

Significance
Poly (ADP-ribose) (PAR) participates in initiating parthanatos, cell death mediated by apoptosis-inducing factor (AIF), following genotoxic stress. PAR translocation from the nucleus to cytoplasm is an essential step for AIF-mediated cell death. Here, we report that poly (ADP-ribose) glycohydrolase (PARG) enhances PAR translocation by releasing protein-free PAR fragments from nuclear acceptor proteins. ADP-ribosyl-acceptor hydrolase (ARH) 3 lowers PAR levels in the nucleus and cytoplasm, preventing PAR translocation and then parthanatos. Elucidation of the sequential action of PARG and ARH3 in the PAR-mediated pathway provides a better understanding of mechanisms underlying oxidative stress and may lead to new therapeutic targets.
Keywords: posttranslational modification, cytotoxicity
Abstract
Poly (ADP ribose) (PAR) formation catalyzed by PAR polymerase 1 in response to genotoxic stress mediates cell death due to necrosis and apoptosis. PAR glycohydrolase (PARG) has been thought to be the only enzyme responsible for hydrolysis of PAR in vivo. However, we show an alternative PAR-degradation pathway, resulting from action of ADP ribosyl-acceptor hydrolase (ARH) 3. PARG and ARH3, acting in tandem, regulate nuclear and cytoplasmic PAR degradation following hydrogen peroxide (H2O2) exposure. PAR is responsible for induction of parthanatos, a mechanism for caspase-independent cell death, triggered by apoptosis-inducing factor (AIF) release from mitochondria and its translocation to the nucleus, where it initiates DNA cleavage. PARG, by generating protein-free PAR from poly-ADP ribosylated protein, makes PAR translocation possible. A protective effect of ARH3 results from its lowering of PAR levels in the nucleus and the cytoplasm, thereby preventing release of AIF from mitochondria and its accumulation in the nucleus. Thus, PARG release of PAR attached to nuclear proteins, followed by ARH3 cleavage of PAR, is essential in regulating PAR-dependent AIF release from mitochondria and parthanatos.
Poly-ADP ribosylation is a reversible posttranslational modification of proteins, which results from the covalent attachment of branched polymers of ADP ribose moieties to amino acid residues of target proteins, in a reaction catalyzed by poly (ADP ribose) polymerases (PARP) (1–3). PARP1, a well-characterized member of the PARP family, is a nuclear protein that acts as a molecular sensor of DNA-strand breaks. Upon binding to sites of single-strand DNA breaks, PARP1 catalyzes the formation of a branched, long poly (ADP ribose) (PAR) chain attached to glutamate or aspartate residues of acceptor proteins including histones, DNA polymerases, topoisomerases, DNA ligase-2, transcription factors, and PARP1 itself (4–7). Poly-ADP ribosylation of these acceptor proteins alters their physical and biological properties, leading to DNA repair and the maintenance of genomic stability. In contrast, PARP1 overactivation, resulting from widespread DNA damage, accelerates consumption of cellular β-NAD with consequent depletion of ATP, leading to necrotic cell death (8, 9). In addition, PAR, not covalently linked to protein, may have an important role as a mediator of parthanatos, a form of caspase-independent cell death, triggered by release of apoptosis-inducing factor (AIF) from mitochondria (10–13). This type of cell death is seen in neurons after glutamate excitotoxicity and brain ischemia (11, 13). PAR, generated by PARP1 in the nucleus, translocates to the cytoplasm and binds to AIF on mitochondrial membranes, triggering its cleavage and release from mitochondria. In the cytoplasm, the process of AIF movement to the nucleus depends on its nuclear localization signal where it induces large-scale DNA fragmentation (10, 13). The two death pathways that depend on PARP1 have been demonstrated in animal models of brain ischemia, myocardial injury, Parkinson’s disease, and diabetes (14–16). Thus, because of its involvement in important biological processes, PAR metabolism is tightly controlled both spatially and temporally.
