

Significance
Understanding the molecular mechanisms that control secretion from cytotoxic T lymphocytes (CTL) and natural killer (NK) cells is the key for understanding how these cells destroy virally infected and tumourigenic cells. Precisely how mutations in Munc18-2 and syntaxin 11 (Stx11) give rise to loss of CTL and NK function and severe immunodeficiency is poorly understood. In this study we present a crystal structure of human Munc18-2 and analyze the disease-causing mutations. Our findings reveal a mechanism that allows Munc18-2 to selectively bind Stx11 and identify potential surrogate binding partners, which could restore Munc18-Stx function upon IL-2 activation.
Keywords: immunodeficiency, membrane trafficking, secretory lysosomes
Abstract
Mutations in either syntaxin 11 (Stx11) or Munc18-2 abolish cytotoxic T lymphocytes (CTL) and natural killer cell (NK) cytotoxicity, and give rise to familial hemophagocytic lymphohistiocytosis (FHL4 or FHL5, respectively). Although Munc18-2 is known to interact with Stx11, little is known about the molecular mechanisms governing the specificity of this interaction or how in vitro IL-2 activation leads to compensation of CTL and NK cytotoxicity. To understand how mutations in Munc18-2 give rise to disease, we have solved the structure of human Munc18-2 at 2.6 Å resolution and mapped 18 point mutations. The four surface mutations identified (R39P, L130S, E132A, P334L) map exclusively to the predicted syntaxin and soluble N-ethylmaleimide–sensitive factor accessory protein receptor binding sites of Munc18-2. We find that Munc18-2 binds the N-terminal peptide of Stx11 with a ∼20-fold higher affinity than Stx3, suggesting a potential role in selective binding. Upon IL-2 activation, levels of Stx3 are increased, favoring Munc18-2 binding when Stx11 is absent. Similarly, Munc18-1, expressed in IL-2–activated CTL, is capable of binding Stx11. These findings provide potential explanations for restoration of Munc18-Stx function and cytotoxicity in IL-2–activated cells.
Cytotoxic T lymphocytes (CTL) and natural killer (NK) cells perform a crucial role in host defense, destroying virally infected and tumourigenic cells. CTL or NK recognition of a target cell triggers polarization of secretory lysosomes, containing the cytotoxic proteins perforin and granzymes, toward the immunological synapse formed between the two cells. Fusion of secretory lysosomes with the CTL plasma membrane releases the cytotoxic proteins and destroys the targeted cell.
Granule secretion is critical for CTL and NK cytotoxicity. Mutations that disrupt this step give rise to profound immunodeficiencies, including familial hemophagocytic lymphohistiocytosis (FHL), which is characterized by fever, cytopenia, and hepatosplenomegaly, symptoms caused by hyperactive CTL and NK (1, 2). To date, four genetic loci have been associated with FHL. Disease-causing mutations have been mapped onto the genes encoding the pore-forming protein perforin (FHL type 2), the secretory regulator Munc13-4 (FHL type 3), the soluble N-ethylmaleimide–sensitive factor accessory protein receptor (SNARE) protein syntaxin 11 (Stx11, FHL type 4), and Munc18-2 (FHL type 5) (3–7).
The molecular mechanisms underlying the killing defect seen in CTL and NK from FHL2 and FHL3 patients have been relatively straightforward to understand, as loss of perforin (FHL2) or inhibition of granule secretion (FHL3) prevents target cell lysis (3, 4). However, understanding the molecular basis of FHL4 or FHL5 mutations has been complicated by the finding that cytotoxicity is often restored by in vitro activation of NK or CTL with IL-2, required for culture of these cells (5–8), limiting investigations and raising the question as to how a genetic deficiency can be overcome by in vitro culture.
Munc18-2 belongs to the Sec1/Munc18-like (SM) protein family, whose members are all ∼600 residues long and are involved in regulation of SNARE-mediated membrane fusion events (9, 10). The two closest homologs of Munc18-2 are Munc18-1, which is crucial for neurotransmitter secretion in neurons (11, 12), and Munc18-3, which is more widely expressed and is involved in Glut4 translocation (13). Stx11 is a member of the syntaxin-family of SNARE proteins, comprised of an N-terminal peptide (N peptide) followed by an autonomously folded, three-helical bundle (HABC domain) and a single helical SNARE motif. However, although most syntaxins associate with membranes through a C-terminal transmembrane domain, Stx11 is unusual in that it contains a cysteine-rich region at its C terminus, allowing for putative palmitoylation and membrane association (14).
Munc18 family proteins regulate SNARE-mediated membrane fusion by binding syntaxins and SNARE complexes, and this is reflected in their structures. All SM proteins analyzed to date adopt an arch-shaped structure formed by three distinct domains (15–19). The cavity within the arch forms the major interface for syntaxin and most likely SNARE complex binding (15, 17, 20). However, the N-terminal peptide of syntaxins extends from this cavity and binds at a spatially distinct site on Munc18 proteins, formed by an acidic groove, a basic region and a hydrophobic pocket (16, 21, 22).
