained pKa values for Glu286 near 9–10. However, recent microscopic calculations have found significantly higher pKa values of more than 12 (17, 27), making it unclear how a proton could be lost from this site, whereas others do not address the proton affinity of the essential Glu (28, 29). The discrepancy between experiment and simulations may result from technical issues such as the use of static protein structures and limited sampling of protonation states of titratable groups, or it may arise from changes in the protein that have been missed. Thus, a key question remaining is how the proton affinity of this essential Glu is modulated so it can donate a proton to the PLS and the BNC through the reaction cycle.
Fig. 1.

Illustration of key residues near the hydrophobic cavity in CcO and general proton pathways to and from Glu286.
In this work, computational studies show the hydration level of an internal cavity near Glu286 changes substantially without needing global conformational changes. Rather, the structure of an internal loop is controlled or anchored by the protonation state of the D-propionic acid of heme a3. This potentially important motion has not been noted in previous computational studies in which part of the protein structure was constrained (21, 27, 28). Both continuum electrostatics and quantum mechanical/classical mechanical (QM/MM) free-energy simulations show that the resultant changes in Glu286 hydration level and electrostatic interactions significantly affect its pKa (proton affinity). These findings point to a molecular mechanism to modulate the timing of proton transfers in the CcO proton pumping cycle by modifying the proton affinity of this key acid. More generally, the results show that changes in protein internal hydration may occur with only small, distal conformational changes, and these can serve as an important regulatory mechanism in ion transport, thus going beyond being part of generic dielectric response of proteins.
Results and Discussion
Hydration Level of the Hydrophobic Cavity Near Glu286 Depends on the Protonation State of the Heme a3 Propionate D.
The hydrophobic cavity that bridges Glu286 and propionate D of heme a3 (PRDa3), ∼10 Å away, is a functionally important region in CcO (Fig. 1). It is surrounded by the key cofactors: heme a, heme a3, and CuB. The latter two form the BNC, which catalyzes the reduction of molecular oxygen to water, providing the overall thermodynamic driving force for proton pumping. No water molecules are observed in the cavity around the Glu in the various crystal structures of CcO from different organisms (SI Appendix, Table S2), although it is assumed that they will be needed to mediate proton transfers through this region. Disordered and dynamic water molecules are hard to see in crystal structures (30, 31). Molecular dynamics (MD) simulations typically find four to five molecules in the region (27, 29, 32–34), especially following Glu ionization (17). However, most previous simulations sampled relatively short times, often did not include a detailed membrane or solvent environment, and most importantly constrain a significant number of atoms to their crystallographic positions.
Here we have carried out a comparison of the hydrophobic cavity near Glu286 in different CcO chemical states in fairly long timescale (multiple 15–50 ns) unconstrained atomistic MD simulations in an explicit membrane environment. We focus on four key substates in the PR → F transition, which has been extensively characterized by experiments (12, 18, 19). The four states are denoted: PR, 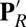 ,
, 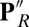 , and ′F. Analyses considering different force field parameters (27, 35), including the effect of electronic polarization (16), and conditions for the MD simulations test the robustness of the results (SI Appendix).
, and ′F. Analyses considering different force field parameters (27, 35), including the effect of electronic polarization (16), and conditions for the MD simulations test the robustness of the results (SI Appendix).
The PR state has the Glu protonated and the PRDa3 ionized. In 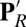 the proton has transferred from Glu, which is now ionized, to the now neutral, protonated PRDa3. In
the proton has transferred from Glu, which is now ionized, to the now neutral, protonated PRDa3. In 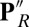 , reprotonation of the Glu leaves both acids protonated. In ′F the acids maintain the
, reprotonation of the Glu leaves both acids protonated. In ′F the acids maintain the 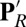 protonation states, but a proton is added to the hydroxyl on CuB, representing proton transfer into the BNC for oxygen reduction chemistry. Thus, PR explores the initial CcO protonation state,
protonation states, but a proton is added to the hydroxyl on CuB, representing proton transfer into the BNC for oxygen reduction chemistry. Thus, PR explores the initial CcO protonation state, 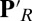 , the one after a proton has moved to the pumping site, and ′F, the one where the proton has transferred to the BNC, before proton release from the PLS. The identity of the proton-loading or pumping site is unknown. Likely candidates are the heme a3 propionates (19, 25) or a His ligand of CuB (36). Here we take PRDa3 as the PLS because it is spatially closer to Glu286 and the D channel, which mediates the transfer of protons taken up from the N side of the membrane (Fig. 1). The BNC is fixed in the specific redox states here (Table 1). However, this should not be critical, as proton pumping is posited to occur via the same mechanism in all CcO redox transitions (18, 19, 37).
, the one after a proton has moved to the pumping site, and ′F, the one where the proton has transferred to the BNC, before proton release from the PLS. The identity of the proton-loading or pumping site is unknown. Likely candidates are the heme a3 propionates (19, 25) or a His ligand of CuB (36). Here we take PRDa3 as the PLS because it is spatially closer to Glu286 and the D channel, which mediates the transfer of protons taken up from the N side of the membrane (Fig. 1). The BNC is fixed in the specific redox states here (Table 1). However, this should not be critical, as proton pumping is posited to occur via the same mechanism in all CcO redox transitions (18, 19, 37).
Table 1.
