

Significance
Biologists frequently overexpress genes to learn about their cellular functions, and biotechnologists do so to construct novel metabolic pathways that produce valuable chemical compounds. However, gene overexpression often leads to deleterious consequences whose cause is unclear. Here, we present a computational method named Expression-Dependent Gene Effects (EDGE) that can successfully predict the deleterious effects resulting from overexpression of either native or foreign (originating in another species) metabolic genes. EDGE relies on genome-scale metabolic models, an emerging computational paradigm for studying metabolism in silico. Beyond its biotechnological significance, gene overexpression also plays an important role in human disease. We show EDGE’s applicability in the latter case by demonstrating its ability to detect toxic genes whose expression tends to be suppressed in cancer cells.
Keywords: systems metabolic engineering, metabolic modeling, constraint-based modeling, flux balance analysis, horizontal gene transfer
Abstract
Gene suppression and overexpression are both fundamental tools in linking genotype to phenotype in model organisms. Computational methods have proven invaluable in studying and predicting the deleterious effects of gene deletions, and yet parallel computational methods for overexpression are still lacking. Here, we present Expression-Dependent Gene Effects (EDGE), an in silico method that can predict the deleterious effects resulting from overexpression of either native or foreign metabolic genes. We first test and validate EDGE’s predictive power in bacteria through a combination of small-scale growth experiments that we performed and analysis of extant large-scale datasets. Second, a broad cross-species analysis, ranging from microorganisms to multiple plant and human tissues, shows that genes that EDGE predicts to be deleterious when overexpressed are indeed typically down-regulated. This reflects a universal selection force keeping the expression of potentially deleterious genes in check. Third, EDGE-based analysis shows that cancer genetic reprogramming specifically suppresses genes whose overexpression impedes proliferation. The magnitude of this suppression is large enough to enable an almost perfect distinction between normal and cancerous tissues based solely on EDGE results. We expect EDGE to advance our understanding of human pathologies associated with up-regulation of particular transcripts and to facilitate the utilization of gene overexpression in metabolic engineering.
Deducing phenotype from genotype is a fundamental goal of modern biology. Traditionally, experimentalists delete or suppress genes to annotate them and to detect phenotypes of interest (1), and such studies are routinely conducted in model organisms. However, it has long been recognized that gene overexpression is a complementary tool for linking genotype to phenotype and usually provides phenotypes that are different from those observed in loss-of-function studies (2).
Beyond basic science, complex human disease is often associated with abnormally up-regulated transcripts (3–5). Studies in murines and in Drosophila have shown that gene overexpression can induce disease on the one hand (6–8) and serve therapeutic purposes on the other (9–11). In addition, phenotypes arising due to overexpression are of prime interest in metabolic engineering, where selected native or heterologous genes are overexpressed to produce novel metabolic pathways in industrial microorganisms (12–16). Metabolic engineers particularly seek to foresee cases in which the up-regulation of a specific gene results in severely reduced fitness (17, 18). Such genes are often referred to as “toxic genes” (19–22).
Nonetheless, there have been only a handful of genome-wide studies addressing gene overexpression to date (2, 23, 24), and this stands in sharp contrast to the wealth of data available from numerous genome-wide KO or knock-down studies (1, 25). For these reasons, in silico approaches to model gene overexpression and studies that apply them genome-wide are highly desirable. Here, we conduct a genome-scale study of the deleterious consequences of inducing metabolic gene overexpression through an in silico algorithm for predicting Expression-Dependent Gene Effects (EDGE). First, we show that EDGE successfully predicts growth retardation phenotypes arising due to inappropriate gene activation in microorganisms, which are a prime concern for metabolic engineers. Second, we show EDGE’s universal applicability by demonstrating across multiple organisms that genes predicted to be nondeleterious when highly expressed are indeed the ones with the highest expression levels. Third, we show EDGE’s applicability to the study of human disease by demonstrating that some aspects of genetic reprogramming in cancer can be explained as an attempt to silence genes whose expression is detrimental toward cancer proliferation.
Our study is conducted within the realm of cellular metabolism, which is particularly amenable to in silico modeling on a genome scale. Genome-scale metabolic models (GSMMs) translate the capabilities of a metabolic network, dictated by an organism’s genome, into a set of mathematical equations (26, 27). They offer a powerful tool for predicting the outcomes of genetic perturbations through testable mechanistic explanations. Notably, GSMMs have been quite successful in predicting phenotypes of loss-of-function mutants (28–30), and therefore hold considerable promise to successfully predict the outcomes of induced gene overexpression as well. A GSMM typically includes a stoichiometric matrix, which represents the network’s topology, constraints (e.g., thermodynamic or environmental constraints) applied to it, and gene–protein associations. EDGE takes a GSMM as its input and quantifies the benefits or losses that a cell incurs by activating a certain metabolic gene in a given environment (Fig. 1, where blue boxes represent EDGE’s inputs and purple boxes represent computation outputs; Materials and Methods). For this, it relies on a hypothesized cellular objective [in this study, we always took the objective to be the commonly assumed maximization of biomass production (31, 32)]. Genes are then classified as (i) beneficial, i.e., contributing toward the realization of the objective (positive score); (ii) detrimental to the objective (negative score); or (iii) neutral with respect to the objective (zero score; SI Appendix).