Munc18 family proteins have been found to regulate SNARE-mediated membrane fusion both positively and negatively. The cocrystal structure of Stx1A with rat Munc18-1 revealed a “closed” Stx1A conformation, with the SNARE helix bound back on the HABC domain, clasped inside the central cavity of Munc18-1 (17, 20). This structural model explains how overexpression of Munc18 proteins might impair secretion by locking syntaxins in an inactive state. Conversely, the ability of the same region in Munc18-1 to bind to fully assembled SNARE complexes explains how Munc18 proteins might also facilitate vesicle docking and catalyze membrane fusion (23–26). Indeed, the yeast homolog Sec1p only binds SNARE complexes and not monomeric syntaxins (27, 28).
The functional importance of the N peptide binding site on the surface of Munc18 proteins is not entirely clear. For syntaxin binding, the N peptide is crucial for the interaction between Munc18-3 and Stx4 (29), and between Munc18-2 and Stx3 (30) but not for the interaction between Stx1A and Munc18-1 (22). It has been proposed that the N peptide initiates contact between syntaxins and Munc18 proteins and in this way may lead to high-affinity binding of the full-length syntaxin molecule (31). In polarized epithelial cells the N peptide has also been found to determine which Munc18 isoform is bound and where the syntaxin localizes (32). However, nothing is known about the role of the N peptide in the selection of syntaxin binding when two different syntaxins are both able to bind the same Munc18 protein.
An essential role for the N peptide in supporting fusion in vitro has been shown in several studies (24, 33, 34). One recent study has proposed that the role of the N peptide is to facilitate the transition of Munc18-bound syntaxins from a “closed” fusion-incompetent to an “open” conformation that allows SNARE complex formation (31, 35), although a subsequent study provides an alternative hypothesis (36). Although gene complementation in Caenorhabditis elegans in vitro studies (37, 38) and studies in cultured neurons (36) support a critical functional role for the N peptide interaction with Munc18 proteins, conflicting reports exist (39, 40).
Although the exact roles of Munc18-2 and Stx11 in CTL and NK are not known, it seems likely that these proteins function together because Munc18-2–deficient patients (FHL5) show decreased levels of Stx11 and the two proteins can be coprecipitated from cell lysates (5, 7, 41).
Studies on FHL4 and FHL5 have been hampered by the fact that CTL and NK, which need to be cultured in IL-2, often show restored cytotoxicity (5, 8, 42), suggesting that IL-2 activation can restore Munc18-Stx function. The molecular basis for this is completely unexplored.
In this study we ask how Munc18-2 and Stx11 function is linked in CTL and NK, and whether the Stx11 N peptide plays a functional role in Munc18-2 binding. We have solved the crystal structure of human Munc18-2 to 2.6 Å resolution and mapped point mutations that lead to FHL5. Our study identifies four disease-causing surface mutations in Munc18-2, all of which map to either the syntaxin or SNARE binding domains. Using biophysical techniques we reveal that the syntaxin N peptide interaction is likely to be important for the selection of Stx11 over Stx3 by Munc18-2. Furthermore, we analyzed changes in syntaxin protein levels that occur upon activation of resting NK with IL-2, and propose a molecular mechanism for the restoration of cytotoxicity in FHL4 and FHL5 upon IL-2 activation.
Results
NHABC Domain of Stx11 Facilitates the Interaction with Munc18-2.
Previous studies showed an interaction between Munc18-2 and Stx11 (5, 7, 41). However, it remained unclear whether that interaction was direct or mediated by a SNARE complex. Therefore, we asked whether Stx11 and Munc18-2 were able to bind each other directly and if so, which domains of Stx11 mediated the interaction. We purified three different Stx11 fusion proteins via a C-terminal or N-terminal GST tag from Escherichia coli (Fig. 1A) and tested for binding of recombinant human Munc18-2 [See Fig. S1 for size-exclusion chromatography (SEC) profiles]. Fig 1B shows that the 66-kDa protein Munc18-2 (lane 6) binds directly to full-length Stx11 (Stx11ΔC) and a construct spanning the N-peptide and HABC domain (Stx11-NHABC, lanes 3 and 4), whereas no significant binding could be observed to the SNARE domain of Stx11 (lane 5). There was no interaction with GST alone (Fig 1B, lane 2) and no Munc18-2 was pulled down in the absence of bait (Fig 1B, lane 1). We further investigated the specificity of the interaction by asking whether Stx11 could bind to human Vps33A, another member of the SM protein family. As shown in Fig 1B lanes 7–12, Vps33A did not bind to any of the Stx11 fusion proteins. These results show that Munc18-2 and Stx11 interact directly via the NHABC domain in a specific fashion.
Fig. 1.

