

Significance
This study characterizes the initially collapsed but not yet folded denatured state ensemble of the large two-domain maltose binding protein (MBP), its contribution to the folding process, and an obligatory on-pathway native-like folding intermediate and probable further intermediates. The results add MBP to the growing list of proteins that fold through a distinct pathway with obligatory intermediates.
Keywords: SAXS, HDX, protein collapse, denatured state ensemble
Abstract
Kinetic folding of the large two-domain maltose binding protein (MBP; 370 residues) was studied at high structural resolution by an advanced hydrogen-exchange pulse-labeling mass-spectrometry method (HX MS). Dilution into folding conditions initiates a fast molecular collapse into a polyglobular conformation (<20 ms), determined by various methods including small angle X-ray scattering. The compaction produces a structurally heterogeneous state with widespread low-level HX protection and spectroscopic signals that match the equilibrium melting posttransition-state baseline. In a much slower step (7-s time constant), all of the MBP molecules, although initially heterogeneously structured, form the same distinct helix plus sheet folding intermediate with the same time constant. The intermediate is composed of segments that are distant in the MBP sequence but adjacent in the native protein where they close the longest residue-to-residue contact. Segments that are most HX protected in the early molecular collapse do not contribute to the initial intermediate, whereas the segments that do participate are among the less protected. The 7-s intermediate persists through the rest of the folding process. It contains the sites of three previously reported destabilizing mutations that greatly slow folding. These results indicate that the intermediate is an obligatory step on the MBP folding pathway. MBP then folds to the native state on a longer time scale (∼100 s), suggestively in more than one step, the first of which forms structure adjacent to the 7-s intermediate. These results add a large protein to the list of proteins known to fold through distinct native-like intermediates in distinct pathways.
Fifty years after Anfinsen’s seminal demonstration that an unfolded protein can refold spontaneously when placed under native conditions, major questions concerning the folding process remain unanswered (1, 2). What is the unfolded state like, its degree of compaction, the reality and character of residual structure before folding begins, and its possible role in guiding the folding process (3–7)? Analogous questions relate to folding intermediates and the folding pathway itself. Do proteins fold through many alternative independent pathways as earlier theoretical investigations have suggested (8–12), or do they fold through necessary intermediates in a distinct pathway (13), as a growing list of experimental observations indicate (14, 15)? To answer these questions, it will be necessary to define experimentally the intermediate forms that proteins move through on their way to the native state. The problem has been that these transient states are beyond the reach of the usual high-resolution crystallographic and NMR structural methods. Most experimental folding studies have therefore relied on low-resolution optical methods that can follow folding in real time but rarely provide the structural information necessary to resolve the basic mechanistic questions.
Recent work has demonstrated an advanced hydrogen-exchange pulse-labeling mass-spectrometry technology (HX MS) that is able to detect and characterize local structure, even when it is only transiently present during the course of kinetic folding (15, 16). The HX pulse-labeling approach provides a snapshot of main chain amide sites that are protected against HX labeling by H bonds present at the time of the labeling pulse (17, 18). HX MS measurements can determine the position, stability, and dynamic behavior of native and nonnative H-bonded structure and whether it persists or dissipates in subsequent folding. In a recent application, the method was able to describe the structure and time-dependent formation of three sequential native-like folding intermediates in the 155-residue ribonuclease H protein (15).
Protein folding studies, whether theoretical or experimental, have been limited to relatively small proteins, with few exceptions. However, biological proteomes and the considerations they raise are dominated by large proteins (19). Here we extend the powerful HX MS technology to the two-domain, 370-residue, maltose binding protein (MBP). MBP is synthesized in the Escherichia coli cytoplasm and transported to the periplasm where it serves as a soluble receptor for the high-affinity capture and import of maltose and maltodextrins (20). The protein folds in vivo after deletion of a signal sequence (21); we study here the mature protein with the signal sequence deleted.
When unfolded MBP is placed into native conditions, we find that it rapidly adopts a dynamic collapsed state, which can lead to aggregation in vitro when the concentration is >1 μM and to inclusion body formation in vivo (22). Folding to the native state occurs much more slowly even in the absence of aggregation, with all molecules moving through one or more intermediate states to the native state. The HX MS experiment provides incisive information on the nature of the initially collapsed state, the slow formation and identity of at least one on-pathway native-like intermediate, and the even slower emergence of native structure.
Results
Equilibrium Stability and Unfolding.
Previous reports have described the global unfolding of wild-type MBP with and without bound substrate analog and various mutations when driven by denaturant or temperature. In our hands denaturation follows a distorted sigmoidal curve with midpoint of the dominant unfolding phase at 0.6 M guanidinium chloride (GdmCl) and substantial posttransition baseline curvature (Fig. 1A). The melting curve can be fit by the six-parameter, two-state, Santoro–Bolen equation (23) leading to equilibrium stability variously reported up to 14.5 kcal/mol. However, the prominent posttransition MBP baseline is not well fit by the two parameters allotted by Santoro–Bolen, making extrapolation and stability analysis problematic.
Fig. 1.

Equilibrium melting and kinetic folding. (A) Equilibrium GdmCl unfolding (20 °C, pH 9), with the posttransition base line compared with kinetic burst phase signals. (B) Fast and slow refolding seen by fluorescence, CD, and ANS binding (1.4 µM 8-anilino-1-naphthalenesulfonic acid). (C) Burst phase amplitude versus GdmCl. (D) Atom pair distances by SAXS after 0.7 s of folding in 0.2 M GdmCl. (Inset) Shape reconstructions. (E) Multiple charge states of burst phase MBP by ESI MS after ∼50 ms of folding compared with unfolded (4 M urea) and native MBP (0.4 M urea). (F) Final folding rate versus solvent viscosity adjusted by glycerol.