Fig. 1.

Schematic workflow of EDGE analysis. Blue boxes represent inputs to the computation, and purple boxes represent computation outputs. EDGE relies on a GSMM, constraints applied to it, an objective function, and an in silico growth medium; all these define a space of feasible metabolic phenotypes. EDGE is formulated as a mixed integer linear program that quantifies the positive or negative effect that small changes in the expression level of a particular gene have on the cellular objective due to the flux rerouting that they induce. The image of the network under the box entitled Metabolic Network was adapted from the work of Montagud et al. (62).
Results
EDGE Algorithm.
A complete description of EDGE is given in Materials and Methods. In short, the EDGE score of a gene, 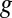 , quantifies the utility of transcribing the gene in small levels compared with not using
, quantifies the utility of transcribing the gene in small levels compared with not using 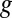 at all. This notion is strongly related to analyzing the sensitivity of a mathematical program to a perturbation of its constraints [sometimes called “shadow pricing” (33)]. Such analyses are fundamental to mathematical programming in general and have previously been applied to GSMMs (27, 34–36). EDGE is an adaptation of these analyses to the special setting of flux balance analysis (FBA) (32). EDGE measures the sensitivity of the optimal objective value to the simultaneous perturbation of multiple constraints that are associated with a particular gene. EDGE scores are not dependent on a particular optimum, which is desirable in the case of GSMMs, whose solution space typically contains multiple optima (37).
at all. This notion is strongly related to analyzing the sensitivity of a mathematical program to a perturbation of its constraints [sometimes called “shadow pricing” (33)]. Such analyses are fundamental to mathematical programming in general and have previously been applied to GSMMs (27, 34–36). EDGE is an adaptation of these analyses to the special setting of flux balance analysis (FBA) (32). EDGE measures the sensitivity of the optimal objective value to the simultaneous perturbation of multiple constraints that are associated with a particular gene. EDGE scores are not dependent on a particular optimum, which is desirable in the case of GSMMs, whose solution space typically contains multiple optima (37).
EDGE simulates the expression of a given gene by enforcing a flux through reactions associated with it; reversible reactions are constrained to carry a flux through either direction. When a gene is associated with more than one reaction, EDGE finds the bottleneck reaction whose limitation has the greatest effect and determines the EDGE score by its utility with regard to the a priori objective function. In each test in this study, we excluded genes whose proteins catalyze blocked reactions (i.e., reactions that cannot carry a flux in steady state in the medium relevant to the test; SI Appendix, Supplementary Methods).
EDGE Predicts Reduced Fitness Due to Overexpression of Native Genes.
We used EDGE to predict which metabolic Escherichia coli genes will prove toxic when overexpressed during growth on glucose-supplemented M9 minimal medium and ranked them by the confidence level of the prediction (SI Appendix). Twenty-six high-ranking genes were chosen for subsequent growth experiments: 12 that were confidently predicted to be toxic and 14 that were confidently predicted not to be toxic (Dataset S1, Table S1). Plasmids (23) containing Isopropyl-β-D-thiogalactoside (IPTG)–inducible constructs of these genes were transformed into a WT K-12 MG1655 E. coli strain, and clones were grown in a minimal (M9) medium supplemented with glucose and 0–1 mM IPTG (Materials and Methods). The growth inhibition resulting from the genes’ experimental overexpression was found to be highly correlated with the genes’ EDGE scores (Spearman ρ = 0.74, P < 7.56e-6; Fig. 2 and SI Appendix, Supplementary Notes and Fig. S1; all P values reported in this paper are against one-sided alternatives unless noted otherwise). These results affirm EDGE’s capability to flag potentially toxic genes, and thus facilitate the design of novel metabolic pathways.
Fig. 2.

EDGE predicts deleterious effects due to gene overexpression in E. coli. We conducted growth experiments in which we measured the growth inhibition resulting from the overexpression of 26 genes in glucose-supplemented M9 minimal medium. The genes were selected by the confidence level assigned by EDGE to the predicted outcome. Twelve genes were confidently predicted to be toxic, and 14 were confidently predicted to be nontoxic. (A) Magnitude of growth inhibition resulting from a gene’s overexpression is strongly correlated with its EDGE-predicted toxicity score (Spearman ρ = 0.74, P < 7.56e-6). (B) Histogram of the growth inhibitions resulting from overexpression of each of the 26 genes. Growth inhibition was quantified as the ratio of the fitness following IPTG-induced overexpression to the fitness with no induced overexpression (SI Appendix). Therefore, 0 denotes growth arrest and 1 denotes no inhibition compared with the WT. Three genes (trpB, ynfJ, and icd) obtained an “inhibition ratio” that was higher than 1.0, which means that the IPTG induction did not inhibit growth but rather contributed to it.
We then turned to conducting a large-scale validation through two existing genome-scale overexpression libraries: the ASKA library for E. coli (23) and the yeast GST-tagged collection for Saccharomyces cerevisiae (24). Both studies carried out systematic, large-scale overexpression of ORFs and listed genes that were toxic (i.e., caused severe growth inhibition when overexpressed) (SI Appendix, Supplementary Notes). The predicted EDGE scores of experimentally toxic genes proved to be significantly lower than those of nontoxic genes (P < 6.1e-4 and P < 7.8e-5 for ASKA and yeast Gal-GST, respectively, for a rank sum test; SI Appendix, Fig. S2). In both datasets, experimentally toxic genes were significantly enriched among the in silico detrimental genes (hypergeometric P < 1.5e-4 and P < 0.018, respectively).