PAR glycohydrolase (PARG) has been thought of as the enzyme primarily responsible for PAR degradation (17). Unlike the multigene PARP family, the single parg gene, via alternative splicing, gives rise to isoforms with different subcellular localizations and activities (18). Full-length, 110-kDa PARG is found in the nucleus, with other forms in the cytoplasm and mitochondria (19–21). In contrast to that of PARP1, the action of PARG in response to DNA damage is still controversial. PARG-knockout mice experience early embryonic death, because of a failure to hydrolyze PAR (22). Mutant Drosophila lacking the conserved catalytic domain of PARG also died in the larval stage at the normal developmental temperature of 25 °C. Although some mutants were viable at 29 °C, they showed extensive accumulation of PAR in the central nervous system and progressive neurodegeneration with reduced locomotor activity (23). Thus, PARG appears to be an enzyme essential in development, especially of the central nervous system, through its effects on PAR degradation. However, PARG inhibitors and siRNA depletion of PARG had protective effects in animal models of stroke and hydrogen peroxide (H2O2)-induced cell death (24–27). Viable and fertile mice deficient in nuclear 110-kDa PARG were generated by targeted deletion of exons 2 and 3 of the parg gene (28). These mice appeared to be protected against renal and intestinal injury induced by ischemia/reperfusion (29, 30), although they exhibited increased sensitivity to alkylating agents and ionizing radiation (28). An explanation for dual roles of PARG in cell death may be that PARG catalyzes both exoglycosidic and endoglycosidic degradation of PAR (31), two activities that can terminate PAR metabolism, but also can cleave PAR chains attached to proteins to generate protein-free PAR, which could serve as signaling molecule, involved in induction of cell death.
Another possible pathway for PAR degradation is catalyzed by ADP ribosyl-acceptor hydrolase (ARH) 3 (32). ARH3 belongs to the ARH family, which comprises three genes, encoding 39-kDa proteins (ARH1-3) that are ubiquitous in mouse and human tissues. ARH3 catalyzes the hydrolysis of PAR to produce ADP ribose in a Mg2+-dependent manner, although its biological role under physiological or pathological conditions remains to be established.
Here, we demonstrate that ARH3 action is crucial in regulating nuclear and cytoplasmic PAR degradation following H2O2 exposure. The protective effect of ARH3 results from its ability to decrease PAR levels in the nucleus and cytoplasm, thereby preventing the release from mitochondria of PAR-driven AIF and its accumulation in the nucleus. Our data also show that PARG is critical to the generation of protein-free PAR from poly-ADP ribosylated proteins, an early step in this process. Thus, the sequential actions of PARG and ARH3 appear to regulate the formation and levels of PAR in the nucleus and cytoplasm and, thus, the cleavage and release of AIF in mitochondria, AIF translocation to the nucleus, and subsequent initiation of a DNA-fragmentation program.
Results
ARH3 Has a Protective Effect Against H2O2-Induced Cell Death.
To investigate pathways that depend on the PAR-degrading activity of ARH3 under oxidative stress induced by H2O2 exposure, we generated mouse embryonic fibroblasts (MEFs) from wild-type (WT) and ARH3−/− littermates (Fig. 1A, Upper). In WT MEFs, although ARH3 has a mitochondrial targeting sequence at its N terminus, most of the ARH3 was found in cytoplasm (65%), followed by mitochondria (25%) and nucleus (10%) (Fig. 1 B and C). Exposure to H2O2 (100–1,000 μM) for 24 h induced greater cytotoxicity with ARH3−/− than WT MEFs (Fig. 1D, Left), and overexpression of ARH3 in ARH3−/− MEFs reduced sensitivity to H2O2-induced cell death (Fig. 1A, Lower and D, Right). Localization of overexpressed ARH3 in ARH3−/− MEFs was similar to that of the endogenous protein (Fig. S1). All findings were consistent with a physiological role for ARH3 in resistance to cell death under oxidative stress induced by H2O2 exposure.
Fig. 1.

ARH3 protected against H2O2-induced cell death. (A) ARH3 expression. Cells were subjected to Western blotting by using anti-ARH3 antibody. GAPDH was used as a loading control. Note that an upper band reactive with anti-ARH3 antibody is nonspecific (n.s.). (B) Intracellular ARH3 distribution. Identity of fractions without cross-contamination was confirmed by localization of histone H3 (nucleus), tubulin, (cytoplasm), and manganese superoxide dismutase (MnSOD, mitochondria). (C) Quantification of intracellular ARH3 distribution (means ± SEM, n = 7). (D) H2O2-induced cell death. Cells were exposed to H2O2 (24 h) at indicated concentrations before assessment of cell viability (means ± SEM, n = 3). These representative data (A and B) have been replicated three times with similar results.
H2O2-Induced Cell Death in ARH3−/− MEFs Results from Caspase-Independent Apoptosis.
Because oxidative stress induced by H2O2 exposure can result in cell death by necrosis and/or apoptosis, we assessed the mode of H2O2-induced cell death in ARH3−/− MEFs. After 3- or 6-h exposure to 300 μM H2O2, ARH3−/− MEFs, but not WT MEFs and ARH3−/− MEFs expressing ARH3, exhibited nuclear shrinkage, chromatin condensation, and exposure of phosphatidylserine on the cell surface (Figs. S2 A–C and Figs. S3 A–C), which are common characteristics of apoptotic cells. Incubation with the caspase inhibitor zVAD-fmk for 1 h before H2O2 exposure did not improve viability of WT or ARH3−/− MEFs (Fig. S2D). Furthermore, H2O2 exposure did not enhance PARP1 cleavage, a hallmark of an early stage of caspase-dependent apoptosis (Fig. S2E). These results indicate that H2O2-induced death of ARH3−/− MEFs resulted from caspase-independent apoptosis.
