

Significance
Beta-amyloid plaque accumulation, glucose hypometabolism, and neuronal atrophy are hallmarks of Alzheimer’s disease. However, the regional ordering of these biomarkers prior to dementia remains untested. In a cohort with Alzheimer’s disease mutations, we performed an integrated whole-brain analysis of three major imaging techniques: amyloid PET, [18F]fluro-deoxyglucose PET, and structural MRI. We found that most gray-matter structures with amyloid plaques later have hypometabolism followed by atrophy. Critically, however, not all regions lose metabolic function, and not all regions atrophy, even when there is significant amyloid deposition. These regional disparities have important implications for clinical trials of disease-modifying therapies.
Keywords: neuroimaging, aging, dementia, neurodegeneration, DIAN
Abstract
Major imaging biomarkers of Alzheimer’s disease include amyloid deposition [imaged with [11C]Pittsburgh compound B (PiB) PET], altered glucose metabolism (imaged with [18F]fluro-deoxyglucose PET), and structural atrophy (imaged by MRI). Recently we published the initial subset of imaging findings for specific regions in a cohort of individuals with autosomal dominant Alzheimer’s disease. We now extend this work to include a larger cohort, whole-brain analyses integrating all three imaging modalities, and longitudinal data to examine regional differences in imaging biomarker dynamics. The anatomical distribution of imaging biomarkers is described in relation to estimated years from symptom onset. Autosomal dominant Alzheimer’s disease mutation carrier individuals have elevated PiB levels in nearly every cortical region 15 y before the estimated age of onset. Reduced cortical glucose metabolism and cortical thinning in the medial and lateral parietal lobe appeared 10 and 5 y, respectively, before estimated age of onset. Importantly, however, a divergent pattern was observed subcortically. All subcortical gray-matter regions exhibited elevated PiB uptake, but despite this, only the hippocampus showed reduced glucose metabolism. Similarly, atrophy was not observed in the caudate and pallidum despite marked amyloid accumulation. Finally, before hypometabolism, a hypermetabolic phase was identified for some cortical regions, including the precuneus and posterior cingulate. Additional analyses of individuals in which longitudinal data were available suggested that an accelerated appearance of volumetric declines approximately coincides with the onset of the symptomatic phase of the disease.
The pathological mechanisms underlying nondominantly inherited late onset Alzheimer’s disease (LOAD) remain an active area of investigation (1). According to the amyloid cascade hypothesis, the precipitating event in LOAD is an alteration of the balance between production and clearance of the metabolites of amyloid precursor protein (APP) (2). Abnormalities in APP metabolism then lead to β-amyloid (Aβ) deposition in the cerebral cortex, the formation of neurofibrillary tangles (NFTs) containing hyperphosphorylated tau protein, neuronal dysfunction, cell loss, and, ultimately, dementia. In vivo biomarkers of LOAD include cerebrospinal fluid (CSF) Aβ42, CSF tau, amyloid deposition imaged with Pittsburgh compound B PET (PiB PET) and other amyloid tracers, altered glucose metabolism imaged with fluro-deoxyglucose PET (FDG PET), and structural atrophy assessed by volumetric MRI. A theoretical model of biomarker changes has been proposed by Jack et al. (3) that links these biomarkers in relation to the amyloid cascade hypothesis. This model has two main predictions. First, each biomarker should exhibit a sigmoidal trajectory characterized by an initial period of little change, then rapid progression and finally a plateau. Second, amyloid markers (CSF Aβ42 and PiB PET) are predicted to become abnormal first, followed by neurodegenerative markers such as elevated levels of CSF tau and decreased FDG PET, and finally concurrently brain atrophy (volumetric MRI). Cognitive symptoms become evident relatively late in the disease course. Based on this model, the National Institutes of Health (National Institute on Aging) and the Alzheimer’s Association have developed guidelines for incorporation of biomarkers into practice and research to support the clinical diagnosis of symptomatic Alzheimer’s disease (AD) (defined here as mild cognitive impairment due to AD and AD dementia).
There has been relatively little in vivo work examining the temporal order of multiple AD biomarkers within a single cohort (4–6). Accurate determination of the temporal order of biomarker changes in LOAD requires long-term longitudinal studies with a large cohort of individuals. In the absence of such data, it is impossible to predict whether an individual will develop symptomatic AD, let alone where they are on a theoretical biomarker timeline. In contrast, autosomal dominant AD (ADAD) caused by mutations in the APP (7), presenilin 1 (PSEN1) (8), and presenilin 2 (PSEN2) (9) genes has virtually complete penetrance with a predictable age of onset (4). Within early-onset ADAD families, the parental age of onset tends to be conserved between family members (5, 6, 10). The preserved age of onset within families is one of the key advantages of studying ADAD, because it allows approximation of how far from symptom onset an individual is at the time of study.
The Dominantly Inherited Alzheimer Network (DIAN) has established a large cohort of individuals from families with ADAD mutations (11). In an initial report from DIAN, Bateman et al. (12) took advantage of the correlation between the parental and actual age of onset within the cohort to estimate the timing and order of all of the major AD biomarkers, including specific examination of PiB and FDG in the precuneus and hippocampal volume in the first 128 participants. Overall, the results are largely consistent with the model proposed by Jack et al. (3) but were limited to testing specific regions in a smaller cohort. Now we greatly expand upon the Bateman et al. (12) report in two distinct ways. First, we examine biomarker trajectories across the entire brain and compare our findings to proposed models of AD. Second, we analyze a subset of participants with longitudinal imaging to gain insight into intraindividual biomarker changes. This study directly integrates the spatial dynamics of all three major imaging biomarkers. To do this, we used the known age of onset within each DIAN family group to estimate where and when pathological changes in amyloid accumulation, glucose metabolism, and atrophy occur across the whole brain.
Results
Biomarker Changes in the Cerebral Cortex.