The interpretation of equilibrium melting baselines has long been unclear. The results in Fig. 1A compare fluorescence and circular dichroism (CD) signals of the posttransition unfolded state with the signals produced in the initial burst phase refolding step described below. Similar burst phase signals have been thought to represent either a rapidly formed folding intermediate or, more simply, the still unfolded state as it exists under native conditions (24). Results described below characterize this condition for MBP and find it to represent a dynamic heterogeneously compacted condition that has little effect on the subsequent folding process except perhaps in a negative sense.
Whole Protein Folding Data.
MBP unfolded in 2 M GdmCl was diluted into native conditions (0.2 M GdmCl, pD 9, 20 °C, 0.5 μM protein) and kinetic folding was observed by a variety of optical methods (Fig. 1B). Within the 4- to 20-ms dead time of the various spectroscopic observations, MBP exhibits a burst phase increase in tryptophan fluorescence and ANS binding and the formation of 40% of its native CD corresponding to 20% helical content, followed by much slower folding to the native state. The CD result suggests the development of significant helical content, which can be observed independently by HX protection (below). Fig. 1C shows that the amplitude of the burst phase (CD and fluorescence) decreases slowly and linearly with increasing denaturant rather than sigmoidally. This result is against a specific barrier-crossing process to some defined intermediate structure.
Fig. 1D shows P(r), the distribution of pairwise atom-to-atom distances obtained by small angle X-ray scattering (SAXS) after 0.7 s of folding. The pairwise distribution function displays a form that is more compact than the unfolded state but more extended than the globular native protein, indicated by the tailing to longer interatom distances. The averaged parameter, radius of gyration (Rg), after 0.7 s of folding is 36 Å (0.2 M GdmCl) compared with 22 Å for native and 73 Å for unfolded MBP in 2 M GdmCl. Computed shape reconstructions (25) repeatedly produce envelopes like those in Fig. 1D, Inset, which we refer to as polyglobular rather than globular structure.
Fig. 1E shows the charge state distribution (CSD) produced by injecting MBP by electrospray ionization (ESI) into a mass spectrometer within ∼50 msec of initiating folding. The spectrum is shifted from the high charge state pattern characteristic of unfolded protein toward the much lower charge state distribution of the native protein, consistent with a significant compaction and reduction in surface exposure to solvent (26, 27). A small population fraction with CSD similar to that of the unfolded protein is also seen but it can be noted that the populations measured in this way are greatly biased toward exaggerating the more unfolded component (26). All of these results point to a fast molecular compaction into an extended polyglobular condition.
The fast molecular collapse is followed by unusually slow folding, with final native state acquisition on a ∼100-s time scale. Double jump experiments (Fig. 1B) designed to maintain proline residues in their native isomeric configuration (unfold for 3 s, immediately refold at 0.5 μM MBP) show modestly faster folding (approximately two times) indicating that the slow folding is affected but not determined by misisomerized proline barriers. CD and fluorescence experiments show essentially no dependence of the folding rate on solvent viscosity (glycerol; Fig. 1F). This observation suggests that polypeptide reconfiguration during the conformational searching that ultimately organizes the native structure is not limited by diffusional searching of the polypeptide chain through free solvent but rather by the difficulty of conformational reorganization within and between condensed polyglobular regions. This behavior studied in smaller proteins is commonly attributed to so-called internal friction (6, 28). Significant differences should be appreciated. The present time scale is seven orders of magnitude slower than is observed in small molecules, in part due to the degree of chain collapse, and also because the folding event requires a specific nearly simultaneous multipoint interaction rather than a general two-point interaction as for example in a FRET experiment.
In summary, upon dilution from unfolding denaturant, MBP experiences a fast molecular collapse into an ensemble of compact polyglobular forms and then folds much more slowly in a way that is limited by the difficulty of chain reconfiguration. However, these widely used methods only monitor whole molecule behavior. They provide little detailed information about structure in the compact unfolded state or the folding mechanism that produces the native state.
HX Pulse Labeling and Fragment Separation Mass Spectrometry.
To study the fast and slow stages of MBP folding at higher structural resolution, we used a quench-flow HX pulse-labeling experiment with analysis by fragment-separation mass spectrometry (Fig. 2A) (15). MBP was unfolded and spontaneously deuterated by H-to-D exchange in D2O, then diluted into folding conditions still in D2O to avoid confounding D-to-H exchange during the long prefolding time. At a series of time points during kinetic folding, the deuterated protein was exposed to a short D-to-H labeling pulse (12–43 ms, pH 9, 20 °C, where average D-to-H exchange lifetime for unprotected amides is ∼1 ms). Amides in already formed H-bonded structure tend to be protected and remain deuterated, whereas unprotected amides become protonated. The H-labeled protein was immediately quenched into a slow HX condition (pH 2.5, 0 °C), injected into an online analysis system, cleaved into small peptide fragments by acid proteolysis (pepsin), the peptide fragments were quickly separated by HPLC and mass spectrometry (LTQ Orbitrap XL) (29), and then identified and analyzed for carried D label (30).
