

Significance
Multiple sclerosis (MS) is a debilitating neurodegenerative disease characterized by inflammation and demyelination. Immunomodulatory agents slow, but do not prevent, disease progression and offer only indirect neuroprotection. The estrogen receptor β (ERβ) ligand diarylpropionitrile (DPN) has direct neuroprotective effects in a mouse model of MS by stimulating endogenous myelination. Here we elucidate the cell type(s) mediating DPN’s effects. Conditional knockout of ERβ in oligodendrocyte (OL) lineage cells prevented DPN-induced improvement in clinical disease and myelination, partially prevented callosal conduction improvement, and prevented activation of a pathway implicated in OL survival/axon myelination. Our results indicate that DPN-conferred neuroprotection in a mouse model of MS is mediated by ERβ in OLs and inform highly targeted pharmacotherapeutic approaches to MS treatment.
Keywords: remyelination, differentiation, proliferation
Abstract
Treatment of experimental autoimmune encephalomyelitis (EAE) mice with the estrogen receptor (ER) β ligand diarylpropionitrile (DPN) has been shown to have neuroprotective effects via stimulation of endogenous myelination. The direct cellular mechanisms underlying the effects of this ERβ ligand on the central nervous system are uncertain because different cell types in both the peripheral immune system and central nervous system express ERs. ERβ is the target molecule of DPN because DPN treatment fails to decrease EAE clinical symptoms in global ERβ-null mice. Here we investigated the potential role of ERβ expression in cells of oligodendrocyte (OL) lineage in ERβ ligand-mediated neuroprotection. To this end, we selectively deleted ERβ in OLs using the well-characterized Cre-loxP system for conditional gene knockout (CKO) in mice. The effects of this ERβ CKO on ERβ ligand-mediated neuroprotective effects in chronic EAE mice were investigated. ERβ CKO in OLs prevented DPN-induced decrease in EAE clinical disease. DPN treatment during EAE did not attenuate demyelination, only partially improved axon conduction, and did not activate the phosphatidylinositol 3-kinase/serine-threonine-specific protein kinase/mammalian target of rapamycin signaling pathway in ERβ CKO mice. However, DPN treatment significantly increased brain-derived neurotrophic factor levels in ERβ CKO mice. These findings demonstrate that signaling through ERβ in OLs is essential for the beneficial myelination effects of the ERβ ligand DPN in chronic EAE mice. Further, these findings have important implications for neuroprotective therapies that directly target OL survival and myelination.
Multiple sclerosis (MS) is an inflammatory, demyelinating neurodegenerative disease characterized by physical, and often cognitive, deficits that can progress to severe debilitation. Although current MS treatments exist in the form of immunomodulatory or immunosuppressive agents, these treatments fail to halt disease progression and are not directly neuroprotective.
Building on a wealth of research supporting a role for estrogens in neuroprotection, we have demonstrated that treatment of experimental autoimmune encephalomyelitis (EAE) mice with the estrogen receptor (ER) β ligand 2,3-bis(4-Hydroxyphenyl)-propionitrile (DPN) attenuates clinical disease, neurodegeneration, and axon demyelination and improves axon conduction (1–4). Notably, these effects were observed with both prophylactic and therapeutic treatment regimens, and they occurred in the presence of peripheral cytokine production and central nervous system (CNS) inflammation. Evidence of direct neuroprotection by an ERβ ligand is welcomed, because it circumvents ERα-mediated adverse effects of synthetic estrogens [i.e., increased breast and uterine endometrial growth in females and feminizing effects in males (5)].
Because ERs are present in various cell types in the peripheral immune system and CNS, including cells of oligodendrocyte (OL) lineage, it is difficult to assess which cell type(s) mediate ERβ ligand-conferred neuroprotection (6). ERβ is the target molecule of DPN: DPN treatment fails to decrease EAE clinical symptoms in global ERβ-null mice (4). Although informative, such studies do not elucidate the cell type(s) on which DPN acts. We have recently demonstrated that therapeutic treatment with DPN increases mature OL numbers, remyelination-induced callosal conduction, and phosphorylated ERβ levels in OLs, and that it activates the phosphatidylinositol 3-kinase (PI3K)/serine-threonine-specific protein kinase (Akt)/mammalian target of rapamycin (mTOR) signaling pathway, a pathway implicated in OL survival, differentiation, and axon myelination (1, 7, 8). Taken together with the pronounced, functional improvement in endogenous myelination observed in DPN-treated chronic EAE mice, a direct effect on ERβ in OLs may mediate ERβ ligand-induced myelination/remyelination improvement and neuroprotection. To test this hypothesis, ERβ was selectively deleted from OLs using the well-characterized Cre-loxP recombination system for conditional gene knockout (CKO) in mice (9–12). The Olig2,ERβ CKO mice that resulted from crossing ERβflox/flox and Olig2-tva-Cre mice were viable and displayed normal behavior and gross CNS anatomy; hence, the effect of ERβ CKO in OLs on ERβ ligand-induced neuroprotection in chronic EAE mice was investigated. ERβ CKO in OLs prevented DPN-induced attenuation of clinical disease and demyelination and failed to activate the PI3K/Akt/mTOR signaling pathway. However, DPN treatment in Olig2,ERβ CKO mice partially improved axon conduction and reduced axonal loss and, as in WT mice, significantly increased BDNF levels. These findings reveal a direct action of the ERβ ligand DPN on ERβ in OLs in DPN-induced myelination/remyelination effects within a chronic mouse model of MS.