Computed Glu286 proton affinities (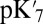 and pKa) using continuum electrostatics (SCCE, MCCE) and microscopic QM/MM thermodynamic integration
and pKa) using continuum electrostatics (SCCE, MCCE) and microscopic QM/MM thermodynamic integration
| Cavity | Input structures* | State† | QM/MM 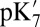
|
SCCE 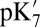 ‡ ‡
|
MCCE 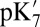 ‡ ‡
|
MCCE titration pKa‡ |
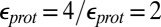 |
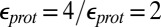 |
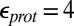 |
||||
| Small | 1M56 | XDD–RO | 18.5 ± 0.6 | 10.2 ± 0.7/15.1 ± 1.6 | 11.4 ± 1.0/18.0 | 14.1 ± 1.5 |
| Small | 1M56 | XPD–RO | – | −/− | 9.7 ± 0.9 /15.9 | 12.4 ± 1.8 |
| Small | 1M56 + 9w | XDD–RO | 14.0 ± 0.6 | −/− | −/− | |
| Small | 1M56 + 9w | XPD–RO | 11.2 ± 0.9 | −/− | −/− | |
| Large | PBC′F | XDD–RO | 14.3 ± 0.8 | 8.7 ± 0.8/11.8 ± 1.6 | 9.3 ± 0.7/12.8 | 11.1 ± 1.4 |
| Large | PBC′F | XPD-RO | 10.6 ± 0.7 | 6.5 ± 1.2/8.3 ± 2.7 | 7.7 ± 0.8/10.7 | 7.5 ± 1.0 |
| pK Changes§ | ||||||
| Dependence on PRDa3 protonation | ||||||
| 1M56 | – | −/− | 1.7/2.1 | 1.7 | ||
| 1M56+9w | 2.8 | −/− | −/− | – | ||
| PBC′F | 3.7 | 2.2/3.5 | 1.6/2.1 | 3.6 | ||
| Dependence on cavity hydration | ||||||
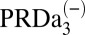
|
4.2 | 1.5/3.3 | 2.1/5.2 | 3.0 | ||
| PRDa3H | – | −/− | 2.0/5.2 | 4.9 | ||
| Combined effect | 7.6 | 3.7/6.8 | 3.7/7.3 | 6.6 | ||
QM/MM and SCCE 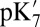 values are calculated with the protonation states for all other titratable groups fixed at their equilibrated protonation states found in MCCE calculations (23) at pH 7 (SI Appendix, Table S4); the MCCE
values are calculated with the protonation states for all other titratable groups fixed at their equilibrated protonation states found in MCCE calculations (23) at pH 7 (SI Appendix, Table S4); the MCCE 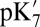 and pKa calculations allow the protonation states for all titratable groups other than those specified in SI Appendix, Table S1, to equilibrate at each pH. SI Appendix provides additional computational details and analyses.
and pKa calculations allow the protonation states for all titratable groups other than those specified in SI Appendix, Table S1, to equilibrate at each pH. SI Appendix provides additional computational details and analyses. 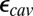 ,
, 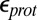 , dielectric constants for the cavity and protein; GSBP, generalized solvent boundary potential; MCCE, multiple-conformer continuum electrostatics; PBC, periodic boundary condition; PRDa3, propionate D of heme a3; SCCE, single-conformer continuum electrostatics; w, water molecules.
, dielectric constants for the cavity and protein; GSBP, generalized solvent boundary potential; MCCE, multiple-conformer continuum electrostatics; PBC, periodic boundary condition; PRDa3, propionate D of heme a3; SCCE, single-conformer continuum electrostatics; w, water molecules.
Local GSBP simulations start with different initial coordinates. 1M56: the crystal structure; 1M56 + 9w: nine additional water molecules are included near the cavity; PBC′F: an equilibrated snapshot from PBC simulation for the ′F state.
The states are labeled with a five-character notation. The first three letters indicate the protonation state (protonated or deprotonated) of Glu286, propionate D of heme a3 (PRDa3), the ligand of CuB [hydroxide (D) or water (P)]. The last two letters indicate the reduction state (reduced or oxidized) of heme a and CuB, respectively. “X” indicates 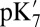 simulations in which the protonation state of Glu286 is varied.
simulations in which the protonation state of Glu286 is varied.
The 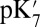 values before and after the slashes are computed with
values before and after the slashes are computed with 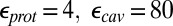 and
and 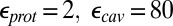 , respectively. Results with other values for the dielectric constants are in SI Appendix, Table S12–S13. For the MCCE pKa calculations,
, respectively. Results with other values for the dielectric constants are in SI Appendix, Table S12–S13. For the MCCE pKa calculations, 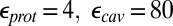 is always used.
is always used.
The effects of cavity size and protonation of PRDa3 are calculated based on the computed 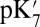 and pKa values. The combined effect is obtained by taking the difference between pK values computed with a small cavity (low hydration) with
and pKa values. The combined effect is obtained by taking the difference between pK values computed with a small cavity (low hydration) with 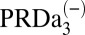 and a large cavity (high hydration) with PRDa3H.
and a large cavity (high hydration) with PRDa3H.
Hydration of the Cavity Near Glu286.
The hydration levels of the cavity in different chemical states of CcO are compared in several ways. It is very small in the unconstrained MD trajectories for the PR state, with Glu286 protonated and PRDa3 deprotonated, similar to that found in CcO crystal structures (SI Appendix, Fig. S3 and Table S2). The free volume found by a probe sphere of 1.4-Å radius is near zero. The continuum solvation energy penalty for Glu ionization in MD snapshots is 6.0 ± 0.5 kcal/mol. Although the MD structure starts with five water molecules in the cavity, they diffuse away in nanoseconds. In independent, 15–50-ns trajectories, there are typically only two water molecules left in the cavity by the end of the simulation. By contrast, the cavity is full of water molecules in both the 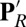 and ′F simulations, each of which have moved a proton off Glu286 onto PRDa3 (Fig. 2). The hydration level is slightly higher in the ′F state where the BNC has an extra proton. The MD trajectories now keep 8–10 water molecules in the cavity, whose volume has expanded to ∼155 ± 21Å3 (SI Appendix, Fig. S3, for an illustration). The Glu continuum solvation penalty for ionization is now only 3.9 ± 0.5 kcal/mol. Thus, the protonation of the Glu and PRDa3 changes the hydration level (Fig. 3) with a small, dry cavity in the PR state and a large, hydrated cavity in the
and ′F simulations, each of which have moved a proton off Glu286 onto PRDa3 (Fig. 2). The hydration level is slightly higher in the ′F state where the BNC has an extra proton. The MD trajectories now keep 8–10 water molecules in the cavity, whose volume has expanded to ∼155 ± 21Å3 (SI Appendix, Fig. S3, for an illustration). The Glu continuum solvation penalty for ionization is now only 3.9 ± 0.5 kcal/mol. Thus, the protonation of the Glu and PRDa3 changes the hydration level (Fig. 3) with a small, dry cavity in the PR state and a large, hydrated cavity in the 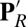 and ′F states. These are referred to as the small- and large-cavity structures in the following.
and ′F states. These are referred to as the small- and large-cavity structures in the following.
Fig. 2.

Snapshots from unconstrained PBC–MD simulations illustrating the hydration level and local conformational changes of the hydrophobic cavity near Glu286 in different chemical states. (A) PR; (B) 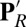 ; (C) ′F; (D) Superposition of snapshots from PR (loop165–177 in cyan with Glu286 protonated, PRDa3 deprotonated) and
; (C) ′F; (D) Superposition of snapshots from PR (loop165–177 in cyan with Glu286 protonated, PRDa3 deprotonated) and 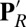 (purple loop with Glu286 deprotonated, PRDa3 protonated) showing that the overall structure does not undergo any major changes, whereas the loop that bears Trp172 moves significantly in response to the protonation of PRDa3. SI Appendix, Fig. S5, provides data from additional CcO states and SI Appendix, Table S1, for the protonation or oxidation states of key groups in the various enzyme states.