ARH3 Alters Effects of H2O2 on Nuclear and Cytoplasmic PAR Content.
Oxidative stress induced by H2O2 exposure causes DNA damage, followed by PAR synthesis. We investigated whether ARH3 deficiency altered cellular PAR metabolism after H2O2 exposure. Before H2O2 exposure, reaction of MEFs with anti-PAR antibodies appeared faint and confined to the cytoplasm (Fig. 2 A, 0 min). At baseline, nuclear and cytoplasmic PAR levels in WT and ARH3−/− MEFs did not differ significantly (Fig. 2 A and B, 0 min). Exposure to 300 μM H2O2 increased PAR content primarily in nuclei of WT and ARH3−/− MEFs, as soon as 10 min after addition of H2O2. PAR content of nuclei reached a peak at 20 min and gradually decreased to basal levels within 1 h (Fig. 2 A and B). At all times after H2O2 exposure, however, nuclear PAR content was much greater in ARH3−/− than WT MEFs. After 20 min, cytoplasmic PAR levels of ARH3−/− MEFs appeared to increase somewhat, concomitant with decreasing nuclear PAR, whereas PAR distribution in WT MEFs did not change. Overexpression of ARH3 protein in ARH3−/− MEFs suppressed the early increase in nuclear PAR as well as its slower accumulation in cytoplasm, effects not seen in ARH3−/− MEFs transfected with EV (Fig. S4 A and B). Western blot analysis confirmed that H2O2 exposure induced greater and more prolonged elevation of PAR in ARH3−/− than WT MEFs or ARH3−/− MEFs overexpressing ARH3 (Fig. 2C and Fig. S4C). On Western blots, the greatest increase in PAR immunoreactivity was seen at ∼120 kDa, which may represent PARP1 automodified with PAR. In addition, PAR modification was more widespread in ARH3−/− than WT MEFs or ARH3−/− MEFs overexpressing ARH3. Expression of an ARH3 (D83N/D84N) inactive mutant in ARH3−/− MEFs did not alter PAR content in response to H2O2 compared with ARH3−/− MEFs transfected with EV (Fig. S4D), suggesting that ARH3 catalytic activity is required. After 2-h exposure to H2O2, PAR fluorescence appeared to overlap with the signal of the mitochondrial marker Tom20 in ARH3−/− MEFs, but not WT MEFs or ARH3−/− MEFs expressing ARH3 (Fig. 2D and Fig. S4E), indicating that ARH3 deficiency may enhance PAR accumulation in cytoplasm and mitochondria. As PARP1 and PARG are believed to be responsible for PAR synthesis and turnover, respectively, under conditions without and with oxidative stress, we questioned whether the higher PAR levels in ARH3−/− MEFs after H2O2 exposure was due to differences in activities of PARP1 and/or PARG. β-NAD consumption reflects PARP activity, which uses β-NAD to synthesize PAR. Although H2O2 exposure decreased cellular β-NAD concentration, there were no significant differences in β-NAD consumption among WT and ARH3−/− MEFs with or without overexpression of the ARH3 protein (Fig. S2F and Fig. S4F). PARP1 protein levels and PARG mRNA levels did not differ significantly among these cell lines (Fig. S2 G–I and Fig. S4 G–I). All data are consistent with the notion that ARH3 participates in nuclear and cytoplasmic PAR degradation following H2O2 exposure, although it does not appear to regulate basal PAR levels.
Fig. 2.

ARH3 regulated nuclear and cytoplasmic PAR content in response to H2O2. (A) Time-dependent PAR localization after 300 μM H2O2 exposure for indicated times. Cells were subjected to immunocytochemistry by using anti-PAR antibody (red in merged images) and DAPI staining (blue in merged images). (B) Mean PAR fluorescence in nuclei and cytoplasm (means ± SEM, n = 6–40 cells). (C) Time course of H2O2-induced PAR accumulation after 300 μM H2O2 exposure for indicated times. Cells were subjected to Western blotting by using anti-PAR antibody. GAPDH was used as a loading control. (D) PAR localization after 2-h exposure to 300 μM H2O2. Cells were subjected to immunocytochemistry by using anti-PAR (red in merged images) and Tom20 antibodies (green in merged images) and DAPI staining (blue in merged images). These representative data (A, C, and D) have been replicated three times with similar results. (Scale bars: 20 μm.)