Methods
Animals.
Olig2-tva-Cre mice [kind gift from David Rowitch, University of California, San Francisco (9)] were generated by insertion of an avian retrovirus receptor (TVA) and IRES-Cre cassette into the endogenous Olig2 locus as previously described (5), allowing for Cre-mediated recombination in OL lineage cells. CKO mice with OL-specific deletion of ERβ were obtained by crossing previously described ERβL2/L2 mice [kind gift from Rhonda Voskuhl, University of California, Los Angeles (UCLA) (13) and Pierre Chambon, Institut de génétique et de biologie moléculaire et cellulaire, Strasbourg, France (10–12)] with the Olig2-tva-Cre line, resulting in ERβflox/flox Olig2-Cre mice (henceforth referred to as Olig2,ERβ CKO or ERβ CKO mice). In each case, Cre-negative homozygous floxed (fl) mice were used as littermate controls. Additionally, age- and sex-matched transgenic WT mice with enhanced green fluorescent protein on the proteolipid protein promoter (PLP_EGFP; generously donated by Wendy Macklin, University of Colorado, Denver, CO) were used. Littermate control mice were found to be no different from control PLP_EGFP WT mice. PLP_EGFP and ERβ CKO mice were bred and housed in separate facilities at UCLA. All mice were held on a 12-h light–dark cycle with access to food and water ad libitum. Procedures were conducted in accordance with the National Institutes of Health and approved by the Animal Care and Use Committee of the Institutional Guide for the Care and Use of Laboratory Animals at UCLA. Genotyping was performed as previously described (11). Also refer to SI Methods.
Number of Mice.
Four treatment groups (normal, vehicle-treated EAE, 17β-estradiol (E2)-treated EAE, and DPN-treated EAE) were used per strain (ERβ CKO, littermate controls, and PLP_EGFP WT), with 10–12 animals per treatment group [3 animals for electrophysiology recording/peripheral cytokine analysis + 3 animals for immunohistochemistry + 3–4 animals for Western blot + 2 animals for electron microscopy (EM)]. Each experiment was repeated three times.
Reagents.
Male or female mice, 8–10 wk old, were administered 8 mg/kg per 48 h DPN (Tocris Bioscience), 0.04 mg/kg per day E2 (Sigma-Aldrich), or vehicle, made up of 10% ethyl alcohol (EM Sciences) and 90% Miglyol 812N oil (kind gift from Sasol North America, Houston), s.c. beginning at EAE postinduction day 0 and continued until day 36–42 (end of experiment). The DPN dose is based on uterine weight measurements for biological response and previous EAE experiments (2, 4). Myelin oligodendrocyte glycoprotein (MOG; amino acids 35–55) was obtained from Mimotopes.
EAE.
Active EAE was induced using MOG35–55 as previously described (1, 2, 4).
Peripheral Immune Response.
Splenocyte isolation, stimulation, and cytokine level determination by Searchlight (Aushon Biosciences) were performed according to standard published protocols (4).
Uterus.
Female mice were weighed after perfusion. The uterus was dissected, and uterus/body weight ratios were calculated.
Histopathology, Immunohistochemistry, and Fluorescent and Electron Microscopy.
Formalin-fixed coronal spinal cord sections were examined by immunohistochemistry using various series of cell type-specific antibodies (4). In parallel, the corpus callosum (CC) was dissected and subjected to EM (2). Antibody sources and concentrations used, as well as stereological and g-ratio analysis, are explained in detail SI Methods.
Electrophysiological Recording.
Two slices per animal containing midline-crossing CC and dorsal hippocampus [plates 40–48; as previously described (2)] were used for electrophysiology recordings of compound action potential (CAP) and refractoriness. CAP amplitude and axon refractoriness were measured, calculated, and graphed (2).
Western Blot Analysis.
Whole spinal cord, including cervical and thoracic regions, was dissected from individual animals, rapidly frozen in dry ice, and stored at −80 °C. Polyacrylamide gel electrophoresis, electrophoretic transfer of protein bands to nitrocellulose, and immunoblotting were performed to detect cell-signaling proteins (1).
Statistical Analysis.
All subjects were coded, and analyses were conducted by multiple researchers, all blind to experimental conditions. Electrophysiology recording and immunohistochemical staining results were quantified and analyzed (1). Potential differences in EAE clinical scores between treatment groups were probed using one-way ANOVA and Friedman test or Bonferroni’s multiple comparisons posttest. *P < 0.05 and **P < 0.001 constituted the criteria for statistical significance. Statistical analyses were performed using Microcal Origin or Prism 4 (GraphPad Software). Western blot data are presented as mean ± SEM and analyzed by t test for independent samples or two-way ANOVA followed by Tukey post hoc test.
Results
Selective Deletion of ERβ from OLs in Olig2,ERβ CKO Mice Presents No Gross Phenotype.