Stx11 NHABC domain facilitates Munc18-2 interaction. (A) Schematic representation of truncation mutants of human Stx11 and localization of the GST tag. The amino acid sequence of the N peptide is written in brackets at the top. C, Cysteine-rich region (amino acids 272–287); NHABC, a folded domain comprising the N peptide and helices HA, HB, and HC. (B) Coomassie-stained gel from GST pull-down experiment of human Munc18-2 (lanes 1–6) or human Vps33A (lanes 7–12) using the fusion proteins described in A as bait. Dashes at the side refer to the respective heights of the proteins as described in the text. Pull-down of Munc18-2 was performed at least three times; pull-down using Vps33A was performed twice.
Crystal Structure of Munc18-2.
To better understand this interaction and how mutations found in FHL5 patients might affect Munc18-2 function, we crystallized human Munc18-2 and determined its 3D structure. Crystals diffracted to a resolution of 2.6 Å and the structure was solved by molecular replacement, revealing one molecule of Munc18-2 per asymmetric unit. The structure was manually rebuilt using one molecule of rat Munc18-1 [PDB ID code 3C98 (22)] as a starting model and refined to 2.6 Å, with final residuals R/Rfree 0.243/0.275 and excellent stereochemistry (Table S1; see Fig. S2 for typical electron density).
The structure of human Munc18-2 (Fig. 2A) resembles its mammalian orthologs rat Munc18-1 (64% sequence identity, 1.4 Å rmsd across 514 Cα atoms) and Munc18-3 (49% sequence identity, 1.7 Å rmsd across 484 Cα atoms) (Fig. 2B) in that the protein can be segregated into three domains: domain 1 (blue in Fig. 2A) comprises the N terminus and together with domain 2 (green in Fig. 2A) forms one-half of the arch-shaped structure, whereas the other half of the arch is predominantly formed by domain 3 (pink in Fig. 2A). Importantly, two structural areas that were previously reported to be crucial for Munc18 protein function are also conserved: the central cavity formed by domains 1 and 3 at the top of the molecule and the N peptide binding site just beneath domain 1, comprising an acidic region and a large hydrophobic pocket (Fig. 2A, Inset). Strikingly, we found an enlarged hydrophobic patch on the peptide binding surface of Munc18-2, compared with Munc18-3, adjacent to the L8 anchoring residue binding pocket (see, for example, Fig. S3A).
Fig. 2.

Structure of human Munc18-2. (A) Cartoon representation of the structure of human Munc18-2 with domain 1 colored in blue, domain 2 in green, and domain 3 shown in pink. The arrow points at the conserved central cavity at the top of the molecule. Inset to the right shows an electrostatic surface representation of the N peptide binding site with a modeled peptide of mouse Stx4. The acidic groove (red, −5 kT), basic patches (blue, +5 kT), and the hydrophobic pocket (white) are highlighted. (B) Structural similarity of rat Munc18-1 (PDB ID code 3C98, Right) (22) and mouse Munc18-3 (PDB ID code 3PUK, Left) (16) to human Munc18-2 (Center). Calculated sequence identity, rmsd, and number of aligned Cα atoms are annotated.
Disease-Causing FHL5 Mutations Predominantly Affect Protein Folding.
Because loss of Munc18-2 causes severe and often lethal immunodeficiency, the structure presented in Fig. 2A offered a unique opportunity to map FHL-causing mutations directly onto the affected protein and to analyze how they could disrupt protein function. We mapped 18 FHL5-causing point mutations, including two newly identified mutations (Table S2), onto the structure of human Munc18-2 (Fig. 3A, pink spheres) and found that 14 lay buried within the core of the structure [Table S2, accessible surface area (ASA) and buried (B) values, columns 4 and 5]. Using our structure, we examined how the buried mutations would change intramolecular interactions, such as hydrogen bonds, salt bridges, or hydrophobic interactions. We further analyzed the stereochemical properties of the mutations, considering whether the introduced side chain could be accommodated or created a void and whether the backbone dihedral angles remained favored (Table S2, structural features). This analysis suggested that the mutations mapped to buried residues within the core of human Munc18-2 would most likely disrupt its structural stability and result in loss of function. However, our mapping also identified four mutations, which lay on the surface of Munc18-2, R39P, L130S, E132A, and P334L (Table S2, ASA values of 52.8%, 31.9%, 38.6%, and 79.3%, respectively), suggesting that these might affect critical interactions with other proteins. We investigated three of these mutations further.
Fig. 3.

Mutation P334L does not affect Stx11 binding. (A) Single point mutations from Table S2 mapped as pink spheres onto a tube representation of human Munc18-2. Insets show magnification of the area around residue P334 (Left) or L130 (Right), the molecular surface of Munc18-2 being shown in the latter. (B) Surface representation of human Munc18-2 (cyan) with residue R39P highlighted in pink. A model of Stx1A bound to Munc18-2, generated by superposition of the Munc18-1:Stx1A complex (22) onto Munc18-2 is shown as an orange tube. Inset to the right shows a magnification of the mutated site, highlighting the intramolecular hydrogen bond formed within the α-helix and to D148, as well as the close proximity to E234 of the modeled Stx1A molecule. (C) Coomassie-stained gel from GST pull-down experiment of WT (lanes 1–6) or P334L (lanes 7–12) Munc18-2 with the fusion proteins described in Fig. 1A as bait. Dashes at the side refer to the respective heights of the proteins. Pull-down of WT Munc18-2 was performed at least three times, pull-down using P334L Munc18-2 was performed twice. The lower molecular weight band in lane 12 (*) is most likely the result of degradation of P334L Munc18-2, and we note that this fragment is also pulled-down by full-length and the NHABC domain of Stx11.