(purple loop with Glu286 deprotonated, PRDa3 protonated) showing that the overall structure does not undergo any major changes, whereas the loop that bears Trp172 moves significantly in response to the protonation of PRDa3. SI Appendix, Fig. S5, provides data from additional CcO states and SI Appendix, Table S1, for the protonation or oxidation states of key groups in the various enzyme states.
Fig. 3.
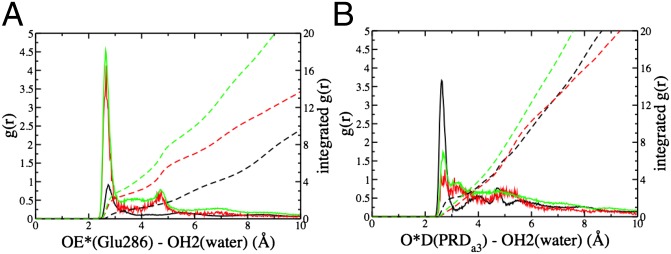
Radial distribution (solid lines) and integrated radial distribution (dashed) of water oxygens in PBC simulations for different CcO states (black: PR; red: 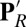 ; green: ′F): (A) around carboxylate oxygens of E286; (B) around carboxylate oxygens of PRDa3. SI Appendix, Fig.S5, provides data from additional CcO states.
; green: ′F): (A) around carboxylate oxygens of E286; (B) around carboxylate oxygens of PRDa3. SI Appendix, Fig.S5, provides data from additional CcO states.
The cavity contacts explain how its size and hydration level are changed. As in the crystal structures, in the PR state, the charged PRDa3 engages in stable hydrogen-bonding interactions with the side chains of Arg481 and Trp172 (Figs. 2A and 4). Moreover, Glu286 is charge-neutral and so does not stabilize the accumulation of a significant number of water molecules. These features lead to a dehydrated compact cavity. In contrast, in 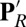 and ′F, PRDa3 is protonated and overall neutral, with weaker interactions with Arg481 and Trp172. This Trp is in a loop with a highly conserved sequence motif GxGxGWxxYxPL (SI Appendix, Fig. S2). The PLS region is constantly more mobile in MD trajectories with a protonated PRDa3 as monitored by the distribution of the distance between PRDa3–Arg481 and PRDa3–Trp172 (Fig. 4). Previous MD simulations where Arg481 has been mutated to a Lys show similar changes in this loop (32). Simulation of a
and ′F, PRDa3 is protonated and overall neutral, with weaker interactions with Arg481 and Trp172. This Trp is in a loop with a highly conserved sequence motif GxGxGWxxYxPL (SI Appendix, Fig. S2). The PLS region is constantly more mobile in MD trajectories with a protonated PRDa3 as monitored by the distribution of the distance between PRDa3–Arg481 and PRDa3–Trp172 (Fig. 4). Previous MD simulations where Arg481 has been mutated to a Lys show similar changes in this loop (32). Simulation of a 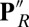 state where both Glu286 and PRDa3 are protonated also leads to an expanded, solvated cavity (SI Appendix, Table S1 and Fig. S5), supporting a model where the protonation of PRDa3, rather than deprotonation of Glu286, triggers changes in the cavity.
state where both Glu286 and PRDa3 are protonated also leads to an expanded, solvated cavity (SI Appendix, Table S1 and Fig. S5), supporting a model where the protonation of PRDa3, rather than deprotonation of Glu286, triggers changes in the cavity.
Fig. 4.

Comparison of key distance distributions for residues near the hydrophobic cavity calculated for different chemical states of CcO with PBC simulations (black: PR; red: 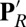 ; green: ′F). (A) E286–PRDa3; (B) R481–PRDa3; (C) W172 side chain-PRDa3; (D) W172 side chain-PRDa. The arrows indicate the distances in the crystal structure (PDB code 1M56). SI Appendix, Fig.S5, provides data from additional CcO states.
; green: ′F). (A) E286–PRDa3; (B) R481–PRDa3; (C) W172 side chain-PRDa3; (D) W172 side chain-PRDa. The arrows indicate the distances in the crystal structure (PDB code 1M56). SI Appendix, Fig.S5, provides data from additional CcO states.
A comparison of structures from these unconstrained simulations with explicit membrane indicates that the significant changes in the hydration of the hydrophobic cavity do not need global conformational transitions. The RMSD of subunit 1 that contains the active centers is less than 1.4 Å. Local structural flexibility, however, is important. In particular, rearrangement of the loop that bears Trp172 (32) is essential for water penetration into the cavity (Fig. 2D). For example, in local, generalized solvent boundary potential (GSBP) MD simulations where part of this loop is constrained to the crystallographic position, no water molecules penetrate or leave the cavity on the nanosecond timescale in either 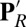 or ′F simulations (SI Appendix, Figs. S6–S8).
or ′F simulations (SI Appendix, Figs. S6–S8).
pKa and 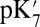 of Glu286.
of Glu286.
To understand the functional implication of the hydration level differences in the large and small-cavity structures, the proton affinity of Glu286 is computed with protonation state of PRDa3 and the BNC fixed to define the states of interest. The free energy of ionization of an acid (A) when the ionization states of all other titratable groups are equilibrated at pH 7 is
Thus, 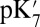 , reported here, represents a transient energy for deprotonating the Glu, as the other groups remain out of equilibrium with the change in Glu charge. The Glu286
, reported here, represents a transient energy for deprotonating the Glu, as the other groups remain out of equilibrium with the change in Glu charge. The Glu286 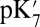 is estimated with multiple computational approaches (Table 1) that include both microscopic [QM/MM–thermodynamic integration (TI) (27, 38)] and continuum electrostatic methods [single-conformer continuum electrostatics (SCCE) (39, 40) and multiple-conformer continuum electrostatics (MCCE) (41)]. The range of
is estimated with multiple computational approaches (Table 1) that include both microscopic [QM/MM–thermodynamic integration (TI) (27, 38)] and continuum electrostatic methods [single-conformer continuum electrostatics (SCCE) (39, 40) and multiple-conformer continuum electrostatics (MCCE) (41)]. The range of 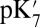 determined with the different methods highlights the difficulty of computing the absolute proton affinity of a deeply buried group in large transmembrane proteins like CcO (17). Therefore, our approach is to compare the results of the very different computational methodologies and identify consistent trends (SI Appendix provides additional details and analyses). Finally, the true pKa is also determined with MCCE titration, a process that keeps the protonation states of all residues at equilibrium with the imposed solution pH.
determined with the different methods highlights the difficulty of computing the absolute proton affinity of a deeply buried group in large transmembrane proteins like CcO (17). Therefore, our approach is to compare the results of the very different computational methodologies and identify consistent trends (SI Appendix provides additional details and analyses). Finally, the true pKa is also determined with MCCE titration, a process that keeps the protonation states of all residues at equilibrium with the imposed solution pH.