Previously characterized Olig2-tva-Cre and ERβfl/fl mice were crossed to produce ERβ CKO mice. Resulting mice were born in the expected Mendelian frequency and were of normal size and weight. Tissue samples were acquired and tested for the presence of Olig2-specific KO of ERβ using a PCR strategy. Homozygous ERβ+/+ (WT), homozygous ERβfl/fl (loxP/loxP; floxed), and heterozygous ERβfl/+ subjects exhibited a 180-, 230-, and both 230- and 180-bp fragment, respectively (Fig. 1A). Separately, presence of Olig2-specific Cre recombinase gene was evidenced by a ∼100-bp fragment (Fig. 1A; ∼320-bp fragment within bottom gel represents internal positive control). When Olig2-tva-Cre+/− mice were crossed to ERβfl/fl mice, the majority (98%) of Olig2+ cells lacked ERβ expression throughout development. A representative section of spinal cord gray matter reveals that Olig2Cre+,ERβfl/fl mice lacked ERβ expression in Olig2+ cells, compared with robust ERβ expression in Olig2Cre-,ERβfl/fl mice (Fig. 1B and Fig. S1). However, no difference in myelin basic protein (MBP) fluorescence intensity and axon label 200-kDa neurofilament (NF200) costained white matter tracts in the cingulum and CC were observed between ERβ CKO and littermate control mice (Fig. S2A). CC axons were further examined using EM. ERβ CKO and littermate control mice showed no difference in myelinated axon numbers or g-ratio of 0.86 ± 0.006 and 0.87 ± 0.002, respectively (Fig. S2B). Additionally, no difference in number or appearance of OLs (immunostained with OL transcription factor, anti-olig2, to label OL lineage cells and adenomatous polyposis coli, anti-CC1, to label mature OLs) was observed in the spinal cord dorsal column (Fig. S2C). In summary, examination and measurement of CNS myelination revealed no gross phenotype in naïve ERβ CKO and littermate control mice.
Fig. 1.
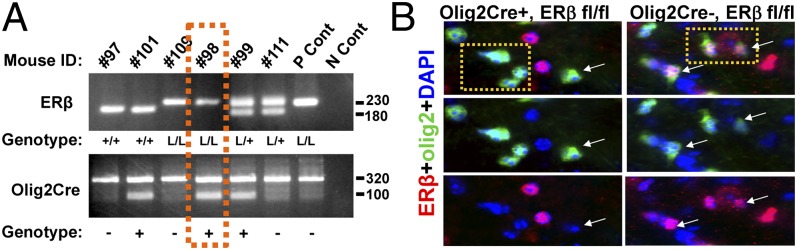
Generation of mice with a conditional knockout of ERβ in OLs. Mice in which ERβ was selectively deleted from OLs (Olig2,ERβ CKO) were generated by crossing ERβfl/fl mice with Olig2Cre mice. (A) ERβ+/+, ERβfl/fl, and ERβfl/+ subjects exhibit a 180-, 230-, and both 230- and 180-bp fragment, respectively (Upper). Presence of Olig2-specific Cre recombinase gene is indicated by a ∼100-bp fragment (Lower). The ∼320-bp fragment within this gel represents internal positive control. Gel area within orange dashes represents a sample from an Olig2,ERβ CKO (Olig2-Cre,ERβfl/fl) mouse. (B) Verification of selective deletion of ERβ in OLs by immunohistochemistry reveals no colocalization of ERβ (red) and cells of OL lineage (olig2; green) in Olig2,ERβ CKO mice, compared with littermate controls. Thoracic spinal cord gray matter of postnatal day 25 (P25) female mice imaged at 40× magnification.
Deletion of ERβ in OLs Prevents DPN-Induced Improvement in EAE Clinical Disease.
To examine the effect of ERβ CKO in OLs on DPN-induced neuroprotection in EAE mice, active EAE was concurrently induced in ERβ CKO, littermate control, and transgenic (PLP_EGFP) WT mice using MOG35–55. Within each genotypic cohort, mice were treated with DPN, E2, or vehicle beginning at EAE postinduction day 0. Mice were scored regularly according to the standard EAE grading scale. Normal animals of all genotypes received complete Freund’s adjuvant (CFA) and Pertussis toxin (PTX), but no MOG35–55, and did not display clinical signs of EAE. MOG35–55-induced EAE in C57BL/6 mice assumes a chronic, severe trajectory, evidenced by persistently high clinical scores (∼3) in all vehicle-treated groups at ∼day 15 (Fig. 2). Importantly, ERβ CKO in OLs has no effect on active EAE induction and disease progression. E2 treatment prevented EAE clinical disease in all genotypes (Fig. 2; **P < 0.001). DPN treatment reduced EAE clinical disease severity in littermate control and WT animals (Fig. 2 A, ii and B, ii; *P < 0.05, **P < 0.001; 2, 4). However, DPN treatment had no effect on EAE clinical disease in ERβ CKO mice (Fig. 2 A, i and B, i). These results strongly suggest that deletion of ERβ in OLs prevents DPN-induced improvement in EAE clinical disease.
Fig. 2.

Selective deletion of ERβ in OLs prevents DPN-induced improvement in EAE clinical disease. All treatments were initiated on active EAE postinduction day 0 and continued until ∼day 30. Clinical disease was evaluated regularly using the standard EAE grading scale. Olig2,ERβ CKO and PLP_EGFP C57BL/6 normal animals were injected with CFA and PTX, but no MOG35–55, and exhibited no clinical disease (Fig. 4) (A, i and A, ii; black). Vehicle-treated animals of all genotypic backgrounds displayed a chronic disease course typical of active EAE induction (red). In all genotypic cohorts, E2 prevented onset of clinical disease (orange). DPN treatment (blue) improved clinical disease later in the course of EAE in both WT (A, ii) and littermate control (B, ii) mice. DPN treatment in Olig2,ERβ CKO animals failed to improve clinical disease (A, i and B, i). n = 5–8 per treatment group. E2 treatment was used as a positive control within each genotypic cohort, **P < 0.001, *P < 0.05; ANOVA Friedman test. Data are representative of experiments repeated three times.