A sample of 229 participants with MRI [noncarriers (NC) = 92, mutation carriers (MC) = 137, from the fourth DIAN Data Freeze] was analyzed (Table 1), with a subset having PiB (NC = 86, MC = 121) and FDG (NC = 84, MC = 116) imaging. For each vertex on the cortical surface, MCs and NCs were tested for differences in PiB, FDG, and cortical thickness at 5-y estimated years from symptom onset (EYO) intervals from −25 to +10 y (Methods). Differences that were P < 0.01 after correction for multiple comparisons across the brain using a false discovery rate (FDR) (13) of 0.05 were considered statistically significant. The difference in PiB uptake between MCs and NCs reached statistical significance at least 15 y before the estimated age of onset (EAO) (Fig. 1A). At EAO = −15, amyloid deposition was widespread in MCs throughout the cerebral cortex, with relative initial sparing of entorhinal, precentral, and postcentral cortices that reached statistical significance at approximately EYO = −10 y. Movie S1 shows PiB accumulation from EYO = −25 to EYO = 10.
Table 1.
Demographics for DIAN participants who completed MRI assessments
| Demographic | NC | MC | P value |
| n | 92 | 137 | — |
| Age (SD), y | 39.3 (9.46) | 38.8 (10.4) | 0.718 |
| EYO (SD), y | −7.49 (11.8) | −7.85 (9.91) | 0.807 |
| Education (SD), y | 14.6 (2.61) | 14.2 (2.6) | 0.215 |
| Male (%) | 38 (41.3) | 56 (40.9) | 0.949 |
| CDR >0 (%) | 5 (5.43) | 54 (39.4) | 3.28E-09 |
| APOE4+ | 23 (25) | 35 (25.5) | 0.926 |
| Mutation type (%) | — | PS1 = 105 (76.6), PS2 = 11 (8.03), APP = 21 (15.3) | — |
Fig. 1.

Statistical significance (P value) maps on medial and lateral left cortical gray surface showing differences between carriers and noncarriers in PiB (A), FDG (B), and cortical thickness (C) at −15, −10, −5, and 0 y before predicted symptom onset. Regions with significant (P < 0.01 after correction for multiple comparisons) increases are shown in red/yellow and decreases in blue/cyan. The medial wall was not analyzed. Significant increases in amyloid PiB PET were detected at least 15 y before the estimated age of symptom onset in across the cerebral cortex. Decreased glucose metabolism was detected 10 y before predicted symptom onset using FDG PET, primarly involving the precuenus, posterior cingulate, and lateral parietal lobes. Cortical thinning on MRI was detected ∼5 y before the estimated onset of symptoms, initially in the precuneus and posterior cingulate as well as portions of the occipital lobe and anterior temporal lobe. Results were consistent for both hemispheres for all modalities.
Using FDG PET, differences in regional hypometabolism between carriers and noncarriers were significant (FDR corrected, P < 0.01) 10 y before the onset of symptoms in the precuneus/posterior cingulate and lateral parietal cortex (Fig. 1B). By EYO = 5, hypometabolism was significant in the middle temporal gyrus and the lateral prefrontal cortex as well. Similar effects appeared in the occipital and medial prefrontal cortices concurrently with the onset of symptoms (i.e., at EYO = 0). Unexpectedly, we found that MCs ∼25 y from their EAO had statistically significantly higher levels of glucose uptake than NCs. Regional hypermetabolism was found in many of the same regions (e.g., lateral parietal cortex precuneus/posterior cingulate) that were hypometabolic later on in the disease (Fig. S1 and Movie S2). This finding should be interpreted with caution, however, because only 11 subjects with EYO values less than or equal to −25 were analyzed. Furthermore, our analysis assumes both that the brainstem is a stable reference region and that the relationship between EYO and glucose metabolism is approximately linear.
Significant (FDR corrected, P < 0.01) cerebral cortical atrophy was not detected until ∼5 y before EAO (Fig. 1C). Although most extensive in the precuneus, decreased cortical thickness was also found in entorhinal, lateral temporal, and lateral parietal cortices. We also found that at EYO = −5, MCs have greater cortical thickness than NCs in the anterior cingulate cortex (ACC) (Fig. 1C). Unlike the hypermetabolism found at EYO = −25, these increases in thickness were not transient (Movie S3).
Biomarker Dissociations in Subcortical Gray Matter.
A similar analysis of each biomarker was also performed within subcortical regions of interest (ROIs). Owing to the smaller number of subcortical regions, an FDR corrected P value of 0.05 was considered significant for subcortical regional analysis. MCs showed statistically significant PiB binding in every subcortical region of interest 15 y before the EAO (Fig. 2A). However, in contrast to cerebral cortex, subcortical gray matter generally did not exhibit decreased glucose metabolism. Glucose hypometabolism was observed only in the hippocampus, and then not until EYO = 0 (Fig. 2B). However, MCs showed volume loss in multiple subcortical structures well before symptom onset. Atrophy of accumbens, amygdala, hippocampus, putamen, and thalamus were all statistically significant compared with NCs by EYO = −10 (Fig. 2C). However, caudate and pallidum did not exhibit significant volume loss, despite marked PiB uptake.
Fig. 2.

Comparison of [11C]Pittsburgh compound B (PiB) (A), FDG (B), and volume (C) between carriers and noncarriers in subcortical gray matter. Significant increases (FDR corrected P < 0.05) are indicated in red/orange and decreases in blue/cyan. Elevated PiB uptake was detected by EYO = −15 in all of the subcortical gray matter structures. Excluding the pallidum and caudate, all subcortical structures exhibited volumetric decline ten years before the expected age of onset. Metabolic decreases were only seen in the hippocampus, and only after the estimated onset of symptoms.
Regional Variability in Cross-Sectional Biomarkers.
The results presented so far concern the cross-sectional sequence of biomarker changes. We now address regional differences in each biomarker with respect to EYO. To estimate such differences, a linear mixed model compared each regional cross-sectional slope against every other region. Matrices showing FDR corrected P values (thresholded at P < 0.01) for each regional comparison are presented in Figs. S2–S4. For visualization purposes, first-degree locally weighted regression method (LOESS) curves were fit to select cortical and subcortical regions (Fig. 3).
Fig. 3.