EDGE Predicts Reduced Fitness Due to Overexpression of Foreign Genes Within E. coli.
So far, we were concerned with the overexpression of genes within their native host. Can EDGE predict in a similar manner the consequences of expressing foreign genes within an organism? It was previously observed that gaps in Sanger-based genome sequencing are often caused by toxic genes that cannot be expressed in an E. coli host (20). To study EDGE’s ability to predict failed gene transfer between organisms due to toxic effects, we used the recently published PanDaTox dataset (21) of genes found to be unclonable into E. coli. We simulated in silico the process of gene transfer from 50 different Gammaproteobacteria into E. coli, and then used EDGE to predict which of the heterologous genes should be toxic to the E. coli host (SI Appendix). Comparing the results with PanDaTox’s experimental data, we found that EDGE scores were highly predictive of the experimental outcome, with a median area under the ROC curve (AUC) of 0.71 when distinguishing unclonable genes from clonable genes based on their EDGE scores (median P < 0.00048; the result is significant for 42 of the Gammaproteobacteria using a 5% false discovery rate level; Dataset S1, Table S2). The focus on Gammaproteobacteria (the class to which E. coli belongs) was due to the nature of the PanDaTox data. When the source organism’s gene promoters are not recognized by the E. coli transcription machinery, little or no gene product is produced; therefore, no toxicity (i.e., “inclonability”) can be observed (20). EDGE, on the other hand, assumes by definition that the gene in question is successfully transcribed. Indeed, a clear inverse correlation was observed between EDGE’s success rate and the phylogenetic distance of the source organism from E. coli (Fig. 3; Spearman ρ = −0.32, P < 9e-5), which reflects transcriptional compatibility. Nonetheless, even when considering all 138 bacteria in the PanDaTox dataset for which data were available (SI Appendix), EDGE’s predictive power remained highly significant (combined Fisher P < 5.5e-203, median AUC = 0.67, median P value <0.0085; median AUC is larger than expected by chance with P < 1e-308; Dataset S1, Table S2). In conclusion, these results support the hypothesis that the transfer of metabolic genes (both by natural horizontal gene transfer and in metabolic engineering efforts) can be hindered by deleterious effects of transcribing them in the recipient organism, and that many of these events can be successfully predicted by EDGE.
Fig. 3.

EDGE predicts whether the transfer of foreign genes into E. coli would be successful. Each point represents one bacterial species. For each of its metabolic genes, EDGE predicted whether its transfer to E. coli would be successful, and the results were compared with previously published experimental data (20, 21). The predictor’s quality was quantified by the area under the ROC curve (AUC) (y axis). A random predictor achieves AUC = 0.5. Filled circles and empty squares represent significant and nonsignificant results, respectively, following correction for multiple hypotheses using a 5% false discovery rate level. Color codes denote the phylogenetic relatedness of the bacterium in question to E. coli according to the National Center for Biotechnology Information’s taxonomy tree (i.e., most recent common ancestor node of the bacterium and E. coli in that tree). Clearly, EDGE’s predictive power is inversely correlated with the bacterium’s phylogenetic distance (63) from E. coli (x axis; Spearman ρ = −0.32, P < 9e-5; eight bacteria that are not part of this phylogenetic tree are omitted from the figure). This is to be expected, because if the transferred genes cannot be transcribed by the host cell due to promoter dissimilarity, they would be clonable even if they are toxic.
Genes Whose Expression Impedes Growth (According to EDGE) Are Lowly Expressed Across a Wide Variety of Species and Tissues.
Next, we expected genes that EDGE predicts to be disadvantageous in a given environment to be lowly expressed in vivo when the organism grows in that environment. We tested this hypothesis by comparing extensive microarray data for E. coli and S. cerevisiae, spanning multiple growth conditions whose medium composition was recreated in silico, with EDGE predictions. In all cases, EDGE-predicted detrimental genes were significantly lowly expressed compared with genes predicted to be beneficial (Fig. 4A; rank sum test, P < 8.8e-18 for all 20 E. coli microarrays except for growth on LB with P < 1.6e-5 and P < 0.0027 for all seven yeast arrays except for one with P < 0.0165; SI Appendix, Supplementary Notes and Dataset S1, Table S3). We believe that the relatively weaker result for E. coli growth on LB medium stems from the incomplete characterization of its chemical contents (leading to its inexact definition in silico) in comparison to the synthetic M9 medium that was used as the basis for all other E. coli arrays. Similarly, the yeast data were obtained on YP medium whose exact chemical composition is unknown.
Fig. 4.