Deletion of ERβ in OLs Does Not Prevent Estradiol-Induced Uterine Weight Increase During EAE.
Uterine weight was assessed by calculating the postperfusion uterus/body weight ratio. For vehicle-treated mice, they were as follows: ERβ CKO, 0.003; littermate controls, 0.004; WT, 0.003. For E2-treated mice, the ratios were nearly two to three times those of vehicle-treated mice. They were as follows: ERβ CKO, 0.009; littermate controls, 0.01; WT, 0.01. Ratios of DPN-treated mice were similar to those of vehicle-treated controls: ERβ CKO, 0.002; littermate controls, 0.002; WT, 0.004. Normal WT mice displayed a mean uterus/body weight ratio of 0.003. Consistent with existing literature, these preliminary data suggest that E2 increased uterine weight to the same degree in females of all genotypes (4, 14) and that DPN had no effect on uterine weight.
DPN-Treated Olig2,ERβ CKO and WT EAE Mice Display Similar Peripheral Cytokine Responses.
Peripheral immune responses play a primary role in the induction of CNS inflammation in EAE. Cytokine production by splenocytes upon ex vivo stimulation by MOG35–55 assessment showed no differences in resulting cytokines [IFN-γ, TNF-α, IL-17, matrix metallopeptidase 9 (MMP9), and IL-5 levels] in DPN-treated ERβ CKO EAE mice compared with DPN-treated WT EAE mice. In addition, similar cytokine levels were found in vehicle-treated ERβ CKO and WT EAE mice (Fig. 3A).
Fig. 3.

DPN-treated Olig2,ERβ CKO and WT EAE mice show similar peripheral cytokine response and CNS inflammation. (A) Cytokine production by MOG35–55-stimulated splenocytes was evaluated. Treatment of Olig2,ERβ CKO and WT EAE mice with DPN had no effect on measured cytokine levels. Data are representative of experiments repeated two times, n = 3 per treatment group. Dorsal column of thoracic spinal cords was imaged at ×10 and ×40 magnification from DPN-treated Olig2,ERβ CKO EAE mice (B, i) and WT EAE mice (B, ii). Both DPN-treated genotypic cohorts exhibit a similar increase in GFAP+ (B, iii), CD45+ (B, iv), and CD3+ (B, v) intensity. *P < 0.05 ANOVAs; Bonferroni’s multiple comparison posttest; n = 8 mice per treatment group.
DPN-Treated Olig2,ERβ CKO and WT EAE Mice Exhibit Comparable Levels of CNS Inflammation.
Extent of CNS inflammation and infiltration by peripheral immune cells was examined in the dorsal column of thoracic spinal cord sections (Fig. 3B; *P < 0.05). As previously observed, hypertrophic, reactive astrocytes immunolabeled with glial fibrillary acidic protein (anti-GFAP), activated microglia (leukocyte antigen marker, anti-CD45), and T cells (anti-CD3) were seen in vehicle- and DPN-treated mice (1, 2). Quantification of GFAP+, CD45+, and CD3+ staining intensity showed no significant differences between vehicle- and DPN-treated ERβ CKO and WT EAE mice. Together, these findings suggest that ERβ CKO in OLs alters neither peripheral immune response nor CNS inflammation in vehicle- or DPN-treated EAE mice.
Deletion of ERβ in OLs Prevents DPN-Induced Neuroprotection of Spinal Cord Axons.
To investigate the effect of ERβ CKO in OLs on DPN-induced improvement in myelination, the thoracic spinal cord dorsal column was examined via immunohistochemistry. Myelin integrity was assessed by immunostaining for MBP (anti-MBP; Fig. 4A). As previously reported, a global myelination loss, specifically at sites of inflammatory cell infiltrates, was observed in vehicle-treated mice (1, 2, 4) (Fig. 4 A, i). Regardless of genotype, vehicle-treated mice displayed a ∼30% reduction (*P < 0.05) in myelin density (Fig. 4 A, i and iii). Comparable to previous results, DPN treatment of WT EAE mice resulted in increased MBP intensity (Fig. 4 A, i and iii) (1, 2, 4). In ERβ CKO mice, however, DPN-treatment had no effect on myelin density (Fig. 4 A, iii). This finding strongly suggests that deletion of ERβ in OLs prevents DPN-induced improvement of CNS myelination in EAE mice.
Fig. 4.