Dependence of the Glu286 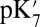 on the Cavity Size.
on the Cavity Size.
Calculations will first be described in the PR-like XDD–ROg state (defined in Table 1 and SI Appendix, Table S1), with a deprotonated PRDa3, which has a small cavity in both local (GSBP) and unconstrained [periodic boundary condition (PBC)] simulations. These structures remain close to the crystal structure 1M56 (42), and so the results can be more readily compared with previous calculations (17, 27). Regardless of the simulation technique used, ionization of Glu286 is very unfavorable at pH 7. The 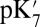 ranges from 18.5 with the QM/MM–TI technique to 10.2 using SCCE with a protein dielectric constant
ranges from 18.5 with the QM/MM–TI technique to 10.2 using SCCE with a protein dielectric constant 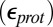 of 4, whereas MCCE calculates a value of 11.4. MCCE calculates a true pKa of 14.1. This value is substantially higher than the MCCE
of 4, whereas MCCE calculates a value of 11.4. MCCE calculates a true pKa of 14.1. This value is substantially higher than the MCCE 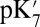 because pKa is calculated with all residues remaining in equilibrium with the pH, so the protein is much more negative overall. Using an
because pKa is calculated with all residues remaining in equilibrium with the pH, so the protein is much more negative overall. Using an 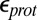 of 2, which has been recommended when multiple conformations from MD simulations are used (40), gives a
of 2, which has been recommended when multiple conformations from MD simulations are used (40), gives a 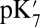 of 15–18 in MCCE or SCCE calculations, closer to that found with the microscopic QM/MM–TI technique.
of 15–18 in MCCE or SCCE calculations, closer to that found with the microscopic QM/MM–TI technique.
The 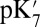 is also calculated imposing the same XDD–ROg charge in structures generated by unconstrained simulations in the ′F state, which result in large cavities. The calculated
is also calculated imposing the same XDD–ROg charge in structures generated by unconstrained simulations in the ′F state, which result in large cavities. The calculated 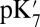 is lowered significantly with all methods (Table 1). The drop is 3.3 pH units (4.5 kcal/mol) using SCCE calculations with
is lowered significantly with all methods (Table 1). The drop is 3.3 pH units (4.5 kcal/mol) using SCCE calculations with 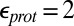 and ∼2 pH units (2.7 kcal/mol) with MCCE,
and ∼2 pH units (2.7 kcal/mol) with MCCE, 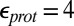 . The MCCE titration pKa drops by ∼3 pH units to 11.1. Thus, opening the cavity moves the free energy required to deprotonate the Glu to near the functional, experimental value (20). The high pKa indicates the Glu will be neutral at physiological pH.
. The MCCE titration pKa drops by ∼3 pH units to 11.1. Thus, opening the cavity moves the free energy required to deprotonate the Glu to near the functional, experimental value (20). The high pKa indicates the Glu will be neutral at physiological pH.
Microscopic, QM/MM–TI 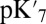 Calculations and the Effect of Cavity Hydration.
Calculations and the Effect of Cavity Hydration.
In local MD simulations of the crystal structure (GSBP–1M56), the cavity remains occupied with approximately five water molecules throughout the thermodynamic integration simulations, giving a high 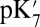 of 18.5. Glu286 becomes better solvated as it becomes increasingly negative (as the titration coordinate λ approaches 1), drawing in water molecules from both the cavity and top of the D channel (SI Appendix, Fig. S11). The TI
of 18.5. Glu286 becomes better solvated as it becomes increasingly negative (as the titration coordinate λ approaches 1), drawing in water molecules from both the cavity and top of the D channel (SI Appendix, Fig. S11). The TI 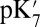 drops by 4.2 pH units to ∼14 in large-cavity structures, a shift that is similar to that found in the SCCE or MCCE continuum electrostatic calculations (Table 1). The importance of cavity water molecules is seen when nine extra water molecules (9w) are added to the small-cavity structures (1M56 + 9w). In the short (∼1–3 ns) local MD simulations, the water molecules cannot escape but relax as best they can in the small cavity. The QM/MM–TI
drops by 4.2 pH units to ∼14 in large-cavity structures, a shift that is similar to that found in the SCCE or MCCE continuum electrostatic calculations (Table 1). The importance of cavity water molecules is seen when nine extra water molecules (9w) are added to the small-cavity structures (1M56 + 9w). In the short (∼1–3 ns) local MD simulations, the water molecules cannot escape but relax as best they can in the small cavity. The QM/MM–TI 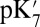 in the overly hydrated small cavity are now close to that found in the equilibrated, well-hydrated large cavity, showing the cavity decreases the
in the overly hydrated small cavity are now close to that found in the equilibrated, well-hydrated large cavity, showing the cavity decreases the 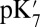 primarily by solvating the ionized Glu286. The
primarily by solvating the ionized Glu286. The 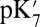 value of ∼14 is similar to that obtained by Chakrabarty and Warshel using a novel approach that adds more water molecules to the cavity as Glu286 titrates (17).
value of ∼14 is similar to that obtained by Chakrabarty and Warshel using a novel approach that adds more water molecules to the cavity as Glu286 titrates (17).
Dependence of the Glu286 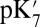 on the PRDa3 Ionization State.
on the PRDa3 Ionization State.