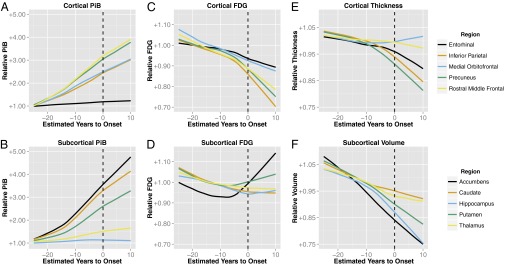
Biomarker trajectories for PiB (A and B), FDG (C and D), cortical thickness (E), and subcortical volume (F) estimated with cross-sectional first-degree LOESS curves in mutation carriers. To account for baseline differences between regions, carrier data were first divided by the mean of noncarriers for that region. Trajectories for five cortical gray matter regions (entorhinal, inferior parietal, medial orbitofrontal, precuneus, and rostral middle frontal) are shown in the top row (A, C, and E). The bottom row (B, D, and F) shows curves for five subcortical gray matter regions (accumbens, caudate, hippocampus, putamen, and thalamus).
Within ADAD MCs, we observed considerable regional variability in the cross-sectionally estimated slopes of amyloid accumulation (Fig. 3A and Fig. S2). Across all ROIs, the largest PiB slopes were observed in nucleus accumbens, caudate, rostral middle frontal gyrus, and precuneus. The greatest glucose hypometabolism was found in the inferior parietal and supramarginal cortices (Fig. 3C and Fig. S3). Atrophy also was spatially heterogeneous. The greatest cortical thinning was observed in the precuneus (Fig. 3E and Fig. S4); volume loss in subcortical gray matter was significantly greater in both the accumbens and hippocampus in comparison with the thalamus (P < 0.01).
Initial Estimates of Longitudinal Change.
Longitudinal imaging was examined on a subset (MRI = 51, PiB = 31, FDG = 40) of the sample with the goal of examining intraindividual biomarker changes (Table S1). For longitudinal analysis participants were placed into one of three groups: NCs, asymptomatic MCs [clinical dementia rating (CDR) 0, indicating cognitive normality], and symptomatic MCs (CDR > 0, indicating cognitive impairment).
Visually, the cross-sectional trajectories seemed to underestimate individual changes, particularly after the age of onset (Fig. 4 B–D). Quantitatively, precuneus PiB binding was not statistically significant between noncarriers, asymptomatic carriers (CDR < 0), and symptomatic carriers (CDR > 0), probably because of limited statistical power (Table S2). However, we did find that declines in glucose metabolism in the precuneus, precuneus thickness, and hippocampal volume were significantly greater in symptomatic carriers than in noncarriers (P < 0.05, uncorrected). Using an alpha value of 0.05, no statistically significant differences were found between asymptomatic and symptomatic carriers. However, declines in hippocampal volume and precuneus thickness were statistically greater in symptomatic carriers than in asymptomatic carriers using a more liberal threshold of threshold of P < 0.10. This result is consistent with the acceleration of changes observed in the qualitative longitudinal analysis.
Fig. 4.

Longitudinal data for PiB (A), FDG (B), cortical thickness (C), and hippocampal volume (D). Asymptomatic (CDR 0) participants are shown in blue. Symptomatic participants (CDR 0.5 or higher) are shown in red. Overlaid on each plot is the LOESS estimated cross-sectional trajectory for carriers with 95% confidence intervals of the mean in gray. Note the generally steeper longitudinal vs. cross-sectional slope in symptomatic participants. The x axis is unlabeled and only carriers are shown to protect the mutation status of each participant.
Discussion
Consistent with the amyloid hypothesis predicted for LOAD, ADAD MCs exhibited increased amyloid deposition, followed by decreased glucose metabolism, and finally atrophy, across many cortical regions. Furthermore, as predicted by Jack et al. (3), we found clear regional effects in biomarker trajectories. For example, hypometabolism was significant first in the posterior cingulate/precuneus and lateral parietal regions before progressing to include the frontal and temporal cortices. This result suggests that there may be a greater temporal lag between amyloid accumulation and metabolic decline in frontal/temporal regions than in parietal regions. It is interesting to note that we found very few regional differences in the timing of amyloid accumulation in this cohort, although total amyloid burden did vary by region. This is consistent with a recent cross-sectional report in another large ADAD cohort (14).
One of the surprising results of our previous work was the finding that volumetric and metabolic changes appeared at approximately the same time in ADAD mutation carriers (12). This, along with other work, prompted a recent revision to the predominant AD biomarker model to propose that FDG PET and volumetric MRI changes appear concurrently in AD (15). Critically, our previous study failed to analyze metabolic and volumetric changes within the same region, as was done here. When each biomarker was examined within the same region, the majority of regions followed the predicted path (amyloid accumulation → hypometabolism → atrophy).
Regional exceptions to the general model (amyloid accumulation → hypometabolism → atrophy) were noted, however. Specifically, in the hippocampus, amyloid accumulation (detected at EYO = −15) was followed first by volume loss (EYO = −10) and then hypometabolism (EYO = 0) (Fig. 2). We also identified regions that seem to skip one or more of the biomarker stages. For instance, the palladium and caudate did not show either metabolic or volumetric declines, despite markedly elevated PiB uptake (Fig. 2). Similarly, we did not detect decreases in the thickness of frontal cortical regions, even though MCs had significant levels of amyloid accumulation and hypometabolism in these regions compared with NCs (Fig. 1). These results indicate that the standard biomarker ordering does not hold in all brain regions. Why some regions with high PiB loads are resistant to metabolic and volumetric decline is unclear. One possibility is that the PiB signal in these regions does not represent plaques with typical neurotoxic effects. For example, neuropathological examinations of the globus pallidus in ADAD cases have reported findings consistent with amyloid angiopathy (16, 17). Correlations between pathology and imaging are needed to resolve these discrepancies.
The regional variability of biomarker trajectories found in this report is consistent with previous neuroimaging results that have identified distinct spatial patterns of amyloid accumulation, hypometabolism, and atrophy (18), where biomarker effects vary across brain regions (19). As previously reported (18), the most commonly affected areas across modalities bear a striking resemblance to the default mode network (20) Furthermore, studies in LOAD have found spreading patterns of atrophy (21, 22), hypometabolism (23), and amyloid deposition (24) as a correlate of increasing disease severity. Our results are also consistent with the neuropathological studies that have found differential temporal ordering of the formation of amyloid plaques and neurofibrillary tangles (25, 26).