Fig. 2.

The HX pulse-labeling experiment. (A) Deuterated unfolded MBP was mixed into folding conditions, pulsed with H2O solvent, quenched into slow HX conditions, and injected into an online flow system where peptide fragments produced by proteolysis are separated by HPLC and mass spectrometry. (B) Diagram of the 225 fragments used, placed in the primary sequence.
The 225 unique peptide fragments obtained, often with multiple charge states, are indicated in Fig. 2B. Each peptide fragment monitors the folding behavior of the protein segment that it represents. The pulse-labeling analysis provides a snapshot of the HX protection of amides in each protein segment at the time of the labeling pulse. We associate HX protection with H-bond formation (31, 32). Analysis of the peptide-bound deuterium tracks the position and time dependence of amide H-bond formation throughout the protein. The degree of resistance of D label to exchange in the H-labeling pulse measures the stability and dynamic behavior of the structure that is formed.
The Rapidly Collapsed State: Structure, Stability, Dynamics.
In the initial molecular collapse, sites in all of the peptides throughout MBP become mildly protected. HX pulse-labeling results at the 0.5-s time point (Fig. 3 A and B) show that when the D-to-H labeling pulse is increased from 12 to 43 ms (∼10–40 HX lifetimes), the number of deuterons that resist exchange decreases, indicating relatively unstable H bonding and rapid fluctuational dynamics.
Fig. 3.

HX protection in the fast collapse phase after 0.5 s of folding. (A) The response to the length of the labeling pulse is plotted in terms of fractional increase in centroid mass. (B) Peptides with broad perhaps bimodal spectra are plotted as the protected population fraction versus the pulse length. (C and D) Envelopes of typical peptides (black curves in A and B) illustrating unimodal and bimodal spectra. The blue curves in A and B represent peptides that form the 7-s intermediate (already ∼7% folded at 0.5 s). (E) Comparison of some peptides (from A and B, color coded) with helical propensity, hydrophobicity, and native conformation (Lower bar).
Most peptides appear as unimodal isotopic envelopes with low-level HX protection. Some peptides exhibit broadened envelopes, indicating protein-to-protein heterogeneity in HX protection at those positions. Fig. 3A plots the increment in centroid mass (sum of retained deuterons) as a function of pulse time for a sampling of peptides distributed through the sequence. To fit the broadened spectra, a minimum of two differently labeled fractions is required, one more protected (heavy) and one less protected (light). For these, the fraction “heavy” is plotted against pulse time in Fig. 3B. An example of each type of spectrum is in Fig. 3 C and D.
To display HX protection through the protein, the normalized centroid increment for the 12-ms pulse data are plotted as a black line through the protein sequence in Fig. 3E. Average D-occupancy levels of ∼30% corresponds to HX protection factors of ∼10–20, indicating stability against exchange in the range of 1 kcal/mol and H-bond reformation times, once HX-competent opening occurs, faster than ∼1 msec. Among the broadened spectra, four regions (90–112, 155–180, 235–275, and 340–346) show a large fractional heavy population. For these, the gray bars in Fig. 3E show the heavy fraction at the 12-ms pulse. Two regions, residues 90–112 and 340–346, show especially high fractional protection.
An Obligatory Intermediate.
From the compacted, heterogeneous, dynamic, polyglobular protein, a specific intermediate structure emerges on a longer time scale. The blue curves in Figs. 3 and 4 represent 15 peptides (residues 9–43, 60–62, and 260–278) that experience a concerted bimodal transition to a form that is highly protected against the HX labeling pulse (example in Fig. 4E). All become well protected with exponential time constant of 7 s. The number of protected sites is equal to the sites protected in the native protein. These peptides represent protein segments that are sequentially distant but are adjacent in the native structure (Fig. 4 B–D) where they come together to close the longest site-to-site loop in the native protein, analogous to some previous results (33). They form the two helices and two of the four β-strands pictured in Fig. 4C. Two intervening strands do not clearly show a bimodal transition (four peptides) because too few deuterons are incorporated, but they do protect several sites during the same ∼7-s time period, matching the number of sites expected by H bonding in the native protein. These results point to the concerted formation of the native-like substructure shown in Fig. 4 C and D (in the MBP N domain). MS envelopes for these peptides directly show that these segments undergo the same concerted structural transition in 100% of the protein population (Fig. 4E). The time-dependent folding results show that the structure, once formed, is retained through final folding (e.g., Fig. 4E).
Fig. 4.
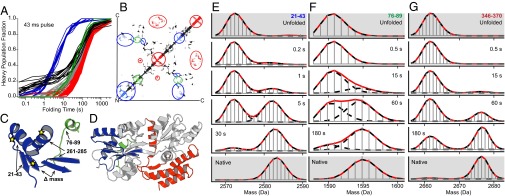
The folding history of different segments. (A) Blue and black segments are from Fig. 3B. Blue segments go on to form the 7-s intermediate, which persists through subsequent folding (E). Black curves track the early protected segments, which do not contribute to the 7-s intermediate. Segments that fold in the very slow broader grouping are graded from early (green), through gray, to late (red). (B) Contact map colored as for A. (C and D) Placement of the blue, green, and red segments in native MBP. (E–G) HX mass spectra comparing the time-dependent folding of some color-marked segments. The blue 7-s structure includes residues 9–43, 60–62, and 260–278. Residues 78–89 are shown in green; residues 180–195 and 315–370 are in red. Pulse time was 43 ms.