(A) E. coli genes predicted by EDGE to be toxic when overexpressed are significantly down-regulated compared with genes predicted to be nontoxic. Results are presented for 13 different growth conditions (64), as detailed in Dataset S1, Table S3A. (B) Magnitude by which EDGE-predicted toxic genes are lowly expressed compared with EDGE-predicted nontoxic genes in the case of human and plant cells (Upper two rows) resembles that observed in microorganisms (Lower two rows). The magnitude of this effect is quantified by the statistic of the Wilcoxon rank sum test, normalized to a standard normal variable (Lower x axis). The P values that match those test statistics, when considering a one-sided alternative, are given as well (Upper x axis), along with a standard normal curve for a sense of scale. Each of the four rows in the y axis represents one organism for which we tested the aforementioned effect using transcriptome data and denotes the mean value and SD of the corresponding test statistics. In the case of microorganisms, we separate experiments conducted in rich and poor, defined media because the former’s composition is uncertain, leading to inexact representation in silico, and thus to less accurate predictions.
The availability of transcriptomic data for human and plant cells allowed us to test EDGE’s applicability in these cases as well. Whereas in the case of microorganisms biomass yield is a common approximation of the cellular objective, the objective of human or plant cells is far more complex and tissue-specific. Nonetheless, even when using biomass yield as a proxy cellular objective (reflecting the need to replenish metabolites continuously due to ongoing metabolic turnover), EDGE-predicted detrimental genes were significantly lowly expressed in comparison to genes predicted to be beneficial across 79 different human tissues, 6 of them cancerous and the rest healthy (38) (rank sum P < 1.4e-5 in all arrays except for superior cervical ganglion, with P = 0.141; Dataset S1, Table S4), and across all the NCI60 cancer cell lines (39) (rank sum P < 8.6e-9 in all cases; Dataset S1, Table S5). We then analyzed in a similar manner transcriptomic data of Arabidopsis thaliana measured in 79 biological contexts and spanning multiple organs and developmental phases of the plant (40), and found a similar trend (rank sum P < 0.05 for 72 of 79 microarrays, median P < 3e-5 across all 79 arrays; SI Appendix, Supplementary Notes and Dataset S1, Table S6). Remarkably, the magnitude of this effect in human and plant transcriptomes is on the order of that we had previously observed in microorganisms (Fig. 4B). We conclude that genes that EDGE predicts to be detrimental toward proliferation are lowly expressed in diverse organisms, both in microbes and in multicellular species. The activation of these genes is thus likely to be highly undesirable and results in reduced fitness that EDGE successfully predicts. Taken together, these findings reflect a universal selection force keeping the expression of potentially deleterious genes in check.
Genetic Reprogramming in Cancer Suppresses the Expression of Proliferation-Inhibiting Genes.
We next turned to study the ability of EDGE to predict genes whose activation impedes proliferation in cancer cells. We first analyzed a dataset of DNA somatic copy number alterations in cancer (41) and found that the set of genes reported to be deleted in at least one of the cancer types in that dataset is significantly enriched with genes that EDGE identifies as detrimental with respect to biomass yield (hypergeometric P < 0.0013), whereas genes that are beneficial with respect to that objective are underrepresented (hypergeometric P < 2.5e-4). We then analyzed three studies that compared mRNA levels of cancer cells and their healthy counterparts. We found that in all three cases, the genes that were down-regulated in the cancerous cells were enriched with genes that EDGE classified as detrimental toward proliferation (SI Appendix and datasets 1–3 in Dataset S1, Table S7). Intriguingly, EDGE was also able to reveal the down-regulation of genes that impede proliferation in histologically normal breast epithelia taken from women held to be at high risk for developing breast cancer (dataset 4 in Dataset S1, Table S7).
Finally, we returned to the aforementioned dataset of gene expression of 79 healthy and cancerous human tissues. In light of the better adherence of cancer cells to the proliferation cellular objective, it was not surprising that the effect we have previously observed (namely, down-regulation of EDGE-predicted toxic genes compared with genes predicted to be nontoxic) was particularly pronounced in cancer tissues. This yields a remarkable ability to separate between cancer and benign tissues in that data based only on the correlation between the gene expression measured in each tissue and EDGE scores (AUC = 0.96, P < 1.64e-22; SI Appendix). Furthermore, when ranking the samples by the magnitude of that effect, we found that the highest ranking healthy tissues were lymphoblasts, adipocytes, and bronchial epithelial cells, all of which are known to be proliferative. In contrast, three of the four lowest ranking tissues (including the only case in which the effect was not statistically significant) were nonproliferative neuronal ganglia cells, with the fourth being another nonproliferative source of rhythmic cells in the cardiac atrioventricular node (SI Appendix, Fig. S3 and Dataset S1, Table S4). We conclude that EDGE has a clear predictive power in identifying genes whose expression potentially impedes proliferation of human cells.
Discussion
Our results that concern gene expression patterns in E. coli are in line with the findings of Lewis et al. (42), who reported that inefficient metabolic pathways are down-expressed in two strains of evolved E. coli. Here, we show that this phenomenon is universal across multiple growth conditions and holds not only in bacteria but also in eukaryotes, including humans. In addition, Lewis et al. (42) showed that the evolution of those E. coli strains was facilitated by up-regulation of optimal genes and, in many cases, by down-regulation of suboptimal genes. We extend these observations and show that the genetic reprogramming in cancer can be partially explained in a similar manner, as alterations intended to down-regulate genes that are detrimental to proliferation. The magnitude of this down-regulation is large enough to enable an almost perfect separation between normal and cancerous microarray samples based solely on EDGE results. Although such separation can be achieved through various computational methods (43), the fact that it can be achieved while relying only on EDGE-based analysis shows that EDGE successfully captures a distinct proliferative signature in the transcriptome of human cells.