Superposing the Munc18-1:Stx1A complex (22) onto Munc18-2 suggested that R39 could form a hydrogen bond to E234 of Stx1A (Fig. 3B). To assess whether this interaction was lost in cells carrying this disease-causing mutation, we expressed the R39P mutant of Munc18-2 as a GST fusion protein. The mutant protein eluted in the void volume of a SEC column (Fig. S4A), suggesting that substituting proline for arginine at this site prevented correct protein folding and most likely the production of functional Munc18-2 protein in vivo.
The P334L mutant of Munc18-2 (Fig. 3A, Left Inset) was readily expressed as a soluble protein in insect cells. However, molecular-weight determination using SEC with in-line multiangle light scattering, which determines the absolute molar mass of a molecule based on the intensity and angle at which polarized light is scattered, revealed that this mutation enhanced the propensity of Munc18-2 to form dimers (Fig. S4B). Intriguingly, the P334L mutation and consequent dimerization did not affect the interaction with Stx11, as shown in Fig. 3C.
Mutation E132A in Munc18-2 Abolishes Stx11 Binding by Destroying the N Peptide Interaction.
Mutations L130S and E132A, like R39P, lie at a syntaxin interaction site, specifically at the site of N peptide binding. Although L130 forms part of the hydrophobic pocket (Fig. 3A, Right Inset), the model in Fig. 4A and previous work suggested that E132 forms a salt bridge to R4 of the bound syntaxin. We decided to concentrate on E132A, as the mutation was identified in a homozygous patient with “full-blown” hemophagocytic lymphohistiocytosis (43), and tested if, and to what extent, mutation E132A would affect syntaxin binding.
Fig. 4.

The N peptide is critical for Stx11 interaction with Munc18-2. (A) Surface representation of the structure of human Munc18-2 (cyan), with mutation E132A highlighted in pink and Stx1A [from PDB ID code 3C98 (22)] modeled in orange (as in Fig. 3). Inset to the right shows a magnification of the mutated site, with residue R4 of the superposed Stx1A molecule predicted to form a salt bridge with E132 of Munc18-2. (B) CD spectra of WT and E132A Munc18-2 showing normal folding of the mutant protein. Measurements were performed twice. (C) Melting curves of WT and E132A mutant Munc18-2 measured by differential scanning fluorimetry. Traces are representative of six repeat experiments. (D–F) ITC quantitating the binding of residues 1–12 of Stx11 to WT (D and circles in F) and E132A (E and triangles in F) Munc18-2. The Stx11 peptide and WT Munc18-2 formed a near unimolar complex (n = 0.78) with a KD of 0.55 ± 0.05 μM calculated as the mean ± SEM of five independent experiments. No measurable interaction was observed between the peptide and the E132A mutant of Munc18-2 (representative of two independent experiments). (G) Coomassie-stained gel of a GST pull-down using Stx11ΔC-GST, GST-Stx11-NHABC, GST-Stx11-SNARE, or GST alone as bait, probing for an interaction with WT (lanes 1–6) or E132A (lanes 7–12) Munc18-2. Image representative for one of three repeat experiments. Sizes of bait and Munc18-2 proteins are indicated by the dashes. The lower molecular weight band in lane 12 (*) is most likely the result of degradation of E132A Munc18-2 but is not pulled-down by full-length or the NHABC domain of Stx11. (H) Amino acid sequence alignment of the N peptides of human Stx3, 4, and 11, rat Stx1A, and mouse Stx4. R4 is highlighted in red and L8 in green. Sequences were aligned with ClustalW2 (EMBL-EBI) and annotated in ALINE (69).
We purified E132A Munc18-2 from insect cells (see Fig. S1B for comparative SEC profile) and confirmed that the E132A mutation did not disrupt protein folding by using circular dichroism (CD) to measure the secondary structural characteristics of WT and E132A Munc18-2. The CD spectra of these two proteins overlaid very well (Fig. 4B), indicating that E132A Munc18-2 was folded like the WT protein. Next we assessed the thermal stability of the mutant protein using differential scanning fluorimetry (44), where the “melting” of a protein to expose its hydrophobic core is assessed by measuring the binding of a hydrophobic dye across increasing temperatures. We found that E132A had the same melting curve as the WT protein, confirming that this mutation did not destabilize the protein (Fig. 4C).