Previous studies have found several destabilizing MBP mutations that greatly slow folding and increase the tendency toward aggregation (V8G, Y283D, G32/I33 to D32/P33) (34–37). All are contained within the apparent 7-s intermediate structure, represented by stars in Fig. 4C. A similar double mutation at a similar αβ position in the C domain was similarly destabilizing but had no effect on folding rate (35). These results support the conclusion that the 7-s structure formation represents an obligatory, on-pathway, native-like folding intermediate. Interestingly, this first formed intermediate is placed on the solvent-exposed protein surface, not buried in the protein core (Fig. 4D).
Given that the pulse time was 43 ms (∼40 HX lifetimes) and the D occupancy is maintained at the native level, the protection of the 7-s intermediate structure against pulse labeling is >100, corresponding to ∼3 kcal/mol of stability, assuming EX2 (38) behavior, and/or a dynamic unfolding rate of less than 150 ms.
Slow Native Transition.
All of the other peptides transition to a protected state on a ∼100-s time scale (Fig. 4A), matching the spectroscopically monitored folding kinetics in Fig. 1B. These segments exhibit a spread of folding rates much broader than is seen for the 7-s class (Fig. 4A; compare also Fig. 4 E–G), suggesting that the spread is significant and that different protein segments may fold sequentially. However, the large number of peptides with halftimes between 60 and 120 s fold too closely in time to resolve them definitively into clearly separate groupings.
It is interesting that peptides that occupy the earliest part of the spread (green in Fig. 4; residues 78–89) represent MBP segments that are adjacent to the 7-s intermediate structure in the native protein. This is as expected from the sequential stabilization mechanism (14) which posits that folding pathways tend to be sequential because already formed structure tends to guide and stabilize the formation of interacting structure. The slowest folding peptides shown in red are remote, in the C-terminal domain of native MBP (Fig. 4; residues 180–195 and 315–370), also as expected for a defined sequential pathway mechanism.
The folding history of the peptides that develop high fractional HX protection in the early collapsed phase (Fig. 3) is indicated by the black curves in Fig. 4A. They do not contribute to the 7-s intermediate. The segments that do produce the 7-s intermediate (blue curves in Figs. 3 and 4) are among the least protected segments in the early condensed protein. Thus, measured protection in the early condensed state does not correlate with subsequent structure formation.
With respect to the question of multidomain folding (19), these results indicate that (parts of) the MBP N domain fold first. Reported single molecule forced unfolding experiments unfold the N domain first but this is because the pulling force is applied at the molecular N and C termini, both of which emanate from the N domain (39).
Discussion
This work used standard optical methods, SAXS, and a developing HX MS pulse-labeling method to study the folding of the large, two-domain, 370-residue maltose binding protein. Upon mixing into folding conditions, the unfolded MBP polypeptide quickly condenses. The results characterize the initially condensed state, the subsequent formation of an obligatory on-pathway intermediate, and the even slower folding to the native state.
Protein Condensation.
A quantity of work has focused on the character of the unfolded state and its possible role in guiding subsequent folding (3, 4, 6, 40–42). Is the denatured state ensemble (DSE) compact at the start of the folding process? Is significant prefolding structure present? If so, does it help to guide or hinder the folding process?
The methodology used here provides some answers. Earlier SAXS studies found that the DSE for small two-state folding proteins under native conditions has the same extended Rg as in high denaturant (43), pointing to the absence of distinct structure (2). However, when unfolded MBP is mixed into folding conditions, the 350-residue polypeptide chain rapidly condenses to a polyglobular form. It seems likely that large proteins like MBP with many more possible hydrophobic interactions will bias more toward an initially collapsed condition (44, 45). Hydrophobic interactions that drive condensation bring together sites that allow ANS-to-protein binding (Fig. 1A) and can similarly promote protein-to-protein aggregation. The same situation promotes the binding of exposed hydrophobic sites of condensed but still unfolded large proteins like MBP to the hydrophobic sites of GroEL and other chaperonins (46, 47), which shield them from aggregation during their vulnerable slow-folding time period (48).
Over time the entire MBP refolding population self-organizes, forming a distinctly structured, obligatory native-like intermediate, and then proceeds to assemble the native state. What is the relationship between structure in the initial collapsed state and subsequent folding? The general character of the collapsed state is indicated by CD and fluorescence results (Fig. 1A), which correlate with the postmelting condition, and by HX results, which indicate widespread, diverse, low-level HX protection among the various sites within each protein molecule. Especially interesting are several unusually well-protected (H-bonded) segments (in Fig. 3E; 90–112, 155–180, 235–275, and 340–346). The detailed interpretation of heterogeneous HX labeling is ambiguous. Different scenarios can be considered, depending on HX options [EX1 (38), local fluctuations, unfolding] and the rate of conformational interconversion. Some other observations are indicative. These segments are differently protected from one molecule to another. They are not selected by helical propensity or by helical conformation in the native protein. Rather, there is a correlation with high hydrophobicity, especially for the segments with the highest protection (104–112 and 340–346), which will promote chain burial (32). Unlike the segments that come together to form the 7-s intermediate, these segments do not interact in the native protein. The segments that do form the intermediate are among the less protected in the initial compaction.
These observations do not favor the possibility that early HX protection presages later structure formation. The compacted condition does contribute to the slow folding in a negative sense because it constrains chain reconfigurational searching. Smaller proteins that do not collapse fold much faster.