Selective deletion of ERβ in OLs prevents DPN-induced improvement in myelin density and mature OL numbers, but partially protects against axonal loss within the spinal cord of EAE mice. Dorsal column of thoracic spinal cord sections from Olig2,ERβ CKO and WT EAE mice were examined for myelin density (A, i; red). Quantification reveals improved MBP+ intensity in DPN-treated WT EAE but not DPN-treated Olig2,ERβ CKO EAE mice (A, iii). Ventral column of thoracic spinal cord sections were imaged at ×40 and depict costain using NF200 (green) and MBP (red; A, ii). Quantification reveals improved NF200+ axon numbers upon treatment with DPN in WT animals and a trend toward this effect in Olig2,ERβ CKO mice (A, iv). Increased numbers of MBP+ and NF200+ axons in DPN-treated WT, but not Olig2,ERβ CKO, EAE mice (A, v) was observed. For both genotypic cohorts, vehicle-treated mice displayed a significant reduction in myelinated axon numbers (normal; A, iii–v). Vehicle-treated Olig2,ERβ CKO and WT animals exhibited a reduction in CC1+ OLs (B, iii). DPN treatment in WT, but not Olig2,ERβ CKO, EAE mice improved mature OL numbers (B, iii). Olig2+ cell numbers were similar across all groups (B, iv). PLP_EGFP normal thoracic spinal cord sections (×4 magnification) were costained with DAPI (red). White square boxes and a, b, and c denote various areas of the spinal cord used for quantification (DC, dorsal column; LF, lateral funiculus; VC, ventral column; B, v). *P < 0.05; ANOVAs; Bonferroni's multiple comparison posttest; n = 8–10 mice per treatment group.
To assess the effect of ERβ CKO in OLs on DPN-induced neuroprotection of axons, thoracic spinal cord sections were immunostained with anti-NF200 and anti-MBP. NF200+ axons were quantified in the ventral column of these sections (Fig. 4 A, ii). Vehicle-treated animals of all genotypes displayed a decrease in axons and myelinated axons compared with healthy control mice (Fig. 4 A, iv, v; *P < 0.05). DPN-treated ERβ CKO EAE mice showed decreases in myelinated axons similar to their respective vehicle group. However, DPN-treated ERβ CKO EAE mice showed a trend toward increased number of spared axons (Fig. 4 A, ii). Thus, DPN treatment in ERβ CKO EAE mice may have partial direct neuroprotective effects on axon health during EAE.
A reduction in myelinated axons was observed in vehicle-treated ERβ CKO and WT EAE groups (Fig. 4 A, v). DPN treatment improved axon myelination in WT animals (Fig. 4 A, v; *P < 0.05). In contrast, DPN failed to improve axon myelination in mice lacking ERβ in OLs. The presence of ERβ in OLs, then, is necessary for the positive effects of DPN on axon myelination in EAE mice.
Deletion of ERβ in OLs Prevents DPN-Induced Increase in Mature OL Population.
The consequence of DPN-treatment on OLs of ERβ CKO EAE mice was investigated in the dorsal column of thoracic spinal cords of various groups. Vehicle-treated ERβ CKO EAE mice displayed a reduction in CC1+ mature OL cells, whereas DPN-treated ERβ CKO mice did not exhibit significant recovery of CC1+ cells. This was in contrast to DPN-treated WT EAE mice, which showed significant recovery of CC1+ cell numbers as compared to vehicle-treated WT EAE mice (Fig. 4 B, i and iii; *P < 0.05). WT mice displayed no difference in olig2+ cell numbers regardless of treatment, and the same was true of ERβ CKO mice (Fig. 4 B, ii and iv). Therefore, DPN-induced increase in mature OL numbers in EAE mice is, at least in part, attributable to ERβ presence in OLs.
Deletion of ERβ in OLs Prevents DPN-Induced Improvement in Myelinated Callosal Axon Conduction.
Remyelination-induced improvement in axon conduction as a result of prophylactic and therapeutic DPN treatment during EAE has been demonstrated (1). We performed CAP recordings of CC axons in ERβ CKO EAE mice treated with vehicle or DPN. Representative recordings from CC show two downward phases of the N1 and N2 CAPs (Fig. 5A). N1 CAPs represent mostly fast depolarizing, myelinated axons, whereas N2 CAPs represent mostly slower depolarizing, nonmyelinated axons (15). Vehicle-treated EAE mice display lower N1 and N2 CAP amplitudes compared with healthy control mice. DPN treatment during EAE improved N1 and N2 amplitudes (Fig. 5 A–C; *P < 0.05; **P < 0.001). DPN-treated ERβ CKO mice showed attenuated improvement in the N1 component compared with vehicle-treated groups (Fig. 5B; *P < 0.05). No significant difference in N2 component was observed between DPN-treated ERβ CKO mice and WT controls (Fig. 5C). Axon refractoriness, a measure of axonal deficits, was evaluated via interpulse intervals (IPIs; see (1, 2, 15)). A small but significant decrease in N1 refractoriness latency was observed in DPN-treated littermate control mice compared with vehicle-treated groups (Fig. 5D). In the normal (healthy control) ERβ CKO group, the N1 component evoked by the second pair of pulses was 50% of the amplitude of a single pulse presentation at an IPI of 2.6 ± 0.1 ms, similar to those of WT mice (1). The IPI for the vehicle-treated EAE group had a slower response of 4.7 ± 0.1 ms. DPN-treated WT controls had a faster N1 IPI of 3.7 ± 0.05 ms. DPN-treated ERβ CKO EAE mice were similar to the vehicle-treated EAE group, with an IPI of 4.1 ± 0.06 ms. There was no difference in IPIs for the N2 component: 3.3 ± 0.1 ms (healthy control), 3.9 ± 0.1 ms (vehicle-treated ERβ CKO EAE), 4.2 ± 0.1 ms (DPN-treated ERβ CKO EAE), and 4.4± 0.1 ms (DPN-treated WT EAE). These data are consistent with DPN-induced neuroprotection being mediated by ERβ in OLs.