We asked whether the N peptide of Stx11 bound to WT Munc18-2 and if E132A had an effect on that interaction by measuring the binding of the Stx11 peptide to WT Munc18-2 or the E132A mutant using isothermal titration calorimetry (ITC). The peptide bound to WT Munc18-2 in an exothermic reaction of near unimolar stoichiometry and a binding affinity of 0.55 μM (Fig. 4D and circles in Fig. 4F). Strikingly, we could not detect any binding of the Stx11 peptide to E132A Munc18-2 (Fig. 4E and triangles in Fig. 4F), suggesting that the salt bridge formed to R4 is crucial for the binding of the entire N peptide of Stx11.
To determine whether abolition of the N peptide interaction was sufficient to disrupt the binding to Stx11ΔC or the NHABC domain, we performed pull-downs of WT or E132A Munc18-2 using Stx11ΔC-GST, GST-Stx11-NHABC, or GST-Stx11-SNARE as bait. As shown earlier, WT Munc18-2 predominantly bound Stx11 via the NHABC domain (Fig. 4G, lanes 1–6) in a specific fashion. E132A Munc18-2, however, bound neither Stx11ΔC-GST nor GST-Stx11-NHABC (Fig. 4G, lanes 9 and 10). Just as the WT protein, E132A Munc18-2 also failed to bind the SNARE domain alone (Fig. 4G, lane 11) and no binding was observed for the GST control (Fig. 4G, lane 8). These results show that the N peptide binding is a key determinant for complex formation between Stx11 and Munc18-2.
Earlier work identified R4 and L8 as the key anchor points for N peptide binding (16) (Fig. 4H). However, when we looked at the framing residues among different syntaxins, we noticed a striking difference between Stx11 and Stx4/Stx1A. Instead of a polar acidic residue in position 9 (+1 position), Stx11 has a nonpolar leucine residue (see alignment in Fig. 4H), which will be better accommodated by the extended hydrophobic region adjacent to the binding pocket for L8 found in Munc18-2 compared with Munc18-3 (Fig. S4A).
Up-Regulation of Stx3 in Activated NK.
CTL and NK cytotoxicity depend on both Stx11 and Munc18-2, as loss of either protein causes immunodeficiency. However, in vitro activation with IL-2 often rescues cytotoxicity, implying that the Munc18-Stx function might somehow be restored. We therefore asked whether compensatory mechanisms were initiated in both FHL4 and FHL5 CTL and NK upon IL-2 activation.
We started by looking at the changes in Stx3, Stx4, and Stx11 expression upon activation of NK. We purified resting NK from healthy individuals and harvested cells either directly after purification (d0) or incubated them with human IL-2 for 4 d (d4). Cells from each time point were lysed and the level of different syntaxin proteins was assessed by Western blotting. We found that stimulation of human NK with IL-2 led to an increase of Stx3 protein level (Fig. 5A, Left), whereas the protein levels of both Stx4 and Stx11 itself were unaffected (Fig. 5A, Center and Right). Quantitation of band intensity relative to calnexin confirmed a 10-fold increase (P < 0.05) in expression of Stx3 following IL-2 treatment (Fig. 5B). Because this observation was made using NK from healthy individuals, we analyzed Stx3 protein levels in FHL4 patient cells to ascertain that Stx3 was not affected by loss of Stx11. We found that Stx3 levels, like Stx4 and Munc18-2 levels, were unchanged in IL-2–activated CTL from an FHL4 patient (Fig. 5C).
Fig. 5.
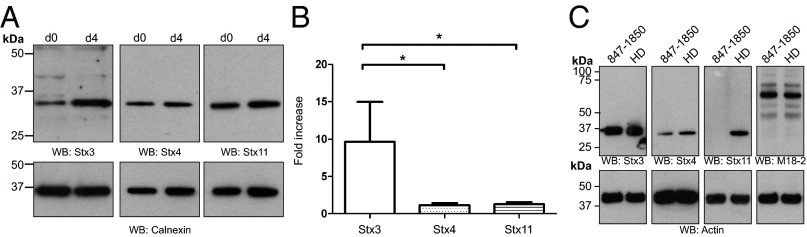
Stx3 is up-regulated in activated NK and not affected by loss of Stx11 in FHL4 patient CTL. (A) Western blot of different syntaxins in resting (d0) and activated (d4) human NK from healthy individuals. Proteins from lysates were separated by SDS/PAGE, transferred onto nitrocellulose and probed with protein-specific antibodies against Stx3, Stx4, Stx11 (Upper) or Calnexin (Lower). (B) Statistical analysis of syntaxin levels presented in A. Protein band intensity was measured using ImageJ and statistical analysis was performed in GraphPad using a one-way ANOVA test, followed by a Tukey’s multicomparison test. Statistical significance with P < 0.05 is indicated by the asterisk. Data were from four unrelated, healthy individuals, each assayed twice. Presented data are the mean ± SEM. (C) Protein abundance of Stx3, Stx4, Stx11, Munc18-2, and actin in CTL from a healthy donor (HD) and an FHL4 patient (847-1850; homozygous AL135917:g.25561-44749_del) after activation with IL-2 was assessed by Western blot using protein-specific antibodies.