Nature of Protein Folding Pathways.
In the early history of the protein folding field, it was assumed that proteins fold through discrete intermediates in discrete pathways, like other biochemical pathways. Early theoretical efforts to study folding mechanisms led to a view of a heterogeneous unfolded state, reminiscent of the initial condensed state studied here. It was inferred that proteins then fold through multiple pathways guided by a funnel-shaped energy landscape (8, 10–12, 41, 49–54). Despite a dearth of experimental verification, this view is still current, and experimental as well as theoretical folding results are often phrased in this language. Some spectrometry-based experiments have been taken to suggest more than one folding pathway (55–59), but it has been shown that this kind of data cannot distinguish alternative parallel pathways from a given pathway with alternative misfolding barriers (60, 61).
A quantity of more recent experimental work using HX and associated methods has found much more organized folding behavior. Many proteins have been found to form at least one specifically structured native-like on-pathway intermediate (16–18, 62–74) and even more impressively, an organized stepwise folding sequence that progressively assembles the native protein (2, 13, 15, 75–78). The present work used an advanced mass spectrometry analysis to extend folding studies to a larger protein with multistate folding where multiple pathways, if they exist, should be more evident. Instead, the results clearly define the formation in the whole protein population of the same obligatory native-like intermediate and the subsequent formation of adjacent structure apparently in a stepwise sequential stabilization way.
This work adds to the growing list of experimental demonstrations that proteins tend to fold through distinct intermediates in distinct pathways. Recent progress in computer simulations of the course of native structure formation now finds similar folding pathways for a number of small proteins (79–81).
Materials and Methods
The E. coli apo-MBP (Protein Data Bank ID: 1OMP) used here is the wild-type mature protein without its 26-residue leader sequence. Expression was described previously (36). To ensure maltose-free protein, unfolded MBP was dialyzed exhaustively, refolded, bound to a strong cation exchange column, washed exhaustively, and tested by binding to a maltose affinity column. In equilibrium melting experiments (0.8 μM protein, 20 µM borate, pD 9.0), each sample was allowed to equilibrate to the new GdmCl concentration for 20 min before signal acquisition.
Small angle X-ray scattering experiments used the BioCAT beamline at the Argonne Advanced Photon Source (APS) as previously described (82). Kinetic SAXS measurements were performed on a home-built continuous flow mixer hooked directly to the observation capillary (folding time 0.7 s; 11 s exposure to increase signal/noise).
Previously described HX MS methods were used (29, 30, 83). Unfolded MBP, initially fully deuterated at exchangeable hydrogen sites in D2O, was allowed to fold for some predetermined time, then probed by a brief pulse of D-to-H labeling to obtain a snapshot of the structure that had been formed to that point. Initial dilution to start folding was into D2O instead of the usual H2O to avoid loss of D during the lengthy (many seconds) prepulse period. For the H-labeling pulse, the refolding protein was diluted by fivefold into H2O buffer at pH 9. Sites already protected by structure remain deuterated, dependent on their degree of structural protection relative to the strength of the labeling pulse (12–43 ms; average unprotected HX time constant ∼1 msec). Labeling was terminated by dilution into low pH (pH 2.5, 1.2 M GdmCl, ∼0 °C) where HX is slow. The quenched D:H-labeled protein was immediately injected into an online flow system (29) where the protein was cleaved into many peptide fragments in an immobilized pepsin column, the fragments were caught in a trap column, washed, roughly separated by fast HPLC (shaped gradient), and injected by ESI into the mass spectrometer (LTQ Orbitrap XL). The resulting mass spectra were analyzed by the ExMS program (30) to identify the many peptides and by an in-house program (HDpop) to measure their D label.
For the unfolded control, 2 M GdmCl was added to the refolding and pulse buffers. For the native control, refolding was allowed to continue for 1 h before being pulsed and quenched as just described. To estimate back exchange, a fully deuterated sample was used. Spectra for multiple charge states of the same peptide were added to increase signal/noise. Reproducibility of measured D/peptide in replicate experiments was ± 0.01 D/peptide. Comparison of data for multiple overlapping peptides allows many internal consistency checks. For structural analysis, we used subsets of peptides with high abundance and signal/noise (116 of the 255 in Fig. 2B).
Acknowledgments
This work was supported by National Institutes of Health (NIH) Research Grants R01 GM031847 (to S.W.E.), R01 GM055694 (to T.R.S.), a Structural Biology Predoctoral Training Grant GM08275 (to B.T.W.), and a National Science Foundation Research Grant MCB1020649 (to S.W.E.). Use of the Advanced Photon Source, an Office of Science User Facility, operated for the Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the DOE under Contract No. DE-AC02-06CH11357. This project was supported by grants from the National Center for Research Resources (2P41RR008630-18) and the National Institute of General Medical Sciences (9 P41 GM103622-18), both under NIH.
Footnotes
The authors declare no conflict of interest.