Fig. 5.

Selective deletion of ERβ in OLs prevents DPN-induced improvement in myelinated CC axon conduction. (A) Typical CAPs from various treatment groups at postinduction day 30 are shown. (B) An improvement in N1 CAP amplitude is evident in DPN-treated littermate control but not DPN-treated Olig2,ERβ CKO CC axons. (C) N2 CAP amplitude measurement reveals no difference. Average C2/C1 ratios were fitted to Boltzmann sigmoid curves (see ref. 1). (D) DPN-treated littermate control CC axons displayed a small but significant leftward shift. (E) No difference in N2 refractoriness was detected. n = 6. Statistically significant compared with normal controls at 1.1 ± 0.15 mA stimulus strength (*P < 0.05; **P < 0.001; ANOVAs; Bonferroni’s multiple comparisons posttest).
Deletion of ERβ in OLs Prevents DPN-Induced Activation of the PI3K/Akt/mTOR Pathway but Does Not Prevent Increase in BDNF Levels.
We have recently shown that DPN treatment of EAE animals mediates the phosphorylation of PI3K/Akt/mTOR proteins, and it is known that this pathway is essential for OL survival and proliferation (1, 7, 16, 17). To assess the effect of ERβ CKO in OLs on DPN-induced activation of the PI3K/Akt/mTOR pathway in EAE mice, a few proteins from the pathway were analyzed by Western blot analysis of spinal cord protein lysates, as in ref. 1. Similar to previous observations, DPN-treated WT mice displayed increased phosphorylated (p)-AKT and p-mTOR levels compared with vehicle-treated EAE mice and healthy controls (Fig. 6 A and B; **P < 0.001). However, regardless of treatment, no difference was observed in ERβ CKO EAE mice and normal, healthy controls within this genotypic cohort (Fig. 6 D and E). A significant increase in BDNF (1) was observed in DPN-treated EAE mice. Similarly, vehicle-treated EAE animals expressed lower BDNF levels compared with the DPN-treated group. ERβ CKO EAE mice treated with DPN also showed an increase in BDNF compared with the vehicle-treated ERβ CKO EAE group (Fig. 6 C and F; **P < 0.001). Taken together, these results suggest that the presence of ERβ in OLs is required for DPN-induced activation of the PI3K/Akt/mTOR pathway but is not necessary for DPN-induced BDNF increase.
Fig. 6.

DPN fails to activate the PI3K/Akt/mTOR cell signaling pathway, but does increase BDNF levels, in Olig2,ERβ CKO EAE mice. Representative Western blot gels containing samples from healthy control and vehicle- and DPN-treated WT (A) and Olig2,ERβ CKO (D) EAE mice are shown. DPN treatment in WT mice improved pAKT and pmTOR levels (B). No such improvement is observed in the DPN-treated Olig2,ERβ CKO group (E). Analysis of BDNF levels reveals a significant increase in both WT and Olig2,ERβ CKO DPN-treated EAE animals (C–F). **P < 0.001; ANOVA; n = 3–6 per group.
Discussion
The use of global genetic deletions of ERβ demonstrates the importance of ERβ in DPN-induced neuroprotection during EAE (4) but does not help identify specific cell target populations of this ERβ ligand. In the present study we examined the importance of ERβ in OLs, specifically, in initiating OL survival and myelination in DPN-treated EAE mice.
Olig2 is a basic helix–loop–helix transcription factor (18). Olig2 expression is detected at very early time points in CNS development, within the radial glia of the neural tube that ultimately give rise to motor neurons and OLs (19). When Olig2-tva-Cre+/1- mice were crossed to ROSA26-eYFPflox/flox conditional reporter mice, 98% recombination was achieved, as evidenced by YFP+ cells in P7 cerebellar white matter expressing Olig2 consistent with OL identity (20). Similarly, ERβ CKO mice generated by crossing Olig2-tva-Cre mice with ERβfl/fl mice showed more than 95% loss of ERβ in olig2+ cells. As in global ERβ-null mice (21), no gross difference in developmental myelination was observed in ERβ CKO mice, indicating a minimal role of ERβ in OL proliferation, differentiation, and axon myelination.
Although there was no difference in EAE severity between ERβ CKO and WT mice, ERβ CKO prevented DPN-induced attenuation of EAE clinical disease and demyelination, activation of the PI3K/Akt/mTOR signaling pathway, and only partially prevented axon degeneration. Taken together, these findings suggest that the ERβ ligand DPN improves myelination in EAE mice by acting directly on OLs or OL lineage cells, and that this direct action is responsible for remyelination-induced axon protection and partial functional recovery. The findings do not exclude additional effects of estrogens that may be mediated by ERα in OLs and ERα/ERβ in other CNS cell types, including astrocytes, neurons, and immune cells, all possible estrogen targets depending on nature of injury and treatment timing. ERα CKO in astrocytes, but not neurons, prevents EAE disease attenuation by the ERα ligand 4,4′,4″-(4-Propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT) (13). ERα deletion in OLs was not performed, but PPT failed to protect against spinal cord demyelination in these ERα CKO and WT mice (13). In contrast, ERβ in the cochlea is responsible for the neuroprotective effects of E2 on acoustic trauma (22). It is important to note that mice globally lacking ERβ show dramatic age-related neurodegeneration throughout the brain, suggesting an important role for ERβ in maintenance of neuronal health (23).