Beyond its contribution to obtaining better mechanistic insights into the way gene expression levels are controlled by their potential toxicity, EDGE bears considerable applicative value for biotechnologists: Genome-scale metabolic modeling has already been successfully applied to devise novel pathways for rational strain design (12, 44–51), and gene overexpression has been considered in this framework as a means to produce a desired chemical (52, 53). EDGE complements the existing computational methods by addressing a prime concern of metabolic engineers, who seek to foresee and mitigate the deleterious effects that often accompany the introduction of a foreign metabolic pathway into a host organism, or the overexpression of one of its native genes (17, 18). Because EDGE provides a mechanistic insight for its predicted deleterious effects, it could be used to suggest further perturbations that can mitigate the expected deleterious effects of gene overexpression within an organism, or of gene transfer between organisms. These perturbations may include either media supplements, i.e., nutrients, that can abolish the rerouted fluxes and reinstate normal biomass production, or genetic perturbations (e.g., gene KOs) that may serve the same purpose.
EDGE operates on a GSMM (26, 27) (Fig. 1). As such, EDGE scores are computed only for genes that take part in the metabolic model, and their predictive power is dependent on the model’s quality. Manually curated GSMMs, which have demonstrated their predictive power, exist for many model organisms, including industrial microorganisms (28, 54) and human (55), and undergo constant improvement; plant models have been recently published as well (56). The Model SEED platform was the first to generate GSMMs automatically (57). Although these models do not outperform manually curated models at the present time (57), the Model SEED platform enables studies that require metabolic reconstructions of multiple species, for which no manually curated GSMMs are available, and it was used in this study for testing EDGE against the PanDaTox data.
WT internal flux levels are generally unavailable and cannot be computed to a sufficient precision without additional inputs beyond a GSMM (28, 58, 59). For this reason, EDGE quantifies the difference (with respect to the objective function) between having the relevant fluxes carry epsilontic and zero fluxes, rather than the difference between the WT plus epsilon and WT fluxes. However, EDGE scores are highly predictive of overexpression phenotypes even for genes that are expressed in the WT, as we demonstrate in this paper.
We expect future work to further improve EDGE’s predictive capability on three levels: first, by detecting and removing deleterious futile metabolic cycles (60); second, by taking into account the promiscuous enzymatic functions that are not normally manifested in the metabolic network but could be catalyzed by an overexpressed protein (2); and, third, by incorporating better-tuned, tissue-specific objective functions for human and plant cells. EDGE could actually facilitate the identification of such functions on account of its proven ability to detect correspondence between empirical transcriptome data and a hypothesized cellular objective.
Materials and Methods
Strain Construction.
The ASKA background strain AG1 [recA1 endA1 gyrA96 thi-1 hsdR17(rK−mK+) supE44 relA1] (23) is a thiamine auxotroph, which is unable to grow in M9 medium. We therefore transformed the plasmids into the MG1655 WT strain, enabling work in minimal M9 medium. This was also desirable because the E. coli metabolic model used in this study is based on the latter strain (28).
The ASKA GFP-tagged E. coli strain library (23) was obtained from the National Bioresource Project at the National Institute of Genetics, Japan. Plasmid DNA was isolated from strains overexpressing the required genes, and the GFP tag sequence was removed by NotI restriction followed by self-ligation. The plasmids were then transformed by electroporation to WT E. coli strain MG1655. Correct sequence of the plasmid was verified by amplifying the insert using pCA24N (ASKA backbone)-specific primers (forward: 5′-ATC ACC ATC ACC ATA CGG AT; reverse: 5′-CTG AGG TCA TTA CTG GAT CTA) and sequencing the product using the pCA24N reverse primer.
Growth Experiments.
E. coli MG1655 clones harboring plasmids of choice were picked into LB medium supplemented with chloramphenicol (34 μg/mL) and grown to OD595 = 0.5. Cells were washed once in saline and resuspended at a dilution of 1:100 into M9-glucose-chloramphenicol minimal media (1× M9 salts, 2 mM MgSO4, 0.1 mM CaCl, 0.4% glucose, 34 μg/mL chloramphenicol) supplemented with 0–1 mM IPTG. Growth measurements were performed in a 96-well plate incubated for 19–24 h at 37 °C in a temperature-controlled plate reader with continuous shaking (ELX808IU-PC; Biotek), and OD595 was monitored every 15 min. Each strain/medium combination was loaded into 2 duplicate wells. The entire growth experiment was repeated two to five times for each strain.
EDGE Algorithm.