The Glu286 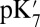 and pKa are calculated with different methods in structures with large and small cavities with the protonation state of PRDa3 (and the BNC) fixed. In all structures, removing the −1 charge from this acid, ∼10 Å from Glu286, reduces its
and pKa are calculated with different methods in structures with large and small cavities with the protonation state of PRDa3 (and the BNC) fixed. In all structures, removing the −1 charge from this acid, ∼10 Å from Glu286, reduces its 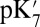 by 1.6–3.7 pH units, indicating the proton affinity has dropped by at least 2 kcal/mol. The nature of the shift is independent of the type of calculation or the size of the cavity near Glu286. The cost of deprotonating the Glu is thus seen to be affected independently and by a similar, significant amount by the opening of the cavity and by the protonation of PRDa3 (Table 1).
by 1.6–3.7 pH units, indicating the proton affinity has dropped by at least 2 kcal/mol. The nature of the shift is independent of the type of calculation or the size of the cavity near Glu286. The cost of deprotonating the Glu is thus seen to be affected independently and by a similar, significant amount by the opening of the cavity and by the protonation of PRDa3 (Table 1).
The results found here support a model where a large cavity will be found when PRDa3 is protonated and a small one when it is ionized. The Glu286 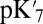 shifts by 3.7–7.6 pH units when the PRDa3 is protonated and the cavity expanded (Table 1), indicating the small change in CcO structure decreases the Glu proton affinity by at least 5 kcal/mol. The MCCE titration pKa shifts to 7.5 in the large-cavity PRDa3 neutral state, indicating that the Glu would be ∼half-ionized at equilibrium at pH 7 under these transiently existing conditions. Now the proton affinity of the BNC does not need to be very high to receive a proton from Glu286 (43).
shifts by 3.7–7.6 pH units when the PRDa3 is protonated and the cavity expanded (Table 1), indicating the small change in CcO structure decreases the Glu proton affinity by at least 5 kcal/mol. The MCCE titration pKa shifts to 7.5 in the large-cavity PRDa3 neutral state, indicating that the Glu would be ∼half-ionized at equilibrium at pH 7 under these transiently existing conditions. Now the proton affinity of the BNC does not need to be very high to receive a proton from Glu286 (43).
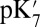 Values Calculated with Different Methods and Input Parameters.
Values Calculated with Different Methods and Input Parameters.
The different methods for calculating 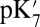 and pKa yield a consistent picture that the hydration and electrostatic properties of the hydrophobic cavity control the proton affinity of Glu286. The ground state structure increases the proton affinity by ∼3 kcal/mol due to the small cavity and by another ∼3 kcal/mol because PRDa3 is ionized. The Glu proton affinity decreases significantly when PRDa3 is protonated, and the cavity expands, as expected when the PLS is ready for pumping.
and pKa yield a consistent picture that the hydration and electrostatic properties of the hydrophobic cavity control the proton affinity of Glu286. The ground state structure increases the proton affinity by ∼3 kcal/mol due to the small cavity and by another ∼3 kcal/mol because PRDa3 is ionized. The Glu proton affinity decreases significantly when PRDa3 is protonated, and the cavity expands, as expected when the PLS is ready for pumping.
The absolute 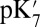 values predicted by different methods differ substantially. For example, given a small cavity and ionized PRDa3, the
values predicted by different methods differ substantially. For example, given a small cavity and ionized PRDa3, the 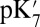 varies by 8.3 pH units, a 11.4 kcal/mol difference in the calculated proton affinity (Table 1). The
varies by 8.3 pH units, a 11.4 kcal/mol difference in the calculated proton affinity (Table 1). The 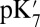 found with SCCE and MCCE methods depends on
found with SCCE and MCCE methods depends on 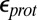 and
and 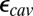 , the dielectric constants for the protein and cavity. As discussed in previous work (40, 44), a lower
, the dielectric constants for the protein and cavity. As discussed in previous work (40, 44), a lower 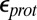 (e.g., 2) may be appropriate for
(e.g., 2) may be appropriate for 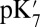 calculations when the protein structure is equilibrated with different protonation states for the titratable group in the linear response framework. The precise value for
calculations when the protein structure is equilibrated with different protonation states for the titratable group in the linear response framework. The precise value for 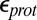 would depend on the degree of sampling (45, 46). In addition, water molecules in protein cavities may be more constrained than in bulk (47), so that
would depend on the degree of sampling (45, 46). In addition, water molecules in protein cavities may be more constrained than in bulk (47), so that 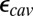 may be less than 80. Using PBC trajectories and the Kirkwood–Fröhlich formalism (48), the local dielectric constants of the hydrophobic cavity and D channel range from 4 to 9 in the chemical states studied here (SI Appendix, Table S3). Although one should be cautious about using such computed local dielectric constants in
may be less than 80. Using PBC trajectories and the Kirkwood–Fröhlich formalism (48), the local dielectric constants of the hydrophobic cavity and D channel range from 4 to 9 in the chemical states studied here (SI Appendix, Table S3). Although one should be cautious about using such computed local dielectric constants in 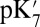 calculations (46), these values suggest the cavity may be surprisingly rigid even in the large, hydrated conformation. As detailed in Table 1 and SI Appendix, Tables S12–S13, as
calculations (46), these values suggest the cavity may be surprisingly rigid even in the large, hydrated conformation. As detailed in Table 1 and SI Appendix, Tables S12–S13, as 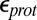 and
and 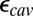 are varied, the Glu286
are varied, the Glu286 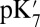 estimated by both SCCE and MCCE change significantly and by similar amounts. Lowering
estimated by both SCCE and MCCE change significantly and by similar amounts. Lowering 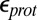 while maintaining a high (80)
while maintaining a high (80) 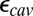 , the estimated
, the estimated 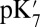 of Glu286 increases by a few pH units, and the impact of the cavity size and protonation of PRDa3 become closer to that predicted by QM/MM–TI. When
of Glu286 increases by a few pH units, and the impact of the cavity size and protonation of PRDa3 become closer to that predicted by QM/MM–TI. When 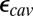 is reduced to 4–9, the impact of the cavity size on the Glu286
is reduced to 4–9, the impact of the cavity size on the Glu286 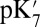 becomes smaller as expected, whereas the effect of PRDa3 protonation remains ∼3–4 pKa units. Thus, whereas it is difficult to establish the absolute
becomes smaller as expected, whereas the effect of PRDa3 protonation remains ∼3–4 pKa units. Thus, whereas it is difficult to establish the absolute 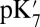 , SCCE/MCCE calculations with
, SCCE/MCCE calculations with 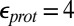 and
and 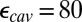 likely lead to the lower limit for this crucial value.
likely lead to the lower limit for this crucial value.
Implication for the CcO Proton-Pumping Mechanism.