PARP1 Catalyzes PAR Synthesis and Thereby Mediates Cell Death in Response to H2O2 in ARH3−/− MEFs.
Because our results strongly suggested PAR participation in the induction of death of ARH3−/− MEFs following H2O2 exposure, we explored the role of PARP in cell viability during oxidative stress. Pharmacological inhibition of PARP by PJ34 significantly increased the number of viable ARH3−/−, but not WT MEFs (Fig. S5A, Left). Because ARH3 also has O-acetyl-ADP ribose (OAADPr) hydrolase as well as PARG activity, and its effects could result from inhibition of SIRT1, a member of the sirtuin family of proteins responsible for OAADPr synthesis, we tested effects of PJ34 on SIRT1 activity. PJ34 did not inhibit recombinant SIRT1 activity (Fig. S5 B and C). In contrast to the effects of PARP inhibition by PJ34, neither PARP2-specific (UPF1035) nor tankyrase-specific (XAV939) inhibitors altered the sensitivity to H2O2-induced cell death (Fig. S5A, Center and Right). Because 90% of PAR accumulation after DNA-damage appeared to be associated with PARP1 activity (33), we assessed the effects of PARP1 depletion with shRNA on ARH3−/− MEFs. Stable expression of PARP1 shRNA in ARH3−/− MEFs reduced PARP1 protein to 20% of that in ARH3−/− MEFs transfected with control shRNA (Fig. 3 A and B). PARG mRNA levels in these cells were only 20% lower than those of ARH3−/− MEFs expressing control shRNA (Fig. S6A). Depletion of PARP1 protein by shRNA reduced ARH3−/− MEF sensitivity to H2O2-induced cell death (Fig. 3C), with significantly less nuclear shrinkage and chromatin condensation than seen in ARH3−/− MEFs transformed with control shRNA (Fig. S6 B and C). Using immunocytochemistry to evaluate localization of intracellular PAR, we found that depletion of PARP1 protein resulted in reduction of nuclear PAR content and decreased accumulation of cytoplasmic PAR (Fig. 3 D and E). Western blot analysis also showed reduced and more transient PAR levels in ARH3−/− MEFs expressing PARP1 shRNA (Fig. S6D). Furthermore, no PAR accumulation was seen in mitochondria after a 2-h exposure to H2O2 (Fig. S6E). Taken together, all results are consistent with the conclusion that PARP1, but neither two other PARP proteins nor SIRT1, is responsible for PAR production and for initiation of cell death in ARH3−/− MEFs after H2O2 exposure. ARH3 appears to confer protection against PARP1-mediated cell death, by preventing accumulation in the nucleus of excess PAR and its translocation to cytoplasm and mitochondria.
Fig. 3.

PARP1-mediated PAR synthesis and cell death in response to H2O2 in ARH3−/− MEFs. (A) PARP1 expression. Cells were subjected to Western blotting by using anti-PARP1 antibody. (B) Quantification of PARP1 expression levels. The amount of PARP1 protein was normalized to that of GAPDH (means ± SEM, n = 3). (C) Effect of PARP1 depletion on ARH3−/− MEFs viability after H2O2 exposure. Cells were exposed to H2O2 (24 h) at indicated concentrations before assessment of cell viability (means ± SEM, n = 3). (D) Time-dependent PAR localization after 300 μM H2O2 exposure. Cells were subjected to immunocytochemistry by using anti-PAR antibody (red in merged images) and DAPI staining (blue in merged images). (Scale bar: 20 μm.) (E) Mean PAR fluorescence in nuclei and cytoplasm (means ± SEM, n = 39–40 cells). These representative data (A and D) have been replicated three times with similar results.
ARH3 Deficiency Enhances Accumulation of Cleaved AIF to the Nucleus After H2O2 Exposure.
After PARP1 activation, excessive PAR production results in parthanatos, which is mediated by release of AIF from mitochondria (10–13). AIF was restricted to mitochondria of WT and ARH3−/− MEFs under resting conditions and, after exposure to 300 μM H2O2 for 3 or 6 h, was accumulated in nuclei of ARH3−/−, but not WT MEFs (Fig. 4). In addition, ARH3 expression or depletion of PARP1 protein in ARH3−/− MEFs prevented AIF accumulation in nuclei (Fig. S7). Longer exposure of higher concentrations of H2O2 (600 μM, 6 h) increased nuclear content of AIF in both WT and ARH3−/− MEFs expressing ARH3 protein (Fig. S8), suggesting that AIF accumulation depends on the intensity of the stimulus and lack of ARH3 protein or activity enhances the magnitude of the effect. It appears that cell death resulting from ARH3 deficiency is associated with parthanatos and involves PARP1 activation and AIF release from mitochondria and translocation to nuclei.