The EDGE score of a gene, 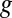 , quantifies the utility in transcribing the gene in epsilontic levels compared with not using
, quantifies the utility in transcribing the gene in epsilontic levels compared with not using 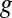 at all. This notion is strongly related to analyzing the sensitivity of a mathematical program to a perturbation of its constraints [sometimes called shadow pricing (33)]. Such analyses are fundamental to mathematical programming in general and have previously been applied to GSMMs (27, 34–36). EDGE is an adaptation of these analyses to the special setting of FBA (32). EDGE measures the sensitivity of the optimal objective value to epsilontic perturbations in a particular gene’s expression, with each gene potentially affecting multiple reactions in the network, and thus multiple constraints. For this, EDGE relies on the gene–protein-reaction mapping embedded in the model. A gene’s EDGE score is uniquely determined and is not dependent on a particular optimum, which is desirable in the case of GSMMs, whose solution space typically contains multiple optima (37).
at all. This notion is strongly related to analyzing the sensitivity of a mathematical program to a perturbation of its constraints [sometimes called shadow pricing (33)]. Such analyses are fundamental to mathematical programming in general and have previously been applied to GSMMs (27, 34–36). EDGE is an adaptation of these analyses to the special setting of FBA (32). EDGE measures the sensitivity of the optimal objective value to epsilontic perturbations in a particular gene’s expression, with each gene potentially affecting multiple reactions in the network, and thus multiple constraints. For this, EDGE relies on the gene–protein-reaction mapping embedded in the model. A gene’s EDGE score is uniquely determined and is not dependent on a particular optimum, which is desirable in the case of GSMMs, whose solution space typically contains multiple optima (37).
EDGE simulates the expression of a given gene by enforcing minimal flux through reactions associated with it; reversible reactions are constrained to carry a minimal flux through either direction. When a gene is associated with more than one reaction, we find the bottleneck reaction, whose limitation has the greatest effect, and determine the EDGE score by its utility with regard to the a priori objective function.
We now present EDGE’s full formulation. Given a gene 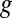 , let
, let 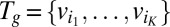 denote the set of reactions in the network that are associated with
denote the set of reactions in the network that are associated with 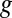 . We define:
. We define:
where  is the optimal objective subject to silencing
is the optimal objective subject to silencing 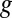 . The minuend
. The minuend 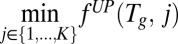 is the optimal objective subject to the most restrictive bottleneck. The difference can be further divided by epsilon for the purpose of normalization, but it was unnecessary in our study because all comparisons reported always involve the same epsilon. We note that this subtraction is prone to numerical “loss of significance” errors; for that reason, we round the result to 10 decimal places.
is the optimal objective subject to the most restrictive bottleneck. The difference can be further divided by epsilon for the purpose of normalization, but it was unnecessary in our study because all comparisons reported always involve the same epsilon. We note that this subtraction is prone to numerical “loss of significance” errors; for that reason, we round the result to 10 decimal places.
Let 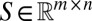 be the stoichiometric matrix of a metabolic network (where m and n are the number of metabolites and reactions in the network, respectively). Let
be the stoichiometric matrix of a metabolic network (where m and n are the number of metabolites and reactions in the network, respectively). Let 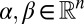 denote the lower and upper bounds, respectively, for reaction fluxes stemming from nutrient availability, thermodynamic constraints, etc.
denote the lower and upper bounds, respectively, for reaction fluxes stemming from nutrient availability, thermodynamic constraints, etc. 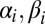 can also be set to
can also be set to 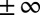 for some
for some 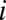 ’s to denote “no bound.” Let
’s to denote “no bound.” Let 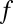 denote a linear cellular objective function to maximize subject to the environmental constraints. In our study,
denote a linear cellular objective function to maximize subject to the environmental constraints. In our study, 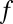 was always the biomass production.
was always the biomass production.
Define 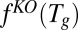 to be the optimal objective value of the following linear program:
to be the optimal objective value of the following linear program:
subject to (i) 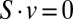 , (ii)
, (ii) 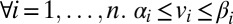 , and (iii)
, and (iii)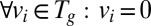 .
.
Define 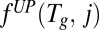 to be the optimal objective value of the following mixed integer linear program:
to be the optimal objective value of the following mixed integer linear program:
subject to (i) 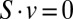 , (ii)
, (ii) 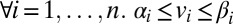 , (iii)
, (iii) 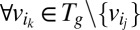 :
: 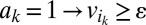 ,
, 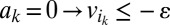 , (iv)
, (iv) 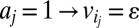 , and (v)
, and (v) 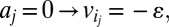 where ε is an infinitesimal constant chosen to reflect the smallest nonnegligible flux possible. However, ε cannot be arbitrarily small due to the finite precision of the floating-point representation. ak variables are binary variables whose purpose is to ensure that the reversible reactions associated with g carry a flux in either direction. They participate in logical constraints that can be transformed into regular integer linear constraints via routine transformations (61). Commercial solvers are sometimes able to branch explicitly on these constraints. We note that we described the algorithm as adding an ak variable for each reaction for the sake of simplicity. In practice, it is unnecessary to introduce an ak variable for irreversible reactions because the respective constraints for those can be simply added as linear constraints. Further implementation considerations are discussed in SI Appendix, Supplementary Methods.
where ε is an infinitesimal constant chosen to reflect the smallest nonnegligible flux possible. However, ε cannot be arbitrarily small due to the finite precision of the floating-point representation. ak variables are binary variables whose purpose is to ensure that the reversible reactions associated with g carry a flux in either direction. They participate in logical constraints that can be transformed into regular integer linear constraints via routine transformations (61). Commercial solvers are sometimes able to branch explicitly on these constraints. We note that we described the algorithm as adding an ak variable for each reaction for the sake of simplicity. In practice, it is unnecessary to introduce an ak variable for irreversible reactions because the respective constraints for those can be simply added as linear constraints. Further implementation considerations are discussed in SI Appendix, Supplementary Methods.