Because Stx3 was expressed in FHL4 patient cells, shares 31% sequence identity with Stx11, and can interact with Munc18-2 (30), we wondered whether Stx3 and Stx11 might show some functional redundancy. We therefore tested whether the N peptide of Stx3 also bound Munc18-2 by measuring the binding affinity of residues 1–12 of Stx3 to both WT and E132A Munc18-2 by ITC. Similar to Stx11, the Stx3 peptide bound to WT Munc18-2 and exhibited the same dependency on the salt bridge to R4 because the peptide no longer bound to E132A Munc18-2 (Fig. 6 A–C). Interestingly, WT Munc18-2 bound the N peptide of Stx3 with an affinity of only 11 μM, ∼20-fold weaker than the binding to the Stx11 peptide.
Fig. 6.

Stx3 also binds to Munc18-2 in a peptide-dependent manner. (A–C) ITC measurement of the binding of residues 1–12 of Stx3 to WT (A and circles in C) and E132A (B and triangles in C) Munc18-2. The peptide and WT Munc18-2 formed a unimolar complex (n = 0.96) with KD of 11 ± 1 μM calculated as the mean ± SEM of three independent experiments. No measurable interaction was observed between the peptide and the E132A mutant of Munc18-2 (representative of two independent experiments). (D) GST pull-down of WT (lanes 1–4) and E132A (lanes 5–8) Munc18-2 using Stx3ΔTM-GST or GST alone as bait. Image shows a Coomassie-stained gel from one out of two independent experiments. The size of the baits and Munc18-2 is indicated by the dashes. (E–H) Munc18-2 binding to the cytosolic domains of Stx11 and Stx3. Shown are sensorgrams for the concentration-dependent binding of either WT (E and F) or mutant (G and H) Munc18-2 to Stx11 or Stx3 captured on a CM5 sensor chip. For E, F, and H, background signal for binding to GST alone was subtracted before fitting a 1:1 Langmuir binding model (thin lines). No signal above background to GST alone could be detected for G. Binding affinities are the mean ± SEM from two independent experiments. Association and dissociation constants are provided in Table S3.
Because E132A Munc18-2 specifically destroyed the N peptide interaction, we used the mutant protein to assess the dependency on N peptide engagement for full-length Stx3 binding. We expressed and purified GST-tagged Stx3 (without the transmembrane region, Stx3ΔTM-GST) in E. coli and performed a pull-down with both WT and E132A Munc18-2. As expected, WT Munc18-2 bound directly to Stx3ΔTM (Fig. 6D, lane 3). However, by abolishing the interaction with the N peptide, via the E132A mutation, Munc18-2 binding to Stx3ΔTM was severely reduced but still detectable (Fig. 6D, lane 7).
To confirm the interaction between Stx3 and E132A Munc18-2, we measured the binding strength of Stx3 and Stx11 to both WT and E132A Munc18-2 by surface plasmon resonance. As shown in Fig. 6 E and F, Stx11 and Stx3 bound to WT Munc18-2 with similar affinities (30 nM and 27 nM, respectively). Performing the same experiment with the E132A Munc18-2; however, no response above background was measured for Stx11 (Fig. 6G), whereas Stx3 still bound E132A Munc18-2, albeit with 1,000-fold weaker affinity than the WT protein (Fig. 6H).
These results show that both Stx11 and Stx3 bind Munc18-2 in an N peptide-dependent manner (see also Fig. S3B), similar to the interaction previously reported for Munc18-3 and Stx4 (29), and are consistent with a model in which the N peptide serves as a selection determinant for syntaxin binding (31, 32). Our results suggest that if complex formation is initiated by N peptide binding Munc18-2 will bind Stx11 in preference to Stx3 when both syntaxins are present. However, because the strength of binding of Munc18-2 to both full-length syntaxins is similar, functional SNARE complexes may be formed equally well with either syntaxin. Thus, in the absence of Stx11, Stx3 could interact with Munc18-2 and form a functional SNARE complex, thereby possibly compensating the missing step required for cytotoxicity.
Munc18-1 Is Expressed in Human CTL and NK and Interacts with Stx3 and Stx11.
Because our data suggest a compensation mechanism for loss of Stx11, we were curious as to how IL-2 activation might compensate the loss of Munc18-2 in cells from FHL5 patients. As Munc18-2 is an ortholog of Munc18-1 and Munc18-3, we asked whether Munc18-1 and Munc18-3 were expressed in CTL. Using quantitative PCR (qPCR) we found that IL-2–activated CTL from both a healthy individual and an FHL5 patient not only produced mRNA for Munc18-3, but also for Munc18-1 (Fig. 7A). Probing for Munc18-1 protein we found low levels expressed in activated human CTL (Fig. 7B). Furthermore, activation of resting NK with IL-2 increased synthesis of Munc18-1 mRNA (Fig. S5A). These results raised the possibility that expression of either Munc18-1 or Munc18-3 in activated CTL might be able to compensate for loss of Munc18-2 in FHL5 patients.