Fig. 4.
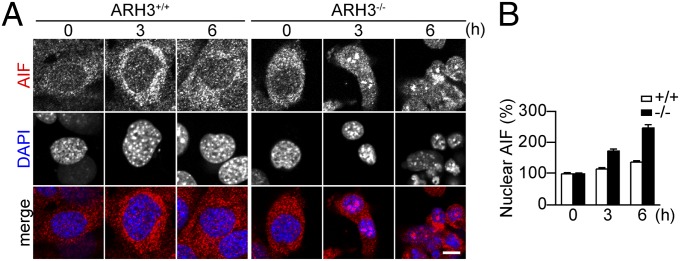
ARH3 deficiency enhanced AIF accumulation in the nucleus after H2O2 exposure. (A) AIF accumulation in nuclei of ARH3−/− MEFs after 3- or 6-h exposure to 300 μM H2O2. Cells were subjected to immunocytochemistry by using anti-AIF antibody (red in merged images) and DAPI staining (blue in merged images). (Scale bar: 10 μm.) These representative data have been replicated three times with similar results. (B) Mean AIF fluorescence in nuclei (means ± SEM, n = 36–40 cells).
Depletion of PARG Protein Protects ARH3−/− MEFs from H2O2-Induced Parthanatos, by Suppressing PAR Release from Poly-ADP Ribosylated PARP1 and its Translocation to the Cytoplasm and Mitochondria.
To explore the relationship between ARH3 and PARG, we generated ARH3−/− MEFs stably expressing PARG shRNA. Stable expression of PARG shRNA in ARH3−/− MEFs suppressed its mRNA to 65% of that seen in ARH3−/− MEFs transfected with control shRNA (Fig. 5A), whereas it did not alter PARP1 protein levels (Fig. S9 A and B). Western blot analysis revealed that PARG depletion in ARH3−/− MEFs led to a slight increase in basal PAR content as well as sustained PAR elevation following H2O2 exposure (Fig. 5B). ARH3−/− MEFs expressing PARG shRNA exhibited reduced sensitivity to H2O2 exposure (Fig. 5C) and less nuclear shrinkage in response to H2O2 than did ARH3−/− MEFs expressing control shRNA (Fig. S9 C and D). Furthermore, ARH3−/− MEFs expressing PARG shRNA failed to accumulate AIF in the nucleus (Fig. 5 D and E). These results indicate that, unlike ARH3 deficiency, PARG depletion by shRNA protected against H2O2-induced parthanatos by preventing AIF release from mitochondria.
Fig. 5.

Partial depletion of PARG enhanced PAR accumulation and decreased AIF-mediated cell death in response to H2O2 in ARH3−/− MEFs. (A) Effect of shRNA on PARG mRNA. PARG mRNA was normalized to GAPDH mRNA (means ± SEM, n = 3). (B) Time course of H2O2-induced PAR accumulation after exposure to 300 μM H2O2 for indicated times. Cells were subjected to Western blotting by using anti-PAR antibody. GAPDH was used as a loading control. (C) Effect of PARG depletion on ARH3−/− MEF viability after H2O2 exposure. Cells were exposed to H2O2 (24 h) at indicated concentrations before assessment of cell viability (means ± SEM, n = 3). (D) AIF accumulation in nuclei of ARH3−/− MEFs after 3- or 6-h exposure to 300 μM H2O2. Cells were subjected to immunocytochemistry by using anti-AIF antibody (red in merged images) and DAPI staining (blue in merged images). (E) Mean AIF fluorescence in nuclei (means ± SEM, n = 40 cells). (F) Time-dependent PAR localization after 300 μM H2O2 exposure for indicated times. Cells were subjected to immunocytochemistry by using anti-PAR antibody (red in merged images) and DAPI staining (blue in merged images). (G) Mean PAR fluorescence in nuclei and cytoplasm (means ± SEM, n = 38–40 cells). (H) Poly-ADP ribosylated PARP1 after 300 μM H2O2 exposure for indicated times. Cells were subjected to immunoprecipitation by using anti-PAR antibody and then Western blotting by using anti-PARP1 antibody to detect poly-ADP ribosylated PARP1. These representative data (B, D, F, and H) have been replicated three times with similar results. (Scale bars: D, 10 μm; F, 20 μm.)