Genes were classified as toxic if they had a negative EDGE score and as nontoxic if they had a positive EDGE score. For the purpose of conducting growth experiments (Results), we used the absolute value of the score as the prediction’s confidence, with higher absolute values denoting the more confident predictions. Genes that were associated with a blocked reaction were excluded from the analysis (SI Appendix, Supplementary Methods).
Supplementary Material
Acknowledgments
We thank Livnat Jerby, Martin Kupiec, Nathan E. Lewis, and Roded Sharan for many fruitful discussions. This research was supported by the Israeli Centers of Research Excellence program of the Israeli Planning and Budgeting Committee and the Israel Science Foundation (Grant 41/11) and by the Seventh Framework Programme of the European Union Microme and Infect Projects. A.W. was supported, in part, by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University. R.S. was supported, in part, by National Institutes of Health Grant R01AI082376-01, Israeli Science Foundation Grant 1303/12, and European Research Council Starting Grants program Grant 260432.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312361110/-/DCSupplemental.
References
- 1.Nichols RJ, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144(1):143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prelich G. Gene overexpression: Uses, mechanisms, and interpretation. Genetics. 2012;190(3):841–854. doi: 10.1534/genetics.111.136911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van’t Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 4.Cooper-Knock J, et al. Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol. 2012;8(9):518–530. doi: 10.1038/nrneurol.2012.156. [DOI] [PubMed] [Google Scholar]
- 5.Oh SC, et al. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut. 2012;61(9):1291–1298. doi: 10.1136/gutjnl-2011-300812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graham M, Shutter JR, Sarmiento U, Sarosi I, Stark KL. Overexpression of Agrt leads to obesity in transgenic mice. Nat Genet. 1997;17(3):273–274. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- 7.Fleming SM, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human α-synuclein. J Neurosci. 2004;24(42):9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. α-Synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol Dis. 2012;47(2):258–267. doi: 10.1016/j.nbd.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinou J-C, et al. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13(4):1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 10.Parkes TL, et al. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19(2):171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 11.Betz AL, Yang G-Y, Davidson BL. Attenuation of stroke size in rats using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist in brain. J Cereb Blood Flow Metab. 1995;15(4):547–551. doi: 10.1038/jcbfm.1995.68. [DOI] [PubMed] [Google Scholar]
- 12.Lee JW, et al. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol. 2012;8(6):536–546. doi: 10.1038/nchembio.970. [DOI] [PubMed] [Google Scholar]
- 13.Keasling JD. Manufacturing molecules through metabolic engineering. Science. 2010;330(6009):1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 14.Martin VJJ, Pitera DJ, Withers ST, Newman JD, Keasling JD. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol. 2003;21(7):796–802. doi: 10.1038/nbt833. [DOI] [PubMed] [Google Scholar]
- 15.Ro D-K, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440(7086):940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 16.Steen EJ, et al. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature. 2010;463(7280):559–562. doi: 10.1038/nature08721. [DOI] [PubMed] [Google Scholar]
- 17.Pitera DJ, Paddon CJ, Newman JD, Keasling JD. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng. 2007;9(2):193–207. doi: 10.1016/j.ymben.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Carothers JM, Keasling JD. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat Biotechnol. 2012;30(4):354–359. doi: 10.1038/nbt.2149. [DOI] [PubMed] [Google Scholar]
- 19.Saïda F, Uzan M, Odaert B, Bontems F. Expression of highly toxic genes in E. coli: Special strategies and genetic tools. Curr Protein Pept Sci. 2006;7(1):47–56. doi: 10.2174/138920306775474095. [DOI] [PubMed] [Google Scholar]
- 20.Sorek R, et al. Genome-wide experimental determination of barriers to horizontal gene transfer. Science. 2007;318(5855):1449–1452. doi: 10.1126/science.1147112. [DOI] [PubMed] [Google Scholar]
- 21.Kimelman A, et al. A vast collection of microbial genes that are toxic to bacteria. Genome Res. 2012;22(4):802–809. doi: 10.1101/gr.133850.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amitai G, Sorek R. PanDaTox: A tool for accelerated metabolic engineering. Bioengineered. 2012;3(4):218–221. doi: 10.4161/bioe.20431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitagawa M, et al. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): Unique resources for biological research. DNA Res. 2005;12(5):291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 24.Sopko R, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21(3):319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Costanzo M, et al. The genetic landscape of a cell. Science. 2010;327(5964):425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol. 2012;10(4):291–305. doi: 10.1038/nrmicro2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palsson BO. Systems Biology: Properties of Reconstructed Networks. Cambridge, UK: Cambridge Univ Press; 2006. [Google Scholar]
- 28.Feist AM, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 2007;3:121. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henry CS, Zinner JF, Cohoon MP, Stevens RL. iBsu1103: A new genome-scale metabolic model of Bacillus subtilis based on SEED annotations. Genome Biol. 2009;10(6):R69. doi: 10.1186/gb-2009-10-6-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szappanos B, et al. An integrated approach to characterize genetic interaction networks in yeast metabolism. Nat Genet. 2011;43(7):656–662. doi: 10.1038/ng.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feist AM, Palsson BO. The biomass objective function. Curr Opin Microbiol. 2010;13(3):344–349. doi: 10.1016/j.mib.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28(3):245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradley SP, Hax AC, Magnanti TL. Applied Mathematical Programming. Reading, MA: Addison–Wesley; 1977. [Google Scholar]