References
- 1.Dill KA, MacCallum JL. The protein-folding problem, 50 years on. Science. 2012;338(6110):1042–1046. doi: 10.1126/science.1219021. [DOI] [PubMed] [Google Scholar]
- 2.Sosnick TR, Barrick D. The folding of single domain proteins—have we reached a consensus? Curr Opin Struct Biol. 2011;21(1):12–24. doi: 10.1016/j.sbi.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haran G. How, when and why proteins collapse: The relation to folding. Curr Opin Struct Biol. 2012;22(1):14–20. doi: 10.1016/j.sbi.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lapidus LJ. Exploring the top of the protein folding funnel by experiment. Curr Opin Struct Biol. 2013;23(1):30–35. doi: 10.1016/j.sbi.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Meng W, Lyle N, Luan B, Raleigh DP, Pappu RV. Experiments and simulations show how long-range contacts can form in expanded unfolded proteins with negligible secondary structure. Proc Natl Acad Sci USA. 2013;110(6):2123–2128. doi: 10.1073/pnas.1216979110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soranno A, et al. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc Natl Acad Sci USA. 2012;109(44):17800–17806. doi: 10.1073/pnas.1117368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Udgaonkar JB. Polypeptide chain collapse and protein folding. Arch Biochem Biophys. 2013;531(1–2):24–33. doi: 10.1016/j.abb.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Brooks CL, 3rd, Gruebele M, Onuchic JN, Wolynes PG. Chemical physics of protein folding. Proc Natl Acad Sci USA. 1998;95(19):11037–11038. doi: 10.1073/pnas.95.19.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14(1):70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Wolynes PG, Onuchic JN, Thirumalai D. Navigating the folding routes. Science. 1995;267(5204):1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 11.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins. 1995;21(3):167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 12.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4(1):10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 13.Maity H, Maity M, Krishna MM, Mayne L, Englander SW. Protein folding: The stepwise assembly of foldon units. Proc Natl Acad Sci USA. 2005;102(13):4741–4746. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Englander SW, Mayne L, Krishna MM. Protein folding and misfolding: Mechanism and principles. Q Rev Biophys. 2007;40(4):287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu W, et al. Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2013;110(19):7684–7689. doi: 10.1073/pnas.1305887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan J, Han J, Borchers CH, Konermann L. Characterizing short-lived protein folding intermediates by top-down hydrogen exchange mass spectrometry. Anal Chem. 2010;82(20):8591–8597. doi: 10.1021/ac101679j. [DOI] [PubMed] [Google Scholar]
- 17.Roder H, Elöve GA, Englander SW. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature. 1988;335(6192):700–704. doi: 10.1038/335700a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Udgaonkar JB, Baldwin RL. NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature. 1988;335(6192):694–699. doi: 10.1038/335694a0. [DOI] [PubMed] [Google Scholar]
- 19.Han JH, Batey S, Nickson AA, Teichmann SA, Clarke J. The folding and evolution of multidomain proteins. Nat Rev Mol Cell Biol. 2007;8(4):319–330. doi: 10.1038/nrm2144. [DOI] [PubMed] [Google Scholar]
- 20.Nikaido H. Maltose transport system of Escherichia coli: An ABC-type transporter. FEBS Lett. 1994;346(1):55–58. doi: 10.1016/0014-5793(94)00315-7. [DOI] [PubMed] [Google Scholar]
- 21.Park S, Liu G, Topping TB, Cover WH, Randall LL. Modulation of folding pathways of exported proteins by the leader sequence. Science. 1988;239(4843):1033–1035. doi: 10.1126/science.3278378. [DOI] [PubMed] [Google Scholar]
- 22.Ganesh C, Zaidi FN, Udgaonkar JB, Varadarajan R. Reversible formation of on-pathway macroscopic aggregates during the folding of maltose binding protein. Protein Sci. 2001;10(8):1635–1644. doi: 10.1110/ps.8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27(21):8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 24.Sosnick TR, Shtilerman MD, Mayne L, Englander SW. Ultrafast signals in protein folding and the polypeptide contracted state. Proc Natl Acad Sci USA. 1997;94(16):8545–8550. doi: 10.1073/pnas.94.16.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42(2):342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konermann L, Rodriguez AD, Liu J. On the formation of highly charged gaseous ions from unfolded proteins by electrospray ionization. Anal Chem. 2012;84(15):6798–6804. doi: 10.1021/ac301298g. [DOI] [PubMed] [Google Scholar]
- 27.Hall Z, Robinson CV. Do charge state signatures guarantee protein conformations? J Am Soc Mass Spectrom. 2012;23(7):1161–1168. doi: 10.1007/s13361-012-0393-z. [DOI] [PubMed] [Google Scholar]