Fig. 7.

Munc18-1 is expressed in CTL and NK and interacts with Stx11. (A) Amplification plot from a qPCR experiment showing the relative fluorescent response plotted against the cycle number of human actin-binding protein (ABP), human Munc18-1, and human Munc18-3 in CTL from a healthy individual and an FHL5 patient. Curves are representative of three independent experiments run as triplicates. Analysis was performed using SDS2.3 software (Applied Biosystems). Controls without water or without reverse transcriptase gave no signal. (B) Expression of Munc18-1 protein in human CTL lysate. Protein from human CTL lysate, as well as Munc18-1, Munc18-2, and Munc18.3 expressed in a cell-free expression system were separated by SDS/PAGE, transferred onto nitrocellulose, and probed with a Munc18-1 or actin-specific antibody by Western blot. Note that the cell-free expression does not contain actin. The additional higher band (marked with an asterisk) is presumed to be nonspecific background binding to a protein from the wheat-germ expression system as it also appears in the mock expressed lysate (no DNA). (C) Anti-myc Western blot from GST pull-down of different myc-tagged Munc18 proteins from HEK293 cells using GST-tagged Stx11, Stx3, and Stx4 as bait. WT Munc18-2 and WT Munc18-1 both bind Stx11 (lanes 2 and 7) and Stx3 (lanes 3 and 8), but not Stx4 (lanes 4 and 9). In contrast, Munc18-3 does not bind to Stx11 or Stx3 (lanes 12 and 13) but instead to Stx4 (lane 14). None of the tested Munc18 proteins showed binding to GST alone (lanes 1, 6, and 11). A sample of the input was loaded in lanes 5, 10, and 15. Pull-down was performed twice. (D) Protein levels of Stx3, Stx4, Stx11, Munc18-2, and actin in CTL from a healthy donor or an FHL5 patient (1976-2) after activation with IL-2 were assessed by Western blot using protein-specific antibodies.
We therefore tested whether either Munc18-1 or Munc18-3 could bind to Stx11. Because Munc18-1 has been shown to bind Stx3 (45) and Munc18-3 interacts with Stx4 (29), we used these as controls. We expressed myc-tagged Munc18 proteins in HEK293 cells to perform pull-downs with Stx11, Stx3, and Stx4 as bait. WT Munc18-2 specifically bound Stx3 and Stx11 but not Stx4, whereas Munc18-3 interacted solely with Stx4 (Fig. 7C). Interestingly, Munc18-1 not only bound to Stx3 as previously shown (45) but also to Stx11, while no binding to Stx4 could be detected (Fig. 7C). These results show that Munc18-1, but not Munc18-3, could compensate for loss of Munc18-2 via a mechanism that involves either Stx11 or Stx3.
Because loss of Munc18-2 in CTL from FHL5 patients has been found to reduce Stx11 protein level (5, 7, 43, 46), we asked whether the abundance of Stx3 or Stx4 were also affected. Western blots of IL-2–activated CTL lysates from an FHL5 patient lacking Munc18-2 (5) and a healthy donor confirmed the reduced level of Stx11. However, levels of Stx3 and Stx4 did not seem to be affected by loss of Munc18-2, with Stx3 being present at normal levels in IL-2–activated CTL from an FHL5 patient (Fig. 7D and Fig. S5B).
Stx3 Is Redistributed by Munc18-2 in the Absence of Stx11.
To test our hypothesis that Stx11 competed with Stx3 for binding to Munc18-2, we investigated the cellular distribution of Stx11 and Stx3 in CTL. We infected primary mouse CTL with a retrovirus expressing Stx11-HA. As shown in Fig. 8A, Stx11 colocalized with the plasma membrane marker CD8. If indeed, by nature of the N peptide, Munc18-2 would selectively bind Stx11 over Stx3, thus stabilizing Stx11 and most likely determining its cellular distribution, we wondered whether in the absence of Stx11 the localization of Stx3 would be affected.
Fig. 8.

Stx3 is redistributed by Munc18-2 in the absence of Stx11. (A) Stx11-HA infected mouse CTL were seeded on multiwell slides, fixed with methanol, and stained with primary antibody against HA-tag and CD8 before incubation with Alexa fluorophore-conjugated secondary antibodies. (B and C) Human CTL from a healthy individual (Top), an FHL4 patient (Middle), and an FHL5 patient (Bottom) were plated and fixed on multiwell slides with methanol. Proteins were stained with primary antibodies against Stx3, CD8, or LAMP2, and visualized using a species-specific Alexafluorophore-conjugated secondary antibody. Colocalization is depicted as gray in the merge images. Nuclei were stained with Hoechst dye. Displayed are single z-slices. (Scale bars, 2 μm.)