Both ERα and ERβ regulate brain cytokine and chemokine levels (24). Estradiol and PPT treatment effects during EAE are associated with down-regulation and up-regulation of pro- and antiinflammatory cytokines, respectively (4, 25). Neuroprotection by DPN is not mutually exclusive with other treatment effects on CNS inflammatory cells, because DPN seems to modulate dendritic cells (26). Although no differences in microglia, T cells, and astrocytes were observed in the present study, a difference in chemokine expression between DPN-treated ERβ CKO and WT brain microglia is possible. A recent study showed that PPT, but not DPN, treatment decreased CCL2 and CCL7 chemokine expression by astrocytes in EAE (27). Unlike DPN, other more selective ERβ ligands with both antiinflammatory and neuroprotective properties during EAE, like Indazole-Cl, have been shown to modulate cytokine and chemokine expression (28). Theoretically, DPN and PPT combination therapy may be most effective during EAE—PPT could optimize the inflammatory state of the system, making it more susceptible to DPN’s beneficial myelination effects. However, ERα, not ERβ, activation is believed to mediate increased risk of breast and uterine cancer; thus, combination with an ERα ligand may not realistically be a safe therapeutic option. The present study supports a direct effect of DPN on (ERβ+) OL/OL progenitor cell populations, making these cell populations a promising target for demyelinating disease therapies, the optimization of which may synergize more effectively with immunomodulatory approaches. Indeed, an additive effect of DPN and antiinflammatory IFN-β combination therapy showed attenuated EAE disease severity (26).
DPN treatment in both WT and ERβ CKO EAE mice increased BDNF levels. BDNF is involved in neuronal survival, myelination, differentiation, and function, as well as axonal growth, modulation of neuronal activity, and activity-dependent synaptic and dendritic plasticity in the CNS (29–31). Tropomyosin receptor kinase B (TrkB) activation by BDNF is a possible protective therapeutic strategy in early EAE, whereas BDNF levels decrease with ongoing chronic EAE (present results and refs. 1 and 32). Our recent study showed a decrease in BDNF levels of CC homogenates during late chronic EAE, without changes in TrkB protein levels (1). The noteworthy increase in BDNF levels observed in DPN-treated ERβ CKO mice implies that ERβ interaction with DPN is also occurring in cell types other than OLs. Even in the absence of ERβ in OLs, DPN-mediated BDNF increase during EAE may be due to its interaction with neurons, astrocytes, and/or immune cells. In support of this observation, a more severe EAE clinical disease course was observed in mice with BDNF CKO in either immune cells (T cells and monocytes/macrophages) or astrocytes/neuronal subpopulations (33). In addition, more axonal damage was detected in these mice, without accompanying alteration of the inflammatory process (33).
OPCs are present in and around MS lesions but remain largely quiescent in the adult CNS. OLs express ERs, and estrogens are known to affect proliferation, differentiation, and axon myelination. The data presented here identify the unique and selective action of estrogens on ERβ-bearing OLs to mediate remyelinating/neuroprotective effects in an inflammatory demyelinating condition. The studies presented here demonstrate a direct effect of the ERβ ligand DPN on ERβ in OLs, elucidating an important step in overcoming the differentiation block observed during MS and EAE. Characterizing the direct targets/actions of neuroprotective agents is paramount to the discovery, optimization, and refinement of potential MS treatments, because it aids in avoiding toxicity, reducing side effects, and increasing efficacy.
Supplementary Material
Acknowledgments
We thank Drs. Rowitch, Chambon, and Voskuhl for providing olig2-cre and ERβfl/fl mice; and the UCLA Division of Laboratory Animal Medicine staff: Mr. Biggs, Ms. Knipe, Mr. Lo, Ms. Sierra, and Mr. Qi for caring for our CKO mouse colonies and administering injections in a blind manner. This work was generously supported by NMSS-RG 4538-A-2 from the National Multiple Sclerosis Society, NIH-R21NS075198 from the National Institutes of Health, and Karo Bio grant (to S.K.T.-W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311763110/-/DCSupplemental.