- 28.Waldauer SA, Bakajin O, Lapidus LJ. Extremely slow intramolecular diffusion in unfolded protein L. Proc Natl Acad Sci USA. 2010;107(31):13713–13717. doi: 10.1073/pnas.1005415107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayne L, et al. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J Am Soc Mass Spectrom. 2011;22(11):1898–1905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kan ZY, Mayne L, Chetty PS, Englander SW. ExMS: Data analysis for HX-MS experiments. J Am Soc Mass Spectrom. 2011;22(11):1906–1915. doi: 10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012;21(7):996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleming PJ, Rose GD. Do all backbone polar groups in proteins form hydrogen bonds? Protein Sci. 2005;14(7):1911–1917. doi: 10.1110/ps.051454805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krishna MM, Englander SW. The N-terminal to C-terminal motif in protein folding and function. Proc Natl Acad Sci USA. 2005;102(4):1053–1058. doi: 10.1073/pnas.0409114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betton JM, Hofnung M. Folding of a mutant maltose-binding protein of Escherichia coli which forms inclusion bodies. J Biol Chem. 1996;271(14):8046–8052. doi: 10.1074/jbc.271.14.8046. [DOI] [PubMed] [Google Scholar]
- 35.Raffy S, Sassoon N, Hofnung M, Betton JM. Tertiary structure-dependence of misfolding substitutions in loops of the maltose-binding protein. Protein Sci. 1998;7(10):2136–2142. doi: 10.1002/pro.5560071010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chun SY, Strobel S, Bassford P, Jr, Randall LL. Folding of maltose-binding protein. Evidence for the identity of the rate-determining step in vivo and in vitro. J Biol Chem. 1993;268(28):20855–20862. [PubMed] [Google Scholar]
- 37.Wang JD, Michelitsch MD, Weissman JS. GroEL-GroES-mediated protein folding requires an intact central cavity. Proc Natl Acad Sci USA. 1998;95(21):12163–12168. doi: 10.1073/pnas.95.21.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hvidt A, Nielsen SO. Hydrogen exchange in proteins. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 39.Bertz M, Rief M. Mechanical unfoldons as building blocks of maltose-binding protein. J Mol Biol. 2008;378(2):447–458. doi: 10.1016/j.jmb.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 40.Bowler BE. Residual structure in unfolded proteins. Curr Opin Struct Biol. 2012;22(1):4–13. doi: 10.1016/j.sbi.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chahine J, Nymeyer H, Leite VB, Socci ND, Onuchic JN. Specific and nonspecific collapse in protein folding funnels. Phys Rev Lett. 2002;88(16):168101. doi: 10.1103/PhysRevLett.88.168101. [DOI] [PubMed] [Google Scholar]
- 42.Orevi T, Rahamim G, Hazan G, Amir D, Haas E. The loop hypothesis: Contribution of early formed specific non-local interactions to the determination of protein folding pathways. Biophys Rev. 2013;5:85–98. doi: 10.1007/s12551-013-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plaxco KW, Millett IS, Segel DJ, Doniach S, Baker D. Chain collapse can occur concomitantly with the rate-limiting step in protein folding. Nat Struct Biol. 1999;6(6):554–556. doi: 10.1038/9329. [DOI] [PubMed] [Google Scholar]
- 44.Wu Y, Kondrashkina E, Kayatekin C, Matthews CR, Bilsel O. Microsecond acquisition of heterogeneous structure in the folding of a TIM barrel protein. Proc Natl Acad Sci USA. 2008;105(36):13367–13372. doi: 10.1073/pnas.0802788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arai M, Iwakura M, Matthews CR, Bilsel O. Microsecond subdomain folding in dihydrofolate reductase. J Mol Biol. 2011;410(2):329–342. doi: 10.1016/j.jmb.2011.04.057. [DOI] [PubMed] [Google Scholar]
- 46.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: The structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12(10):842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 47.Basha E, O’Neill H, Vierling E. Small heat shock proteins and α-crystallins: Dynamic proteins with flexible functions. Trends Biochem Sci. 2012;37(3):106–117. doi: 10.1016/j.tibs.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Houry WA, Frishman D, Eckerskorn C, Lottspeich F, Hartl FU. Identification of in vivo substrates of the chaperonin GroEL. Nature. 1999;402(6758):147–154. doi: 10.1038/45977. [DOI] [PubMed] [Google Scholar]
- 49.Plotkin SS, Onuchic JN. Understanding protein folding with energy landscape theory. Part II: Quantitative aspects. Q Rev Biophys. 2002;35(3):205–286. doi: 10.1017/s0033583502003785. [DOI] [PubMed] [Google Scholar]
- 50.Plotkin SS, Onuchic JN. Understanding protein folding with energy landscape theory. Part I: Basic concepts. Q Rev Biophys. 2002;35(2):111–167. doi: 10.1017/s0033583502003761. [DOI] [PubMed] [Google Scholar]
- 51.Onuchic JN, Nymeyer H, García AE, Chahine J, Socci ND. The energy landscape theory of protein folding: Insights into folding mechanisms and scenarios. Adv Protein Chem. 2000;53:87–152. doi: 10.1016/s0065-3233(00)53003-4. [DOI] [PubMed] [Google Scholar]
- 52.Socci ND, Onuchic JN, Wolynes PG. Protein folding mechanisms and the multidimensional folding funnel. Proteins. 1998;32(2):136–158. [PubMed] [Google Scholar]
- 53.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Theory of protein folding: The energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 54.Wolynes PG. Energy landscapes and solved protein-folding problems. Philos Trans A Math Phys. Eng Sci. 2005;363:453–467. doi: 10.1098/rsta.2004.1502. [DOI] [PubMed] [Google Scholar]
- 55.Radford SE, Dobson CM, Evans PA. The folding of hen lysozyme involves partially structured intermediates and multiple pathways. Nature. 1992;358(6384):302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 56.Wu Y, Matthews CR. Parallel channels and rate-limiting steps in complex protein folding reactions: Prolyl isomerization and the alpha subunit of Trp synthase, a TIM barrel protein. J Mol Biol. 2002;323(2):309–325. doi: 10.1016/s0022-2836(02)00922-1. [DOI] [PubMed] [Google Scholar]