Several of our results were unexpected. Our finding of hypermetabolism on FDG PET ∼25 y before symptom onset does not have, to our knowledge, a direct precedent in the neuroimaging literature. Previous studies in presymptomatic AD typically have reported decreased glucose metabolism (27). However, there have been reports in normal aging (28) and mild cognitive impairment (29) of an association between early PiB uptake and hypermetabolism, albeit of variable focality, as determined by FDG PET. This association has been attributed to compensation for AD pathology (29). Alternatively, this association has been understood as reflecting the fact that metabolism and Aβ deposition both are driven by neuronal activity (30). Our data are more consistent with the latter hypothesis, because the hypermetabolic phase occurred before amyloid deposition or atrophy.
Without further replication, however, this result should be considered provisional. The small number of participants with FDG imaging and EYO values less or equal to −25 (n = 11) prevent drawing strong conclusions. Future study of even younger individuals with ADAD mutations clearly is needed to determine whether or not this abnormality truly precedes fibrillar Aβ deposition (31). Furthermore, our decision to use a linear function to model metabolic change may have also affected our results. Given the evident scatter in the data, we chose to use linear models in all our analyses to limit the number of free parameters, despite recent evidence that biomarker trajectories are nonlinear with respect to EYO (32–37). Artificial imposition of linearity could have created the appearance of increased glucose metabolism 25 y before EYO. If true, this result would be eliminated by appropriate flattening of the initial portion of the curve, in accordance with extant biomarker models. It is theoretically possible to estimate voxelwise nonlinear biomarker trajectories. However, this would require imposing some form of spatial regularization and the rules by which this should be done are presently unclear. Finally it is possible that changes in the metabolism of our reference region (the brainstem) may be responsible for the apparent hypermetabolic phase in MCs. To adequately address this possibility would require fully quantitative metabolic data, which would necessitate arterial blood sampling and estimation of the arterial input function (AIF). This, however, is an invasive procedure that would be difficult to implement in a multisite study such as DIAN. We are currently investigating noninvasive, MRI-based methods of obtaining the AIF and quantitative PET data (38).
We also cannot currently explain the anomalously high cortical thickness estimates in the anterior cingulate at EYO = 0 (Fig. 1C). Previous studies have reported alterations in image contrast properties in both aging (39) and AD (40). Decreases in white matter contrast could create the appearance of an increase in cortical thickness. However, we found had no detectable differences between MCs and NCs in gray, white, or gray/white contrast in the anterior cingulate. We also visually examined the structural images and the FreeSurfer segmentations and did not observe a consistent artifact or segmentation error that could explain this finding. It should be noted that previous studies in aging have reported a positive correlation between age and cortical thickness in the anterior cingulate cortex (41–43). Similarly, studies using voxel-based morphometry (44) and manual segmentation (45) have found a preservation of the anterior cingulate in normal aging. One intriguing possibility for these findings is that the ACC is involved in a compensatory response to AD pathology. In support of this hypothesis, recent studies using resting-state functional MRI have reported an increase in functional connectivity between the ACC and other nodes of the salience network (46, 47). Whether these findings truly represent a compensatory response or are instead reflective of the instinct resistance to AD pathology of the cell types within the ACC is a matter for future work.
Our study has several limitations. First, the majority of our data are cross-sectional; inferences depend on the assumption that biomarker trajectories are similar across participants. We partially compensated for this limitation by including the available longitudinal data. Additional longitudinal studies are needed to fully describe intraindividual biomarker change. Second, our analysis assumes that the parental age of onset is an accurate estimate of symptomatic onset in the affected offspring. We observed a significant correlation between the actual and parental age of onset in the symptomatic participants but there were several outliers. A predictable effect of uncertainty in EYO would be blunting of features in biomarker trajectory curves, as suggested by the results shown in Fig. 4. However, the temporal ordering of biomarker differences is less likely to be affected by inaccuracies in EYO, because all of the participants used in this study were drawn from the same cohort. Our results may also be influenced by the inclusion of multiple mutation subtypes. This design has the possible advantage of increasing applicability to ADAD as a whole, but it may well also increase the heterogeneity of our sample. Additional research is needed to assess how biomarker trajectories differ between mutation subtypes.
The spatial resolution of PET imaging (∼8 mm isotropic in this study) is an inherent limitation in any study attempting to characterize regional PiB and FDG changes in AD. One of the well-known effects of limited spatial resolution is spillover of the PET signal from nearby tissue types. We chose to use a regional-spread function (RSF)-based approach to partial volume correction (PVC) to minimize such effects. Although this method has been reported to be accurate (48), it is biased from misregistrations between the PET and MRI data (49) and by inaccurate segmentation of the MRI data (50). Although we did not observe any registration or segmentation errors by visual inspections, it is possible that some still occurred. Regardless of inaccuracies in registration and segmentation, PVC also propagates the noise inherent in the PET image (48). Therefore, the dissociations we report between amyloid deposition, metabolic dysfunction, and atrophy may be influenced by a PVC-induced decrease in signal-to-noise ratio. To address this possibility, we have included uncorrected versions of Figs. 2–4 in Supporting Information (Figs. S6–S8). The results were largely the same with and without PVC. The only major difference between two analyses was the presence of significant metabolic declines in the thalamus and caudate without PVC. It is unclear whether these declines are truly biological or are artifacts introduced by partial volume effects.
In contrast to prior publications (51, 52), we did not detect caudate atrophy in our ADAD cohort. This discrepancy is possibly attributable to differences in segmentation technique. However, FreeSurfer’s segmentation of the caudate has been shown to have a high correspondence with manual tracing (53), and studies in ADAD cohorts have found caudate atrophy using FreeSurfer (51). An alternative possibility is that caudate atrophy is not a consistent feature of ADAD, but rather is specific to mutation subtype.
Finally it is unclear to what extent ADAD and LOAD are pathologically similar entities. PiB binding in the striatum in ADAD is notably greater than that typically seen in LOAD (54). Similarly, our finding of increased PiB in occipital cortex is not typical of LOAD (17). However, the neuroimaging literature suggests that otherwise ADAD and LOAD are substantially similar. ADAD and sAD are both associated with elevated cortical PiB uptake, especially in the in the precuneus/posterior cingulate and prefrontal cortex (54–56). Reductions in glucose metabolism in the temporal and parietal lobes have also been observed in both diseases (57–60). Finally, both LOAD and ADAD have been shown to result in cortical (51, 61–65) and hippocampal (63, 65–68) atrophy. Further comparisons of LOAD and ADAD are ongoing as part of the DIAN study.