References
- 1.Kumar S, et al. Estrogen receptor β ligand therapy activates PI3K/Akt/mTOR signaling in oligodendrocytes and promotes remyelination in a mouse model of multiple sclerosis. Neurobiol Dis. 2013;56:131–144. doi: 10.1016/j.nbd.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crawford DK, et al. Oestrogen receptor beta ligand: A novel treatment to enhance endogenous functional remyelination. Brain. 2010;133(10):2999–3016. doi: 10.1093/brain/awq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiwari-Woodruff S, Voskuhl RR. Neuroprotective and anti-inflammatory effects of estrogen receptor ligand treatment in mice. J Neurol Sci. 2009;286(1-2):81–85. doi: 10.1016/j.jns.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiwari-Woodruff S, Morales LB, Lee R, Voskuhl RR. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proc Natl Acad Sci USA. 2007;104(37):14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caringella AM, Di Naro E, Loverro G. [Clinical function of estrogen receptors in endometrial cancer] Minerva Ginecol. 2011;63(6):495–504. [PubMed] [Google Scholar]
- 6.Platania P, et al. Differential expression of estrogen receptors alpha and beta in the spinal cord during postnatal development: Localization in glial cells. Neuroendocrinology. 2003;77(5):334–340. doi: 10.1159/000070899. [DOI] [PubMed] [Google Scholar]
- 7.Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29(21):6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyler WA, et al. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29(19):6367–6378. doi: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrington EP, et al. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol. 2010;68(5):703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dupont J, Karas M, LeRoith D. The potentiation of estrogen on insulin-like growth factor I action in MCF-7 human breast cancer cells includes cell cycle components. J Biol Chem. 2000;275(46):35893–35901. doi: 10.1074/jbc.M006741200. [DOI] [PubMed] [Google Scholar]
- 11.Antal MC, Krust A, Chambon P, Mark M. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proc Natl Acad Sci USA. 2008;105(7):2433–2438. doi: 10.1073/pnas.0712029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell L, et al. Estrogen promotes cutaneous wound healing via estrogen receptor beta independent of its antiinflammatory activities. J Exp Med. 2010;207(9):1825–1833. doi: 10.1084/jem.20100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spence RD, et al. Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proc Natl Acad Sci USA. 2011;108(21):8867–8872. doi: 10.1073/pnas.1103833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris HA, et al. Evaluation of an estrogen receptor-beta agonist in animal models of human disease. Endocrinology. 2003;144(10):4241–4249. doi: 10.1210/en.2003-0550. [DOI] [PubMed] [Google Scholar]
- 15.Mangiardi M, et al. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol. 2011;21(3):263–278. doi: 10.1111/j.1750-3639.2010.00444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flores AI, et al. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28(28):7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bibollet-Bahena O, Almazan G. IGF-1-stimulated protein synthesis in oligodendrocyte progenitors requires PI3K/mTOR/Akt and MEK/ERK pathways. J Neurochem. 2009;109(5):1440–1451. doi: 10.1111/j.1471-4159.2009.06071.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Q, Wang S, Anderson DJ. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron. 2000;25(2):331–343. doi: 10.1016/s0896-6273(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 19.Meijer DH, et al. Separated at birth? The functional and molecular divergence of OLIG1 and OLIG2. Nat Rev Neurosci. 2012;13(12):819–831. doi: 10.1038/nrn3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schüller U, Ruiter M, Herms J, Kretzschmar HA, Grasbon-Frodl E. Absence of mutations in the AKT1 oncogene in glioblastomas and medulloblastomas. Acta Neuropathol. 2008;115(3):367–368. doi: 10.1007/s00401-007-0334-2. [DOI] [PubMed] [Google Scholar]
- 21.Krege JH, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA. 1998;95(26):15677–15682. doi: 10.1073/pnas.95.26.15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meltser I, et al. Estrogen receptor beta protects against acoustic trauma in mice. J Clin Invest. 2008;118(4):1563–1570. doi: 10.1172/JCI32796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Andersson S, Warner M, Gustafsson JA. Morphological abnormalities in the brains of estrogen receptor beta knockout mice. Proc Natl Acad Sci USA. 2001;98(5):2792–2796. doi: 10.1073/pnas.041617498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown CM, Mulcahey TA, Filipek NC, Wise PM. Production of proinflammatory cytokines and chemokines during neuroinflammation: Novel roles for estrogen receptors alpha and beta. Endocrinology. 2010;151(10):4916–4925. doi: 10.1210/en.2010-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morales LB, et al. Treatment with an estrogen receptor alpha ligand is neuroprotective in experimental autoimmune encephalomyelitis. J Neurosci. 2006;26(25):6823–6833. doi: 10.1523/JNEUROSCI.0453-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du S, Sandoval F, Trinh P, Voskuhl RR. Additive effects of combination treatment with anti-inflammatory and neuroprotective agents in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;219(1-2):64–74. doi: 10.1016/j.jneuroim.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spence RD, et al. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J Neurosci. 2013;33(26):10924–10933. doi: 10.1523/JNEUROSCI.0886-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saijo K, Collier JG, Li AC, Katzenellenbogen JA, Glass CK. An ADIOL-ERβ-CtBP transrepression pathway negatively regulates microglia-mediated inflammation. Cell. 2011;145(4):584–595. doi: 10.1016/j.cell.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao MV. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 30.Cellerino A, Carroll P, Thoenen H, Barde YA. Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci. 1997;9(5-6):397–408. doi: 10.1006/mcne.1997.0641. [DOI] [PubMed] [Google Scholar]
- 31.Djalali S, et al. Effects of brain-derived neurotrophic factor (BDNF) on glial cells and serotonergic neurones during development. J Neurochem. 2005;92(3):616–627. doi: 10.1111/j.1471-4159.2004.02911.x. [DOI] [PubMed] [Google Scholar]
- 32.Song F, et al. Complexity of trophic factor signaling in experimental autoimmune encephalomyelitis: Differential expression of neurotrophic and gliotrophic factors. J Neuroimmunol. 2013;262(1-2):11–18. doi: 10.1016/j.jneuroim.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linker RA, et al. Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: Therapeutic implications in a model of multiple sclerosis. Brain. 2010;133(Pt 8):2248–2263. doi: 10.1093/brain/awq179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.