- 34.Varma A, Boesch BW, Palsson BO. Stoichiometric interpretation of Escherichia coli glucose catabolism under various oxygenation rates. Appl Environ Microbiol. 1993;59(8):2465–2473. doi: 10.1128/aem.59.8.2465-2473.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edwards JS, Ramakrishna R, Palsson BO. Characterizing the metabolic phenotype: A phenotype phase plane analysis. Biotechnol Bioeng. 2002;77(1):27–36. doi: 10.1002/bit.10047. [DOI] [PubMed] [Google Scholar]
- 36.Reznik E, Mehta P, Segrè D. Flux imbalance analysis and the sensitivity of cellular growth to changes in metabolite pools. PLOS Comput Biol. 2013;9(8):e1003195. doi: 10.1371/journal.pcbi.1003195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng. 2003;5(4):264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Su AI, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101(16):6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JK, et al. A strategy for predicting the chemosensitivity of human cancers and its application to drug discovery. Proc Natl Acad Sci USA. 2007;104(32):13086–13091. doi: 10.1073/pnas.0610292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid M, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37(5):501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 41.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis NE, et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol. 2010;6:390. doi: 10.1038/msb.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fishel I, Kaufman A, Ruppin E. Meta-analysis of gene expression data: A predictor-based approach. Bioinformatics. 2007;23(13):1599–1606. doi: 10.1093/bioinformatics/btm149. [DOI] [PubMed] [Google Scholar]
- 44.Burgard AP, Pharkya P, Maranas CD. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84(6):647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 45.Ranganathan S, et al. An integrated computational and experimental study for overproducing fatty acids in Escherichia coli. Metab Eng. 2012;14(6):687–704. doi: 10.1016/j.ymben.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Lee KH, Park JH, Kim TY, Kim HU, Lee SY. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol Syst Biol. 2007;3:149. doi: 10.1038/msb4100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park JH, Lee KH, Kim TY, Lee SY. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci USA. 2007;104(19):7797–7802. doi: 10.1073/pnas.0702609104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becker J, Zelder O, Häfner S, Schröder H, Wittmann C. From zero to hero—Design-based systems metabolic engineering of Corynebacterium glutamicum for L-lysine production. Metab Eng. 2011;13(2):159–168. doi: 10.1016/j.ymben.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 49.Asadollahi MA, et al. Enhancing sesquiterpene production in Saccharomyces cerevisiae through in silico driven metabolic engineering. Metab Eng. 2009;11(6):328–334. doi: 10.1016/j.ymben.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Izallalen M, et al. Geobacter sulfurreducens strain engineered for increased rates of respiration. Metab Eng. 2008;10(5):267–275. doi: 10.1016/j.ymben.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Oberhardt MA, Palsson BO, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng. 2006;8(1):1–13. doi: 10.1016/j.ymben.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 53.Kim J, Reed JL. OptORF: Optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst Biol. 2010;4:53. doi: 10.1186/1752-0509-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mo ML, Palsson BO, Herrgård MJ. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Syst Biol. 2009;3:37. doi: 10.1186/1752-0509-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duarte NC, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA. 2007;104(6):1777–1782. doi: 10.1073/pnas.0610772104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Oliveira Dal’Molin CG, Quek L-E, Palfreyman RW, Brumbley SM, Nielsen LK. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010;152(2):579–589. doi: 10.1104/pp.109.148817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henry CS, et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28(9):977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 58.Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suthers PF, et al. Metabolic flux elucidation for large-scale models using 13C labeled isotopes. Metab Eng. 2007;9(5-6):387–405. doi: 10.1016/j.ymben.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinchuk GE, et al. Constraint-based model of Shewanella oneidensis MR-1 metabolism: A tool for data analysis and hypothesis generation. PLOS Comput Biol. 2010;6(6):e1000822. doi: 10.1371/journal.pcbi.1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bisschop J. AIMMS—Optimization Modeling. Haarlem, The Netherlands: Paragon Decision Technology; 2011. [Google Scholar]
- 62.Montagud A, Navarro E, Fernández de Córdoba P, Urchueguía JF, Patil KR. Reconstruction and analysis of genome-scale metabolic model of a photosynthetic bacterium. BMC Syst Biol. 2010;4:156. doi: 10.1186/1752-0509-4-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dehal PS, et al. MicrobesOnline: An integrated portal for comparative and functional genomics. Nucleic Acids Res. 2009;38(Database issue):D396–D400. doi: 10.1093/nar/gkp919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lewis NE, Cho B-K, Knight EM, Palsson BO. Gene expression profiling and the use of genome-scale in silico models of Escherichia coli for analysis: Providing context for content. J Bacteriol. 2009;191(11):3437–3444. doi: 10.1128/JB.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.