Our results still have important implications. Biomarker imaging is increasingly important in clinical trials of potential AD therapeutic agents. These trials will most likely be using imaging and methodical methods similar to those we have used in this study. Published trials of antiamyloid treatments have already used global measures of amyloid deposition as a primary outcome measure (69). Whereas global measures likely capture a large portion of intraindividual amyloid change, our results suggest that there is potentially useful information in local changes in amyloid deposition. For instance, measuring changes in already affected regions may be a less important disease marker than determining whether or not pathology has spread to later-affected structures. Our results are also consistent with recent studies reporting that combining multiple biomarkers is more informative than examining a single biomarker alone (51, 70–73). From a clinical perspective, therapeutically induced decreases in global amyloid load may mean little if metabolic and volumetric declines still are occurring. Conversely, monitoring changes in amyloid PET uptake in regions that do not show any other signs of pathology, such as the palladium and anterior cingulate cortex in this study, may be relatively uninformative. These issues will become increasingly relevant as large-scale, multimodal studies including longitudinal follow-up become more common.
Methods
Participants.
Individuals from families with known ADAD mutations were recruited at 11 separate sites as part of the DIAN initiative. All participants with MRI, genetic, and clinical data that passed quality control procedures from the fourth semiannual data cutoff were included in the analysis. Of these participants, 13 with available MRI data were excluded owing to processing failures (n = 9) or excessive pathology (n = 4). After exclusions, a total of 92 NCs and 137 MCs were analyzed (Table 1). A subset of these participants had analyzable PiB (NC = 84, MC = 116) and FDG (NC = 86, MC = 121) scans (13 PiB and 4 FDG scans were excluded for technical failures). A small number of participants also had serial MRI and PET imaging (Table S1). The institutional review board at Washington University provided supervisory review and human studies approval for all study procedures. Each participating institution also obtained local human studies approval. Approvals were obtained from Brown University, Butler Hospital, Columbia University, Edith Cowan University, University of Western Australia, Partners Human Research Committee, Indiana University, University of California Los Angeles, University of Melbourne, Melbourne Health, University of Pittsburgh, University of New South Wales, Neuroscience Research Australia, and University College of London. All participants or their caregivers provided written informed consent approved by their local institutional review board.
Clinical Assessment.
Each participant underwent an extensive clinical assessment including a medical history, family history of AD, and physical and neurological examinations. Dementia status was assessed using the CDR (74). EYO was calculated as the difference between the participant’s age at evaluation and the age at which parental cognitive decline began (12). For the symptomatic MCs in this report, the parental and actual age of onsets were correlated (Pearson correlation coefficient = 0.65, P < 1.4⋅10−07). The presence or absence of an ADAD mutation was determined using PCR-based amplification of the appropriate exon followed by Sanger sequencing (12). Each participant’s apolipoprotein E4 (APOE4) genotype was determined using methods described elsewhere (75). Clinical evaluators were blind to participant mutation status. Unless medically indicated, research data were not shared with the participants.
MRI.
Structural MRI acquisition was performed using the Alzheimer’s Disease Neuroimaging Initiative (ADNI) protocol (76, 77). All participating sites used a 3T scanner and were required to pass initial and regular follow-up quality control assessments to ensure acquisition uniformity. Scans acquired in each participant included accelerated 3D, sagittal T1-weighted images of the head (1.1- × 1.1- × 1.2-mm voxels, approximate scan time 5–6 min). These images were screened for artifacts and protocol compliance by the ADNI Imaging Core before further analysis.
Volumetric segmentation and cortical surface reconstruction was performed using FreeSurfer 5.1 (78). This procedure automatically segments subcortical and cortical structures using a probabilistic atlas. Each voxel in the brain was assigned an anatomical label (53, 79). Segmented volumes were corrected for intracranial volume using an analysis of covariance approach (80). For each vertex on the cortical surface, thickness was calculated as the shortest distance from the gray/white boundary to the gray/CSF boundary (81). To facilitate group analyses, cortical thickness maps were registered to an average cortical surface and geodesically smoothed with a Gaussian 10-mm FWHM kernel. A trained rater visually verified segmentations and surfaces for accuracy. Manual edits were performed when necessary according to the FreeSurfer manual (http://surfer.nmr.mgh.harvard.edu/fswiki/).
PET.
Each site underwent an initial evaluation to ensure compliance with common PiB and FDG PET ADNI protocols. Amyloid imaging was performed with a bolus injection of ∼15 mCi of [11C]PiB. Dynamic acquisition consisted of either a 70-min scan starting at injection or a 30-min scan beginning 40 min postinjection. For analysis, the PiB PET data in the common time frame between 40–70 min was used. Metabolic imaging with [18F]FDG-PET was performed with a 30-min 3D dynamic acquisition beginning 30 min after injection. The last 25 min of each FDG scan was used for analysis purposes. The ADNI PET Core verified that all PET images were acquired using the established protocol and substantially free of artifacts.
Each participant’s PET data were motion-corrected and registered to his or her MRI (82, 83). Differences owing to image resolution across scanners were minimized by smoothing PET images to a common resolution of 8 mm isotropic (84). Using FreeSurfer ROIs, standardized uptake value ratios (SUVRs) were calculated using the brainstem as a reference region. The brainstem was used as the reference tissue as histological studies have found amyloid plaques in the cerebellar cortex of ADAD mutation carriers (85, 86). The brainstem has been shown to be a reliable reference region for PiB (87) and FDG PET (88). To minimize the impact of partial volume effects on the PET signal, an RSF-based approach (48) for partial volume correction was used for all regional PET measurements. Versions of Figs. 2–4 without PVC can be found in Supporting Information (Figs. S6–S8). For vertex-wise group analyses, uncorrected SUVR images were sampled halfway between the pial and white surface, transformed to the average cortical surface, and smoothed with a surfaced-based Gaussian 5-mm FWHM kernel.
Statistical Analysis.