PAR translocation to the cytoplasm and mitochondria appeared essential to trigger release of AIF from mitochondria. Using immunocytochemistry, we found that the accumulation of nuclear PAR content in ARH3−/− MEFs expressing PARG shRNA was greater and more prolonged than those of ARH3−/− MEFs expressing control shRNA (Fig. 5 F and G). ARH3−/− MEFs expressing PARG shRNA, however, failed to show slower accumulation of PAR in the cytoplasm, despite a cytoplasmic PAR content slightly higher in those cells expressing control shRNA under resting conditions (Fig. 5 F and G); thus, PARG appeared to regulate PAR translocation from the nucleus to cytoplasm. In addition, 2-h exposure to H2O2 failed to induce PAR accumulation in mitochondria of ARH3−/− MEFs expressing PARG shRNA (Fig. S9E). Because the majority of PAR is attached to PARP1 itself and PARG catalyzes hydrolysis of more protein-bound than protein-free PAR (34), we assumed a role of PARG in the release of protein-free PAR from poly-ADP ribosylated PARP1. Poly-ADP ribosylation of PARP1 in both WT and ARH3−/− MEFs was seen as soon as 20 min of H2O2 exposure. ARH3−/− MEFs expressing PARG shRNA maintained the levels for more than 1 h, whereas poly-ADP ribosylation of PARP1 in MEFs expressing control shRNA was significantly elevated only at 20 min (Fig. 5H). Our findings indicate that PARG regulates PAR release from poly-ADP ribosylated PARP and, thereby, its levels in nuclei and cytoplasm.
Discussion
The importance of PARP1 in PAR metabolism under both physiological and pathophysiological conditions has been intensively studied. There is accumulating evidence that, after genotoxic injury, activated PARP1 is crucial in initiating parthanatos, a form of caspase-independent cell death (10–13). Here, we show that, first, ARH3 can protect cells from oxidative stress-induced parthanatos, by regulating PAR degradation in nuclei and cytoplasm, and second, PARG participates in the process, by catalyzing the hydrolysis of PAR attached to PARP1 to generate protein-free PAR molecules, which can act as signals to release AIF from mitochondrial membranes (Fig. 6). Thus, both ARH3 and PARG, via their different enzymatic actions, are responsible for the hydrolysis of PAR synthesized by PARP1 in response to oxidative stress induced by H2O2 exposure.
Fig. 6.

Model for a role of ARH3 in PAR degradation and AIF-mediated cell death. Overactivation of PARP1 by widespread DNA damage results in poly-ADP ribosylation of PARP1 and other acceptor proteins in the nucleus. PARG hydrolyzes PAR attached to acceptor proteins such as PARP1 to generate protein-free small PAR molecules, thereby facilitating its translocation to the cytoplasm and mitochondria. ARH3 located in the nucleus and cytoplasm hydrolyzes PAR. PAR may bind to AIF anchored in mitochondrial membrane, releasing it to the cytoplasm. Once in the cytoplasm, AIF translocates to the nucleus via its nuclear localization signal. In nucleus, AIF recruits nucleases such as cyclophilin A and H2AX, resulting in large-scale DNA fragmentation. ARH3 is also located in the mitochondrial matrix.
PARG, which rapidly hydrolyzes PAR synthesized by PARP, had been believed to be the primary catalyst for PAR degradation (17), and complete disruption of the murine PARG gene resulted in robust PAR accumulation with early embryonic lethality (22). Because the specific activity of ARH3 is much lower than that of PARG (32), ARH3 was thought unable to compete with the PAR-hydrolyzing activity of PARG. In fact, ARH3−/− mice are healthy and fertile. ARH3 deficiency, unlike that of PARG, did not cause detectable PAR accumulation in MEFs without introduction of stress conditions. Pharmacological and genetic experiments reported here indicate that PARP1, as expected, is the primary enzyme for PAR synthesis following H2O2 exposure. PAR hydrolysis by ARH3 might have a physiological role under stress conditions when excessive PAR, synthesized by activated PARP1, accumulated in the cell.
Cytoprotective effect of PARG depletion by shRNA seemingly results from an inability to generate protein-free PAR from poly-ADP ribosylated acceptor proteins such as PARP1. PARG releases both terminal ADP ribose moieties from PAR via exoglycosidic activity and generates protein-free PAR via endoglycosidic cleavage. PARG, however, preferentially cleaves long rather than short PAR chains, because the Km value for long PAR chains is approximately ∼1% of that for small ones (35). In addition, PARG hydrolyzes covalently protein-bound PAR more rapidly than it does protein-free ones (34). We suggest that PARG not only regulates PAR metabolism, but also initiates signaling through production of small protein-free PAR molecules that can pass through nuclear pores (10). This hypothesis is consistent with our findings that PARG depletion by shRNA inhibited PAR translocation from the nucleus to cytoplasm, although it induced persistent PAR accumulation in nuclei of ARH3−/− MEFs. In addition, a recent study identified another potential candidate, terminal ADP ribose glycohydrolase TARG (C6orf130), which generates protein-free PAR from poly-ADP ribosylated proteins. In addition to PAR, TARG removes the terminal ADP ribose linked to glutamate in poly-ADP ribosylated proteins (36). TARG by generating protein-free PAR may contribute to parthanatos.