Despite decades of experimental and theoretical analyses, it remains unclear how CcO couples the redox chemistry of O2 reduction to the transport of eight charges across the protein to add to the transmembrane proton gradient. Oxygen chemistry occurs in the R to P transition, with four electrons accumulated by CcO in previous intermediates now transferred to O2, without generating other reactive oxygen intermediates. The oxidized protein is then rereduced back to the R state through donation of four electrons from cytochrome c. Concomitant with each redox reaction, one proton is transferred to the pumping site, here assumed to be PRDa3, to be pumped into the P side of the membrane. Another proton is delivered to the BNC.
The mechanism that controls the branching competition between proton transfer to the pumping site or the BNC is not understood (43). It must reflect changes in the proton affinity of the donor and acceptor sites, particularly Glu286, the pumping site, and the BNC (19, 25), as well as control of the proton transfer pathways (18, 37). Using a kinetic network model for CcO, Hummer and coworkers (49, 50) analyzed trends in the proton transfer rate constants that would lead to efficient pumping. Their analysis supports proton transfer to the BNC being gated by protonation of the pumping site. This is consistent with the results found here where the protonation of PRDa3 increases the thermodynamic driving force for proton transfer to the BNC by reducing the Glu286 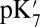 .
.
The proton affinity of the BNC and pumping site will be affected by the chemical states of the enzyme. For example, each CcO reduction step goes through a stage when heme a is reduced and BNC oxidized. Heme a reduction increases the pumping site 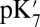 , favoring proton transfer there rather than to the BNC, whereas electron transfer from heme a to the BNC increases the BNC proton affinity, attracting the proton from Glu286. This may be important in some BNC reduction steps such as from ferric to ferrous heme which do not have a strong thermodynamic driving force for coupled proton uptake (43).
, favoring proton transfer there rather than to the BNC, whereas electron transfer from heme a to the BNC increases the BNC proton affinity, attracting the proton from Glu286. This may be important in some BNC reduction steps such as from ferric to ferrous heme which do not have a strong thermodynamic driving force for coupled proton uptake (43).
Despite very different approximations and limitations, our continuum electrostatic and microscopic calculations show that the proton affinity as monitored by the 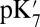 of Glu286 is unusually high when the presumed pumping site (PRDa3), 10 Å away, is deprotonated (e.g., in PR). The high pKa and
of Glu286 is unusually high when the presumed pumping site (PRDa3), 10 Å away, is deprotonated (e.g., in PR). The high pKa and 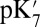 is due to the Glu being in a dehydrated, hydrophobic cavity. In contrast, when PRDa3 is protonated (as in
is due to the Glu being in a dehydrated, hydrophobic cavity. In contrast, when PRDa3 is protonated (as in 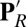 ,
, 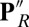 , and ′F states here), its hydrogen-bonding interactions with Arg481 and Trp172 weaken, leading to the displacement of the loop-bearing Trp172 and an expanded, more solvated cavity. Both continuum electrostatics and microscopic calculations indicate that these changes in hydration and local electrostatics lead to substantial depression of Glu286 pKa to the experimentally measured range of 9–10, which was estimated based on a specific kinetic model (20, 26). Thus, it is the proton affinity of Glu286, not the BNC, that is modulated by the loading of the pumping site. This is an attractive model (SI Appendix, Fig. S12) as it requires that proton transfer to the pumping site precede that to the BNC, a feature that would minimize the amount of “slippage,” where chemistry is done without pumping. The model also provides a microscopic framework for the kinetic gating phenomena identified in the kinetic network analysis (50) and discussed previously (51, 52). Although our calculations focus on states implicated in the PR to ′F transition, because the key driving force for the Glu286
, and ′F states here), its hydrogen-bonding interactions with Arg481 and Trp172 weaken, leading to the displacement of the loop-bearing Trp172 and an expanded, more solvated cavity. Both continuum electrostatics and microscopic calculations indicate that these changes in hydration and local electrostatics lead to substantial depression of Glu286 pKa to the experimentally measured range of 9–10, which was estimated based on a specific kinetic model (20, 26). Thus, it is the proton affinity of Glu286, not the BNC, that is modulated by the loading of the pumping site. This is an attractive model (SI Appendix, Fig. S12) as it requires that proton transfer to the pumping site precede that to the BNC, a feature that would minimize the amount of “slippage,” where chemistry is done without pumping. The model also provides a microscopic framework for the kinetic gating phenomena identified in the kinetic network analysis (50) and discussed previously (51, 52). Although our calculations focus on states implicated in the PR to ′F transition, because the key driving force for the Glu286 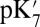 modulation is protonation of the presumed pumping site (PRDa3), it does not depend on the specific chemical state of the BNC. Thus, this mechanism for raising and lowering the Glu286
modulation is protonation of the presumed pumping site (PRDa3), it does not depend on the specific chemical state of the BNC. Thus, this mechanism for raising and lowering the Glu286 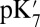 can be repeated each time CcO is reduced.
can be repeated each time CcO is reduced.
A role for changing hydration in determining proton or electron transfer activities has been considered as one general mechanism to modulate the proton affinity of buried charges (16, 44). A specific role of water penetration has been proposed to influence the Glu286 pKa (17). The current study is distinct in that it captures a specific local loop motion coupled to the protonation of a remote (10 Å from Glu286) group that triggers the change of cavity hydration level. This in turn modulates the proton affinity of Glu286, thus potentially establishing the molecular mechanism that controls the hydration level and proton affinity of this key residue.
Conclusion
In the course of the reaction cycle, Glu286 is presumed to donate a proton to the proton-loading site and the BNC. This requires modulation of the acid’s proton affinity twice. The work here presents a hypothesis that provides a solution to half the problem, given that PRDa3 is the PLS. The results of unrestrained MD simulations show that when the loading site is protonated, the 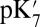 of Glu286 is significantly depressed, making it more favorable to transfer a proton to the BNC. The mechanism provides both a thermodynamic push for the proton transfer and controls the sequence of events so that the BNC will not receive its proton until the pumping site is loaded. The hypothesis provides a challenge to experiment, to detect a cavity of ∼155 Å3 that might be expected to live for the millisecond timescale associated with each pumping step (19). In addition, the importance of this loop suggests a region for mutations near Trp172 (53) that may have a significant impact on the pumping efficiency and/or the rate of proton transfer to the BNC.
of Glu286 is significantly depressed, making it more favorable to transfer a proton to the BNC. The mechanism provides both a thermodynamic push for the proton transfer and controls the sequence of events so that the BNC will not receive its proton until the pumping site is loaded. The hypothesis provides a challenge to experiment, to detect a cavity of ∼155 Å3 that might be expected to live for the millisecond timescale associated with each pumping step (19). In addition, the importance of this loop suggests a region for mutations near Trp172 (53) that may have a significant impact on the pumping efficiency and/or the rate of proton transfer to the BNC.