Statistical analysis of all cross-sectional ROI and vertex-wise measures was performed using the nlme package (89) in R (www.r-project.org) (90). Carriers and noncarriers were tested for differences as a function of EYO to estimate regional differences in biomarker trajectories using a general linear mixed effects model. Histograms of the EYO distribution for each marker are shown in Fig. S5. The statistical model included linear terms for mutation status, EYO, and the interaction between mutation status and EYO. Because some participants were recruited from the same family, a random effect of family was added to allow for possible correlations between family members. An analogous model consisting of fixed effects for region, EYO, and the interaction between EYO and region was used to estimate carrier differences in cross-sectional rates of change across brain regions. To account for baseline differences between regions, all regional values were first divided by the noncarrier mean for that region. A random effect for participant nested within family affiliation was used to account for possible correlations within participant as well as within family group. Both models included covariates for age, sex, education, and the presence of absence of an APOE4 allele. All analyses were corrected for multiple comparisons across space using the FDR.
To estimate the trajectory of biomarker changes, curves were fit to the cross-sectional data using LOESS regression (91). Movies of biomarker change as a function of EYO were also computed by fitting a LOESS curve for each vertex on the cortical surface (Movies S1–S3). For all surface-based analyses, a given participant’s data were included only if they had a defined (nonzero) value at that vertex. Because there was a small amount of coverage loss throughout the study, a vertex was never analyzed with less than 96% of the available participants. The PySurfer library was used for visualization of all surface-based analyses (http://pysurfer.github.io/) (92). All other visualizations were created using the ggplot2 package in R (93).
The quantity of longitudinal data precluded a full analysis across the full EYO range. Thus, participants were stratified into one of three groups based on their mutation and cognitive status. Any carrier with a CDR score greater than 0 at their first longitudinal visit was placed into the mutation positive, symptomatic group (MC/S). The remaining carriers were classified as asymptomatic (MC/AS). Finally, all of the noncarriers were grouped together as a control group (NC). To quantify and statistically evaluate the within participant imaging biomarker rate of change over time, general linear mixed models with random intercepts/slopes (94) at the subject level and random intercepts at the family level were used. All general linear mixed models in the longitudinal analysis were estimated using restricted maximum likelihood estimation, with the approximate F-test denominator degrees of freedom based on the method of Kenward and Roger (95). The model included fixed effects for group membership, time from baseline assessment, and the interaction between group and time. In addition, an unstructured covariance model was used to allow the covariance in each biomarker to vary between MCs and NCs. All longitudinal analysis was performed using SAS version 9.3 (SAS Institute, Inc.).
Supplementary Material
Acknowledgments
We wish to acknowledge the dedication of the participants and their families, without which these studies would not be possible, and all of the participating researchers in the Dominantly Inherited Alzheimer Network (www.dian-info.org/personnel.htm). We also thank Dr. Brian Gordon for his helpful comments on earlier versions of this manuscript. Funding for this research was provided by National Institutes of Health(NIH)/National Institute on Aging (NIA) (U19AG032438). Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially provided through National Center for Research Resources (NCRR) 1S10RR022984-01A1. Research reported in this publication was additionally supported by the Charles F. and Joanne Knight Alzheimer's Research Initiative, Alzheimer's Research UK, Brain Research Trust and the Washington University Institute of Clinical and Translational Sciences grant UL1 TR000448 from the National Center for Advancing Translational Sciences (NCATS) of the NIH.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1317918110/-/DCSupplemental.
References
- 1.Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 2.Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campion D, et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox NC, et al. Clinicopathological features of familial Alzheimer’s disease associated with the M139V mutation in the presenilin 1 gene. Pedigree but not mutation specific age at onset provides evidence for a further genetic factor. Brain. 1997;120(Pt 3):491–501. doi: 10.1093/brain/120.3.491. [DOI] [PubMed] [Google Scholar]
- 6.Acosta-Baena N, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: A retrospective cohort study. Lancet Neurol. 2011;10(3):213–220. doi: 10.1016/S1474-4422(10)70323-9. [DOI] [PubMed] [Google Scholar]
- 7.Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 8.Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 9.Levy-Lahad E, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 10.Houlden H, et al. A novel presenilin mutation (M233V) causing very early onset Alzheimer’s disease with Lewy bodies. Neurosci Lett. 2001;313(1-2):93–95. doi: 10.1016/s0304-3940(01)02254-6. [DOI] [PubMed] [Google Scholar]
- 11.Morris JC, et al. Developing an international network for Alzheimer’s research: The Dominantly Inherited Alzheimer Network. Clin Investig. 2012;2(10):975–984. doi: 10.4155/cli.12.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateman RJ, et al. Dominantly Inherited Alzheimer Network Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15(4):870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- 14.Fleisher AS, et al. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: A cross-sectional study. Lancet Neurol. 2012;11(12):1057–1065. doi: 10.1016/S1474-4422(12)70227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Jr, et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iseki E, et al. Morphological characteristics of senile plaques in familial Alzheimer’s disease. Acta Neuropathol. 1990;80(3):227–232. doi: 10.1007/BF00294638. [DOI] [PubMed] [Google Scholar]
- 17.Koivunen J, et al. PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain. 2008;131(Pt 7):1845–1853. doi: 10.1093/brain/awn107. [DOI] [PubMed] [Google Scholar]