Based on the crystal structure, ARH3 appears to bind the terminal ADP ribose moiety of PAR chains (37) and, in fact, ARH3 hydrolyzes PAR to generate mainly ADP ribose (32). Because most of the ARH3 is located in the cytoplasm, followed by mitochondria and the nucleus, cytoplasmic ARH3 may catalyze degradation of protein-free PAR that is released from poly-ADP ribosylated PARP1 by PARG action. Moreover, because ARH3 deficiency resulted in nuclear accumulation of PAR, ARH3 may participate in the degradation of both protein-free and protein-attached PAR in the nucleus. It has also been postulated that ARH3 may also hydrolyze the terminal ADP ribose moiety attached to an acceptor protein, generating an ADP ribose-free state and an unmodified acceptor protein (38).
PARP1 activation and excessive PAR accumulation caused by severe DNA damage may initiate parthanatos (10–13). AIF release from mitochondria was recently reported to require its binding to PAR (13). After release, AIF translocates to the nucleus where it associates with histone H2AX and cyclophilin A, initiating large-scale DNA fragmentation (39, 40). Although most AIF resides in the inner mitochondrial membrane, 20–30% of it is found on the cytoplasmic surface of the outer mitochondrial membrane where cytoplasmic PAR can bind AIF and enhances its release (13).
Once released from mitochondria, AIF translocates to the nucleus via its nuclear localization sequence. However, recent studies suggest that cytoplasmic PARG isoforms (103 and 99 kDa) might mediate AIF translocation, by their recruitment to DNA damage sites in the nucleus (41, 42). PARG, a protein containing a macrodomain, is also associated with AIF–PAR complexes and may regulate their translocation as well as their composition (31, 43). Thus, cytoplasmic PARGs might be a cargo, which facilitates AIF translocation to the nucleus, along with its translocation. Further studies to define mechanisms involved in AIF translocation to the nucleus are needed.
In summary, ARH3 regulates nuclear and cytoplasmic PAR degradation and confers protection against H2O2-induced parthanatos. ARH3 regulates the levels of PAR following its release from poly-ADP ribosylated acceptor proteins by action of PARG. ARH3 is responsible for modulating cytoplasmic PAR content and localization, thereby regulating AIF release from mitochondria. Our observations are evidence for physiological functions of ARH3 and extend the current knowledge on complexity of PAR metabolism regulated by PARP1 and PARG.
Materials and Methods
Sources of antibodies and other specific reagents are reported in SI Materials and Methods. WT and ARH3−/− MEFs were grown in high-glucose DMEM containing 10% (vol/vol) FBS, and cell viability assay and confocal immunofluorescence microscopy experiments were performed as described in SI Materials and Methods. All data are represented as mean ± SEM. Significance was determined using paired t tests, Student's t test for pairwise comparison or a two-way ANOVA with post hoc Bonferroni test. More detailed information is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
This study was supported by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. V.L.D. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312783110/-/DCSupplemental.
References
- 1.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): Novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 2.Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26(8):882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 3.Ueda K, Hayaishi O. ADP-ribosylation. Annu Rev Biochem. 1985;54:73–100. doi: 10.1146/annurev.bi.54.070185.000445. [DOI] [PubMed] [Google Scholar]
- 4.Masson M, et al. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18(6):3563–3571. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 6.Ogata N, Ueda K, Kawaichi M, Hayaishi O. Poly(ADP-ribose) synthetase, a main acceptor of poly(ADP-ribose) in isolated nuclei. J Biol Chem. 1981;256(9):4135–4137. [PubMed] [Google Scholar]
- 7.Krupitza G, Cerutti P. ADP-ribosylation of ADPR-transferase and topoisomerase I in intact mouse epidermal cells JB6. Biochemistry. 1989;28(5):2034–2040. doi: 10.1021/bi00431a011. [DOI] [PubMed] [Google Scholar]
- 8.Szabó C, Zingarelli B, O’Connor M, Salzman AL. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci USA. 1996;93(5):1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA. 1999;96(24):13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu SW, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103(48):18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrabi SA, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103(48):18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu SW, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos) Sci Signal. 2011;4(167):ra20. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pacher P, Szabó C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: The therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev. 2007;25(3):235–260. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komjáti K, Besson VC, Szabó C. Poly (adp-ribose) polymerase inhibitors as potential therapeutic agents in stroke and neurotrauma. Curr Drug Targets CNS Neurol Disord. 2005;4(2):179–194. doi: 10.2174/1568007053544138. [DOI] [PubMed] [Google Scholar]