However, the mechanism for decreasing the proton affinity of Glu286 does not provide an answer to how the relative proton affinities are modified so that PRDa3 becomes protonated initially. Indeed, QM/MM calculations found that the direct proton transfer from Glu286 to a deprotonated PRDa3 is energetically very unfavorable in a PR-like state. The energetics for an alternative “concerted transfer” mechanism that involves the participation of an additional proton in the D channel (54) may be more consistent with kinetic data for the P → F transition. Previous empirical valence bond calculations also thoroughly discussed direct vs. concerted proton transfers (55) and emphasized the importance of including Glu286 flexibility (25, 26). We hope the impact of the surprising cavity opening reported here stimulates additional experiments and simulations to dissect robust elements that modulate the proton transfers.
Materials and Methods
The MD and QM/MM–TI simulations use the same protocol as in our previous studies (27, 56). The MCCE calculations use parameters and methods fully described in Refs. 23 and 57. SI Appendix provides more complete descriptions and additional simulation results.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health (NIH) Grant R01-GM084028. Computational resources from the National Center for Supercomputing Applications at the University of Illinois and the Center for High Throughput Computing at University of Wisconsin-Madison are greatly appreciated; computations are also supported in part by National Science Foundation through a major instrumentation grant (CHE-0840494). M.R.G. and J.L. are supported by the National Science Foundation Grant MCB 1022208, with infrastructure support from the National Center for Research Resources (2G12RR03060) and the National Institute on Minority Health and Health Disparities (8G12MD007603) from NIH.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313908110/-/DCSupplemental.
References
- 1.Ball P. Water as an active constituent in cell biology. Chem Rev. 2008;108(1):74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg D, Kauzmann W. The Structure and Properties of Water. Oxford, UK: Oxford University Press; 2005. [Google Scholar]
- 3.Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annu Rev Biophys Biomol Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 4.Fenimore PW, Frauenfelder H, McMahon BH, Parak FG. Slaving: Solvent fluctuations dominate protein dynamics and functions. Proc Natl Acad Sci USA. 2002;99(25):16047–16051. doi: 10.1073/pnas.212637899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitkup D, Ringe D, Petsko GA, Karplus M. Solvent mobility and the protein ‘glass’ transition. Nat Struct Biol. 2000;7(1):34–38. doi: 10.1038/71231. [DOI] [PubMed] [Google Scholar]
- 6.Warshel A, Levitt M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976;103(2):227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 7.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solution. New York: Wiley; 1991. [Google Scholar]
- 8.Cui Q, Karplus M. Catalysis and specificity in enzymes: A study of triosephosphate isomerase and comparison with methyl glyoxal synthase. Adv Protein Chem. 2003;66:315–372. doi: 10.1016/s0065-3233(03)66008-0. [DOI] [PubMed] [Google Scholar]
- 9.Silverman DN. Marcus rate theory applied to enzymatic proton transfer. Biochim Biophys Acta. 2000;1458(1):88–103. doi: 10.1016/s0005-2728(00)00061-x. [DOI] [PubMed] [Google Scholar]
- 10.Riccardi D, Yang S, Cui Q. Proton transfer function of carbonic anhydrase: Insights from QM/MM simulations. Biochim Biophys Acta. 2010;1804(2):342–351. doi: 10.1016/j.bbapap.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sham YY, Muegge I, Warshel A. Simulating proton translocations in proteins: Probing proton transfer pathways in the Rhodobacter sphaeroides reaction center. Proteins. 1999;36(4):484–500. [PubMed] [Google Scholar]
- 12.Hosler JP, Ferguson-Miller S, Mills DA. Energy transduction: Proton transfer through the respiratory complexes. Annu Rev Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swanson JMJ, et al. Proton solvation and transport in aqueous and biomolecular systems: Insights from computer simulations. J Phys Chem B. 2007;111(17):4300–4314. doi: 10.1021/jp070104x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garczarek F, Gerwert K. Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature. 2006;439(7072):109–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- 15.Zhu FQ, Hummer G. Pore opening and closing of a pentameric ligand-gated ion channel. Proc Natl Acad Sci USA. 2010;107(46):19814–19819. doi: 10.1073/pnas.1009313107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochim Biophys Acta. 2006;1764(11):1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabarty S, Warshel A. Capturing the energetics of water insertion in biological systems: The water flooding approach. Proteins. 2013;81(1):93–106. doi: 10.1002/prot.24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brzezinski P, Gennis RB. Cytochrome c oxidase: Exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40(5):521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaila VR, Verkhovsky MI, Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110(12):7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 20.Namslauer A, Aagaard A, Katsonouri A, Brzezinski P. Intramolecular proton-transfer reactions in a membrane-bound proton pump: The effect of pH on the peroxy to ferryl transition in cytochrome c oxidase. Biochemistry. 2003;42(6):1488–1498. doi: 10.1021/bi026524o. [DOI] [PubMed] [Google Scholar]
- 21.Popović DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome C oxidase. J Am Chem Soc. 2004;126(6):1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 22.Popovic DM, Stuchebrukhov AA. Two conformational states of Glu242 and pKas in bovine cytochrome c oxidase. Photochem Photobiol Sci. 2006;5(6):611–620. doi: 10.1039/b600096g. [DOI] [PubMed] [Google Scholar]
- 23.Song Y, Michonova-Alexova E, Gunner MR. Calculated proton uptake on anaerobic reduction of cytochrome C oxidase: Is the reaction electroneutral? Biochemistry. 2006;45(26):7959–7975. doi: 10.1021/bi052183d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fadda E, Yu C-H, Pomès R. Electrostatic control of proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2008;1777(3):277–284. doi: 10.1016/j.bbabio.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Pisliakov AV, Sharma PK, Chu ZT, Haranczyk M, Warshel A. Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105(22):7726–7731. doi: 10.1073/pnas.0800580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakrabarty S, Namslauer I, Brzezinski P, Warshel A. Exploration of the cytochrome c oxidase pathway puzzle and examination of the origin of elusive mutational effects. Biochim Biophys Acta. 2011;1807(4):413–426. doi: 10.1016/j.bbabio.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh N, Prat-Resina X, Gunner MR, Cui Q. Microscopic pKa analysis of Glu286 in cytochrome c oxidase (Rhodobacter sphaeroides): Toward a calibrated molecular model. Biochemistry. 2009;48(11):2468–2485. doi: 10.1021/bi8021284. [DOI] [PubMed] [Google Scholar]