- 18.Buckner RL, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25(34):7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.La Joie R, et al. Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J Neurosci. 2012;32(46):16265–16273. doi: 10.1523/JNEUROSCI.2170-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raichle ME, et al. A default mode of brain function. Proc Natl Acad Sci USA. 2001;98(2):676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson PM, et al. Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci. 2003;23(3):994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitwell JL, et al. 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain. 2007;130(Pt 7):1777–1786. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Förster S, et al. Regional expansion of hypometabolism in Alzheimer’s disease follows amyloid deposition with temporal delay. Biol Psychiatry. 2012;71(9):792–797. doi: 10.1016/j.biopsych.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 24.Villain N, et al. AIBL Research Group Regional dynamics of amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: A voxelwise PiB-PET longitudinal study. Brain. 2012;135(Pt 7):2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- 25.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 26.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45(3):358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 27.Reiman EM, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci USA. 2004;101(1):284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oh H, Habeck C, Madison C, Jagust W (2012) Covarying alterations in Aβ deposition, glucose metabolism, and gray matter volume in cognitively normal elderly. Hum Brain Mapp, 10.1002/hbm.22173. [DOI] [PMC free article] [PubMed]
- 29.Cohen AD, et al. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: An example of brain reserve. J Neurosci. 2009;29(47):14770–14778. doi: 10.1523/JNEUROSCI.3669-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cirrito JR, et al. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron. 2005;48(6):913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 31.Reiman EM, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol. 2012;11(12):1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Potter R et al. (2013) Increased in vivo amyloid- 42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med 5(189):189ra77. [DOI] [PMC free article] [PubMed]
- 33.Sabuncu MR, et al. Alzheimer’s Disease Neuroimaging Initiative The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011;68(8):1040–1048. doi: 10.1001/archneurol.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caroli A, Frisoni GB. Alzheimer’s Disease Neuroimaging Initiative The dynamics of Alzheimer’s disease biomarkers in the Alzheimer’s Disease Neuroimaging Initiative cohort. Neurobiol Aging. 2010;31(8):1263–1274. doi: 10.1016/j.neurobiolaging.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jack CR et al. (2012) Shapes of the trajectories of 5 major biomarkers of Alzheimer disease. Arch Neurol 69(7):856–867. [DOI] [PMC free article] [PubMed]
- 36.Jack CR, Jr, et al. Brain β-amyloid load approaches a plateau. Neurology. 2013;80(10):890–896. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villemagne VL, et al. Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013;12(4):357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 38.Su Y, et al. Noninvasive estimation of the arterial input function in positron emission tomography imaging of cerebral blood flow. J Cereb Blood Flow Metab. 2012;33(1):115–121. doi: 10.1038/jcbfm.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salat DH, et al. Age-associated alterations in cortical gray and white matter signal intensity and gray to white matter contrast. Neuroimage. 2009;48(1):21–28. doi: 10.1016/j.neuroimage.2009.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salat DH, et al. Hippocampal degeneration is associated with temporal and limbic gray matter/white matter tissue contrast in Alzheimer’s disease. Neuroimage. 2011;54(3):1795–1802. doi: 10.1016/j.neuroimage.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salat DH, et al. Thinning of the cerebral cortex in aging. Cereb Cortex. 2004;14(7):721–730. doi: 10.1093/cercor/bhh032. [DOI] [PubMed] [Google Scholar]
- 42.Fjell AM, et al. High consistency of regional cortical thinning in aging across multiple samples. Cereb Cortex. 2009;19(9):2001–2012. doi: 10.1093/cercor/bhn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fjell AM, et al. Alzheimer Disease Neuroimaging Initiative Accelerating cortical thinning: Unique to dementia or universal in aging? Cereb Cortex. 2012 doi: 10.1093/cercor/bhs379. 23236213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abe O, et al. Aging in the CNS: Comparison of gray/white matter volume and diffusion tensor data. Neurobiol Aging. 2008;29(1):102–116. doi: 10.1016/j.neurobiolaging.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Raz N, et al. Aging, sexual dimorphism, and hemispheric asymmetry of the cerebral cortex: Replicability of regional differences in volume. Neurobiol Aging. 2004;25(3):377–396. doi: 10.1016/S0197-4580(03)00118-0. [DOI] [PubMed] [Google Scholar]
- 46.Zhou J, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain. 2010;133(Pt 5):1352–1367. doi: 10.1093/brain/awq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brier MR, et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32(26):8890–8899. doi: 10.1523/JNEUROSCI.5698-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rousset OG, Ma Y, Evans AC (1998) Correction for partial volume effects in PET: Principle and validation. J Nucl Med 39(5):904–911. [PubMed]
- 49.Frouin V, Comtat C, Reilhac A, Grégoire M-C. Correction of partial-volume effect for PET striatal imaging: Fast implementation and study of robustness. J Nucl Med. 2002;43(12):1715–1726. [PubMed] [Google Scholar]
- 50.Zaidi H, Ruest T, Schoenahl F, Montandon M-L. Comparative assessment of statistical brain MR image segmentation algorithms and their impact on partial volume correction in PET. Neuroimage. 2006;32(4):1591–1607. doi: 10.1016/j.neuroimage.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 51.Fortea J, et al. Increased cortical thickness and caudate volume precede atrophy in PSEN1 mutation carriers. J Alzheimers Dis. 2010;22(3):909–922. doi: 10.3233/JAD-2010-100678. [DOI] [PubMed] [Google Scholar]
- 52.Lee GJ, et al. Regional brain volume differences in symptomatic and presymptomatic carriers of familial Alzheimer’s disease mutations. J Neurol Neurosurg Psychiatry. 2013;84(2):154–162. doi: 10.1136/jnnp-2011-302087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fischl B, et al. Whole brain segmentation: Automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 54.Klunk WE, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27(23):6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villemagne VL, Ataka S, Mizuno T. High striatal amyloid β-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66(12):1537–1544. doi: 10.1001/archneurol.2009.285. [DOI] [PubMed] [Google Scholar]
- 56.Knight WD, et al. Carbon-11-Pittsburgh compound B positron emission tomography imaging of amyloid deposition in presenilin 1 mutation carriers. Brain. 2011;134(Pt 1):293–300. doi: 10.1093/brain/awq310. [DOI] [PubMed] [Google Scholar]
- 57.Perani D, Grassi F, Sorbi S. PET study in subjects from two Italian FAD families with APP717 Val to Ileu mutation. Eur J Neurol. 1997;4(3):214–220. [Google Scholar]