- 57.Kamagata K, Sawano Y, Tanokura M, Kuwajima K. Multiple parallel-pathway folding of proline-free Staphylococcal nuclease. J Mol Biol. 2003;332(5):1143–1153. doi: 10.1016/j.jmb.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 58.Bieri O, Wildegger G, Bachmann A, Wagner C, Kiefhaber T. A salt-induced kinetic intermediate is on a new parallel pathway of lysozyme folding. Biochemistry. 1999;38(38):12460–12470. doi: 10.1021/bi9909703. [DOI] [PubMed] [Google Scholar]
- 59.Wildegger G, Kiefhaber T. Three-state model for lysozyme folding: Triangular folding mechanism with an energetically trapped intermediate. J Mol Biol. 1997;270(2):294–304. doi: 10.1006/jmbi.1997.1030. [DOI] [PubMed] [Google Scholar]
- 60.Bédard S, Krishna MM, Mayne L, Englander SW. Protein folding: Independent unrelated pathways or predetermined pathway with optional errors. Proc Natl Acad Sci USA. 2008;105(20):7182–7187. doi: 10.1073/pnas.0801864105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krishna MM, Englander SW. A unified mechanism for protein folding: Predetermined pathways with optional errors. Protein Sci. 2007;16(3):449–464. doi: 10.1110/ps.062655907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat Struct Biol. 1996;3(9):782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 63.Raschke TM, Marqusee S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat Struct Biol. 1997;4(4):298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 64.Nishimura C, Dyson HJ, Wright PE. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J Mol Biol. 2006;355(1):139–156. doi: 10.1016/j.jmb.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 65.Fuentes EJ, Wand AJ. Local dynamics and stability of apocytochrome b562 examined by hydrogen exchange. Biochemistry. 1998;37(11):3687–3698. doi: 10.1021/bi972579s. [DOI] [PubMed] [Google Scholar]
- 66.Feng HQ, Vu ND, Bai YW. Detection of a hidden folding intermediate of the third domain of PDZ. J Mol Biol. 2005;346(1):345–353. doi: 10.1016/j.jmb.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Z, Feng H, Bai Y. Detection of a hidden folding intermediate in the focal adhesion target domain: Implications for its function and folding. Proteins. 2006;65(2):259–265. doi: 10.1002/prot.21107. [DOI] [PubMed] [Google Scholar]
- 68.Kato H, Vu ND, Feng HQ, Zhou Z, Bai YW. The folding pathway of T4 lysozyme: An on-pathway hidden folding intermediate. J Mol Biol. 2007;365(3):881–891. doi: 10.1016/j.jmb.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bai Y. Protein folding pathways studied by pulsed- and native-state hydrogen exchange. Chem Rev. 2006;106(5):1757–1768. doi: 10.1021/cr040432i. [DOI] [PubMed] [Google Scholar]
- 70.Bollen YJ, Sánchez IE, van Mierlo CP. Formation of on- and off-pathway intermediates in the folding kinetics of Azotobacter vinelandii apoflavodoxin. Biochemistry. 2004;43(32):10475–10489. doi: 10.1021/bi049545m. [DOI] [PubMed] [Google Scholar]
- 71.Korzhnev DM, Religa TL, Lundström P, Fersht AR, Kay LE. The folding pathway of an FF domain: Characterization of an on-pathway intermediate state under folding conditions by (15)N, (13)C(alpha) and (13)C-methyl relaxation dispersion and (1)H/(2)H-exchange NMR spectroscopy. J Mol Biol. 2007;372(2):497–512. doi: 10.1016/j.jmb.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 72.Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329(5997):1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- 73.Gsponer J, et al. Determination of an ensemble of structures representing the intermediate state of the bacterial immunity protein Im7. Proc Natl Acad Sci USA. 2006;103(1):99–104. doi: 10.1073/pnas.0508667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kulkarni SK, et al. A near-native state on the slow refolding pathway of hen lysozyme. Protein Sci. 1999;8(1):35–44. doi: 10.1110/ps.8.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng H, Zhou Z, Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc Natl Acad Sci USA. 2005;102(14):5026–5031. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yan S, Kennedy SD, Koide S. Thermodynamic and kinetic exploration of the energy landscape of Borrelia burgdorferi OspA by native-state hydrogen exchange. J Mol Biol. 2002;323(2):363–375. doi: 10.1016/s0022-2836(02)00882-3. [DOI] [PubMed] [Google Scholar]
- 77.Sosnick TR, Hinshaw JR. Biochemistry. How proteins fold. Science. 2011;334(6055):464–465. doi: 10.1126/science.1214018. [DOI] [PubMed] [Google Scholar]
- 78.Zheng Z, Sosnick TR. Protein vivisection reveals elusive intermediates in folding. J Mol Biol. 2010;397(3):777–788. doi: 10.1016/j.jmb.2010.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334(6055):517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 80.Piana S, Lindorff-Larsen K, Shaw DE. Protein folding kinetics and thermodynamics from atomistic simulation. Proc Natl Acad Sci USA. 2012;109(44):17845–17850. doi: 10.1073/pnas.1201811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adhikari AN, Freed KF, Sosnick TR. Simplified protein models: Predicting folding pathways and structure using amino acid sequences. Phys Rev Lett. 2013;111(2):028103. doi: 10.1103/PhysRevLett.111.028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pan T, Sosnick TR. Structural analysis of RNA and RNA-protein complexes by small-angle x-ray scattering. In: Hartmann R, Schon A, Westhof E, editors. Handbook of RNA Biochemistry. Weinheim, Germany: Wiley-VCH; 2005. pp. 385–397. [Google Scholar]
- 83.Walters BT, Ricciuti A, Mayne L, Englander SW. Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J Am Soc Mass Spectrom. 2012;23(12):2132–2139. doi: 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]