- 28.Lee HJ, et al. Intricate role of water in proton transport through cytochrome c oxidase. J Am Chem Soc. 2010;132(45):16225–16239. doi: 10.1021/ja107244g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita T, Voth GA. Insights into the mechanism of proton transport in cytochrome c oxidase. J Am Chem Soc. 2012;134(2):1147–1152. doi: 10.1021/ja209176e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews BW, Liu LJ. A review about nothing: Are apolar cavities in proteins really empty? Protein Sci. 2009;18(3):494–502. doi: 10.1002/pro.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin H, Feng G, Clore GM, Hummer G, Rasaiah JC. Water in the polar and nonpolar cavities of the protein interleukin-1β. J Phys Chem B. 2010;114(49):16290–16297. doi: 10.1021/jp108731r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seibold SA, Mills DA, Ferguson-Miller S, Cukier RI. Water chain formation and possible proton pumping routes in Rhodobacter sphaeroides cytochrome c oxidase: A molecular dynamics comparison of the wild type and R481K mutant. Biochemistry. 2005;44(31):10475–10485. doi: 10.1021/bi0502902. [DOI] [PubMed] [Google Scholar]
- 33.Tashiro M, Stuchebrukhov AA. Thermodynamic properties of internal water molecules in the hydrophobic cavity around the catalytic center of cytochrome c oxidase. J Phys Chem B. 2005;109(2):1015–1022. doi: 10.1021/jp0462456. [DOI] [PubMed] [Google Scholar]
- 34.Kaila VRI, Verkhovsky MI, Hummer G, Wikström M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105(17):6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansson MP, Kaila VRI, Laakkonen L. Charge parameterization of the metal centers in cytochrome c oxidase. J Comput Chem. 2008;29(5):753–767. doi: 10.1002/jcc.20835. [DOI] [PubMed] [Google Scholar]
- 36.Quenneville J, Popović DM, Stuchebrukhov AA. Combined DFT and electrostatics study of the proton pumping mechanism in cytochrome c oxidase. Biochim Biophys Acta. 2006;1757(8):1035–1046. doi: 10.1016/j.bbabio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson-Miller S, Hiser C, Liu J. Gating and regulation of the cytochrome c oxidase proton pump. Biochim Biophys Acta. 2012;1817(4):489–494. doi: 10.1016/j.bbabio.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riccardi D, Schaefer P, Cui Q. pKa calculations in solution and proteins with QM/MM free energy perturbation simulations: A quantitative test of QM/MM protocols. J Phys Chem B. 2005;109(37):17715–17733. doi: 10.1021/jp0517192. [DOI] [PubMed] [Google Scholar]
- 39.Antosiewicz J, McCammon JA, Gilson MK. Prediction of pH-dependent properties of proteins. J Mol Biol. 1994;238(3):415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 40.Archontis G, Simonson T. Proton binding to proteins: a free-energy component analysis using a dielectric continuum model. Biophys J. 2005;88(6):3888–3904. doi: 10.1529/biophysj.104.055996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song YF, Mao JJ, Gunner MR. MCCE2: Improving protein pKa calculations with extensive side chain rotamer sampling. J Comput Chem. 2009;30(14):2231–2247. doi: 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svensson-Ek M, et al. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321(2):329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 43.Gunner MR, Amin M, Zhu X, Lu J. Molecular mechanisms for generating transmembrane proton gradients. Biochim Biophys Acta. 2013;1827(8-9):892–913. doi: 10.1016/j.bbabio.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell ST, Warshel A. Calculations of electrostatic energies in proteins. The energetics of ionized groups in bovine pancreatic trypsin inhibitor. J Mol Biol. 1985;185(2):389–404. doi: 10.1016/0022-2836(85)90411-5. [DOI] [PubMed] [Google Scholar]
- 45.King G, Lee FS, Warshel A. Microscopic simulations of macroscopic dielectric constants of solvated proteins. J Chem Phys. 1991;95(6):4366–4377. [Google Scholar]
- 46.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44(4):400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 47.Rasaiah JC, Garde S, Hummer G. Water in nonpolar confinement: From nanotubes to proteins and beyond. Annu Rev Phys Chem. 2008;59:713–740. doi: 10.1146/annurev.physchem.59.032607.093815. [DOI] [PubMed] [Google Scholar]
- 48.Simonson T, Perahia D. Internal and interfacial dielectric properties of cytochrome c from molecular dynamics in aqueous solution. Proc Natl Acad Sci USA. 1995;92(4):1082–1086. doi: 10.1073/pnas.92.4.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim YC, Wikström M, Hummer G. Kinetic models of redox-coupled proton pumping. Proc Natl Acad Sci USA. 2007;104(7):2169–2174. doi: 10.1073/pnas.0611114104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim YC, Hummer G. Proton-pumping mechanism of cytochrome c oxidase: A kinetic master-equation approach. Biochim Biophys Acta. 2012;1817(4):526–536. doi: 10.1016/j.bbabio.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Popović DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. FEBS Lett. 2004;566(1-3):126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 52.Cui Q. Theoretical and computational studies of vectorial processes in biological systems. Theor Chem Acc. 2006;116(1):51–59. [Google Scholar]
- 53.Ribacka C, et al. An elementary reaction step of the proton pump is revealed by mutation of Trptophan-164 to Phenylalanine in cytochrome c oxidase from Paracoccus denitrificans. Biochem. 2005;44:16502–16512. doi: 10.1021/bi0511336. [DOI] [PubMed] [Google Scholar]
- 54.Siegbahn PEM, Blomberg MRA. Energy diagrams and mechanism for proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2007;1767(9):1143–1156. doi: 10.1016/j.bbabio.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 55.Olsson MHM, Warshel A. Monte Carlo simulations of proton pumps: On the working principles of the biological valve that controls proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103(17):6500–6505. doi: 10.1073/pnas.0510860103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang S, Cui Q. Glu-286 rotation and water wire reorientation are unlikely the gating elements for proton pumping in cytochrome C oxidase. Biophys J. 2011;101(1):61–69. doi: 10.1016/j.bpj.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang J, Gunner MR. Multiconformation continuum electrostatics analysis of the effects of a buried Asp introduced near heme a in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2010;49(37):8043–8052. doi: 10.1021/bi100663u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.