- 58.Kennedy AM, et al. Chromosome 14 linked familial Alzheimer’s disease. A clinico-pathological study of a single pedigree. Brain. 1995;118(Pt 1):185–205. doi: 10.1093/brain/118.1.185. [DOI] [PubMed] [Google Scholar]
- 59.Polinsky RJ, et al. Dominantly inherited Alzheimer’s disease: Cerebral glucose metabolism. J Neurol Neurosurg Psychiatry. 1987;50(6):752–757. doi: 10.1136/jnnp.50.6.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mosconi L, Sorbi S, de Leon MJ, Li Y (2006) Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J Nucl Med 47(11):1778–1786. [PubMed]
- 61.Gregory GC, Macdonald V, Schofield PR, Kril JJ, Halliday GM. Differences in regional brain atrophy in genetic forms of Alzheimer’s disease. Neurobiol Aging. 2006;27(3):387–393. doi: 10.1016/j.neurobiolaging.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 62.Ginestroni A, et al. Early structural changes in individuals at risk of familial Alzheimer’s disease: A volumetry and magnetization transfer MR imaging study. J Neurol. 2009;256(6):925–932. doi: 10.1007/s00415-009-5044-3. [DOI] [PubMed] [Google Scholar]
- 63.Schott JM, et al. Assessing the onset of structural change in familial Alzheimer’s disease. Ann Neurol. 2003;53(2):181–188. doi: 10.1002/ana.10424. [DOI] [PubMed] [Google Scholar]
- 64.Knight WD, et al. Acceleration of cortical thinning in familial Alzheimer’s disease. Neurobiol Aging. 2011;32(10):1765–1773. doi: 10.1016/j.neurobiolaging.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 65.Apostolova LG, et al. Cortical and hippocampal atrophy in patients with autosomal dominant familial Alzheimer’s disease. Dement Geriatr Cogn Disord. 2011;32(2):118–125. doi: 10.1159/000330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fox NC, Warrington EK, Stevens JM (1996) Atrophy of the hippocampal formation in early familial Alzheimer's disease. Ann N Y Acad Sci 777:226–232. [DOI] [PubMed]
- 67.Fox NC, et al. Presymptomatic hippocampal atrophy in Alzheimer’s disease. A longitudinal MRI study. Brain. 1996;119(Pt 6):2001–2007. doi: 10.1093/brain/119.6.2001. [DOI] [PubMed] [Google Scholar]
- 68.Ridha BH, et al. Tracking atrophy progression in familial Alzheimer’s disease: A serial MRI study. Lancet Neurol. 2006;5(10):828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- 69.Ostrowitzki S, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207. doi: 10.1001/archneurol.2011.1538. [DOI] [PubMed] [Google Scholar]
- 70.Kohannim O, et al. Alzheimer’s Disease Neuroimaging Initiative Boosting power for clinical trials using classifiers based on multiple biomarkers. Neurobiol Aging. 2010;31(8):1429–1442. doi: 10.1016/j.neurobiolaging.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roe CM, et al. Improving CSF biomarker accuracy in predicting prevalent and incident Alzheimer disease. Neurology. 2011;76(6):501–510. doi: 10.1212/WNL.0b013e31820af900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang D, Wang Y, Zhou L, Yuan H, Shen D. Alzheimer’s Disease Neuroimaging Initiative Multimodal classification of Alzheimer’s disease and mild cognitive impairment. Neuroimage. 2011;55(3):856–867. doi: 10.1016/j.neuroimage.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hinrichs C, Singh V, Xu G, Johnson SC, Initiative TADN. Alzheimers Disease Neuroimaging Initiative Predictive markers for AD in a multi-modality framework: An analysis of MCI progression in the ADNI population. Neuroimage. 2011;55(2):574–589. doi: 10.1016/j.neuroimage.2010.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 75.Cruchaga C, et al. NIA-LOAD/NCRAD Family Study Consortium Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE. 2012;7(2):e31039. doi: 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jack CR, Jr, et al. The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27(4):685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jack CR, Jr., et al. (2010) Update on the Magnetic Resonance Imaging Core of the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dementia 6(3):212–220. [DOI] [PMC free article] [PubMed]
- 78.Fischl B. FreeSurfer. Neuroimage. 2012;62(2):774–781. doi: 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Desikan RS, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 80. Jack CR, et al. (1989) Anterior temporal lobes and hippocampal formations: Normative volumetric measurements from MR images in young adults. 172(2):549–554. [DOI] [PubMed]
- 81.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA. 2000;97(20):11050–11055. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rowland DJ, Garbow JR, Laforest R, Snyder AZ. Registration of [18F]FDG microPET and small-animal MRI. Nucl Med Biol. 2005;32(6):567–572. doi: 10.1016/j.nucmedbio.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 83.Eisenstein SA, et al. Characterization of extrastriatal D2 in vivo specific binding of [¹⁸F](N-methyl)benperidol using PET. Synapse. 2012;66(9):770–780. doi: 10.1002/syn.21566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Joshi A, Koeppe RA, Fessler JA (2009) Reducing between scanner differences in multi-center PET studies. NeuroImage 46(1):154–159. [DOI] [PMC free article] [PubMed]
- 85.Lippa CF, et al. Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology. 1996;46(2):406–412. doi: 10.1212/wnl.46.2.406. [DOI] [PubMed] [Google Scholar]
- 86.Mann DMA, Pickering-Brown SM, Takeuchi A, Iwatsubo T. Members of the Familial Alzheimer’s Disease Pathology Study Group Amyloid angiopathy and variability in amyloid β deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am J Pathol. 2001;158(6):2165–2175. doi: 10.1016/s0002-9440(10)64688-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Edison P, et al. Can target-to-pons ratio be used as a reliable method for the analysis of [11C]PIB brain scans? Neuroimage. 2012;60(3):1716–1723. doi: 10.1016/j.neuroimage.2012.01.099. [DOI] [PubMed] [Google Scholar]
- 88.Minoshima S, Frey KA, Foster NL, Kuhl DE. Preserved pontine glucose metabolism in Alzheimer disease: A reference region for functional brain image (PET) analysis. J Comput Assist Tomogr. 1995;19(4):541–547. doi: 10.1097/00004728-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 89. Pinheiro J, Bates D, DebRoy S, Sarkar D, Team TRDC (2012) Linear and nonlinear mixed effects models. R package version 3, pp 57.
- 90. R Development Core Team (2011) R: A Language and Environment for Statistical Computing (R Foundation, Vienna). Available at www.R-project.org/. Accessed February 5, 2013.
- 91.Cleveland WS. Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74(368):829–836. [Google Scholar]
- 92.Ramachandran P, Varoquaux G. Mayavi: 3D visualization of scientific data. Comput Sci Eng. 2011;13(2):40–51. [Google Scholar]
- 93. Wickham H (2009) ggplot2: Elegant Graphics for Data Analysis (Springer, Dordrecht, The Netherlands)
- 94.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–974. [PubMed] [Google Scholar]
- 95.Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53(3):983–997. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.