- 16.Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 17.Bonicalzi ME, Haince JF, Droit A, Poirier GG. Regulation of poly(ADP-ribose) metabolism by poly(ADP-ribose) glycohydrolase: Where and when? Cell Mol Life Sci. 2005;62(7-8):739–750. doi: 10.1007/s00018-004-4505-1. [DOI] [PubMed] [Google Scholar]
- 18.Meyer RG, Meyer-Ficca ML, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase (PARG) gene and the common promoter sequence it shares with inner mitochondrial membrane translocase 23 (TIM23) Gene. 2003;314:181–190. doi: 10.1016/s0378-1119(03)00738-8. [DOI] [PubMed] [Google Scholar]
- 19.Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res. 2004;297(2):521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 20.Niere M, et al. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose) J Biol Chem. 2012;287(20):16088–16102. doi: 10.1074/jbc.M112.349183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer RG, Meyer-Ficca ML, Whatcott CJ, Jacobson EL, Jacobson MK. Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-ribose) glycohydrolase (PARG) activity. Exp Cell Res. 2007;313(13):2920–2936. doi: 10.1016/j.yexcr.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koh DW, et al. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci USA. 2004;101(51):17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanai S, et al. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci USA. 2004;101(1):82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu XC, et al. Post-treatment with a novel PARG inhibitor reduces infarct in cerebral ischemia in the rat. Brain Res. 2003;978(1-2):99–103. doi: 10.1016/s0006-8993(03)02774-4. [DOI] [PubMed] [Google Scholar]
- 25.Genovese T, et al. Treatment with a novel poly(ADP-ribose) glycohydrolase inhibitor reduces development of septic shock-like syndrome induced by zymosan in mice. Crit Care Med. 2004;32(6):1365–1374. doi: 10.1097/01.ccm.0000127775.70867.0c. [DOI] [PubMed] [Google Scholar]
- 26.Erdélyi K, et al. Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. FASEB J. 2009;23(10):3553–3563. doi: 10.1096/fj.09-133264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blenn C, Althaus FR, Malanga M. Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem J. 2006;396(3):419–429. doi: 10.1042/BJ20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cortes U, et al. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol. 2004;24(16):7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuzzocrea S, et al. PARG activity mediates intestinal injury induced by splanchnic artery occlusion and reperfusion. FASEB J. 2005;19(6):558–566. doi: 10.1096/fj.04-3117com. [DOI] [PubMed] [Google Scholar]
- 30.Patel NS, et al. Mice lacking the 110-kD isoform of poly(ADP-ribose) glycohydrolase are protected against renal ischemia/reperfusion injury. J Am Soc Nephrol. 2005;16(3):712–719. doi: 10.1681/ASN.2004080677. [DOI] [PubMed] [Google Scholar]
- 31.Barkauskaite E, Jankevicius G, Ladurner AG, Ahel I, Timinszky G. The recognition and removal of cellular poly(ADP-ribose) signals. FEBS J. 2013;280(15):3491–3507. doi: 10.1111/febs.12358. [DOI] [PubMed] [Google Scholar]
- 32.Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem. 2006;281(2):705–713. doi: 10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- 33.Schreiber V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277(25):23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 34.Uchida K, et al. Preferential degradation of protein-bound (ADP-ribose)n by nuclear poly(ADP-ribose) glycohydrolase from human placenta. J Biol Chem. 1993;268(5):3194–3200. [PubMed] [Google Scholar]
- 35.Hatakeyama K, Nemoto Y, Ueda K, Hayaishi O. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose) J Biol Chem. 1986;261(32):14902–14911. [PubMed] [Google Scholar]
- 36.Sharifi R, et al. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 2013;32(9):1225–1237. doi: 10.1038/emboj.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mueller-Dieckmann C, et al. The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc Natl Acad Sci USA. 2006;103(41):15026–15031. doi: 10.1073/pnas.0606762103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleine H, et al. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol Cell. 2008;32(1):57–69. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Artus C, et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29(9):1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Candé C, et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23(8):1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- 41.Mortusewicz O, Fouquerel E, Amé JC, Leonhardt H, Schreiber V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 2011;39(12):5045–5056. doi: 10.1093/nar/gkr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haince JF, Ouellet ME, McDonald D, Hendzel MJ, Poirier GG. Dynamic relocation of poly(ADP-ribose) glycohydrolase isoforms during radiation-induced DNA damage. Biochim Biophys Acta. 2006;1763(2):226–237. doi: 10.1016/j.bbamcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Gagné JP, et al. Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 2012;40(16):7788–7805. doi: 10.1093/nar/gks486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.