Using primary CTL from a healthy donor and a Stx11-deficient patient, we looked at the distribution of endogenous Stx3. Interestingly, in the healthy donor control Stx3 predominantly colocalized with the lysosomal-associated membrane protein 2 (LAMP2) (Fig. 8 B and C, Top), whereas in Stx11-deficient cells, Stx3 was found on the plasma membrane and only partly on lysosomal structures (Fig. 8 B and C, Middle). In contrast, the localization of Stx4, a syntaxin that does not bind to Munc18-2 (Fig. 7C), was not affected in Stx11-deficient CTL (Fig. S6A). This finding suggested that in the absence of Stx11, Stx3 bound to Munc18-2 and would thereby be trafficked to a different cellular compartment. To ascertain that the redistribution of Stx3 was indeed dependent on Munc18-2, we assessed the localization of Stx3 in CTL from a Munc18-2–deficient patient. Interestingly, in the absence of Munc18-2, Stx3 still predominantly localized to lysosomal structures (Fig. 8 B and C, Bottom), supporting the model that, when both Stx11 and Stx3 are present, Munc18-2 selectively binds and chaperones Stx11.
Discussion
Munc18-2 and Stx11 play critical roles in CTL and NK-mediated cytotoxicity, as loss of either protein results in profound immunodeficiency. Although Munc18-2 and Stx11 have been shown to interact, little is known about their precise role in CTL and NK, or how mutations give rise to disease. In this study we solved the structure of human Munc18-2 at 2.6 Å resolution. Although Munc18-2 share 64% sequence identity with rat Munc18-1 (and 49% with Munc18-3), the overall structure of Munc18-2 closely resembles that of Munc18-1 and Munc18-3. One striking difference, however, exists in the N peptide-binding site where Munc18-2 has a larger hydrophobic region (Fig. 2A, Inset, and Fig. S3A). Moreover, Munc18-2 was crystallized without a ligand, thus offering analysis without the binding effects a peptide might have. Although many of the analyzed point mutations identified in FHL5 patients are conserved in Munc18-1 and Munc18-3, the structural environment these are located in differ. Therefore, only analysis of these mutations in the affected protein Munc18-2 can give a reliable picture as to how protein function might be affected by the mutations.
Fourteen of the analyzed point mutations are buried within the core of the structure and are predicted to disrupt the stability of the protein and lead to degradation of Munc18-2, in keeping with the loss of protein observed in cell lysates analyzed from patients with these mutations (5, 7, 43, 46). Four mutations mapped to the surface of Munc18-2 and seemed likely to be involved in interactions with other proteins. Strikingly, all of these mutations mapped to either predicted syntaxin or SNARE binding sites, emphasizing the critical role of this interaction for CTL and NK function.
The R39P mutation was identified in an FHL5 patient carrying a deleterious splice site mutation in the other allele (47). Our studies suggest that R39P Munc18-2 does not fold properly (Fig. S4A) and the protein is therefore rapidly degraded in vivo. Interestingly, in Unc18-1–deficient C. elegans an R39C mutant could fully restore locomotion (37), suggesting that R39 itself is not a crucial site for normal Munc18-2 function. However, the FHL5 mutation introduces a proline at this site that seems to affect protein folding, presumably by disrupting the hydrogen-bonding pattern of the α-helix in which R39 resides.
E132A is also located at the surface of Munc18-2 but does not affect protein folding or stability (Fig. 4 B and C). Furthermore, both glutamate and arginine are often conserved among Munc18 and syntaxin homologs, highlighting the functional significance of this interaction site (16) and (Fig. 4H). Here we show that E132A Munc18-2 no longer binds the N peptide of either Stx11 or Stx3 (Figs. 4 E and F, and 6 B and C) and its binding to the full cytosolic domain of these syntaxins is abolished or severely reduced, respectively (Figs. 4G and 6 D–H). In this regard Stx11 and Stx3 resemble Stx4, which also depends on the N peptide:pocket interaction for Munc18 binding, and not Stx1A, which binds to Munc18-1 even in the absence of the Stx1A N peptide (22).
Because the E132A mutation in Munc18-2 was identified in a homozygous FHL5 patient with full-blown FLH (43), this shows that disruption of the N peptide interaction alone causes FHL, suggesting that the binding of the syntaxin N peptide to Munc18 is required for membrane fusion events (33, 36, 48, 49). A recent study suggests that E132A is unable to rescue secretion in a Munc18-2–depleted rat mast cell line (41). However, because Stx11−/− mice do not show a mast cell degranulation defect (50) and there are no reports of mast cell defects from FHL4 and FHL5 patients, the role of Stx11 in mast cells remains unclear.
The third mutation that we investigated, P334L, lies in an extended antiparallel helical hairpin that bends slightly as a result of the proline residue. Earlier studies have shown that this site is highly conserved between different Munc18 proteins and across species (31). Furthermore, mutations in this region have been shown to affect the binding of SNARE complexes rather than monomeric syntaxins (25, 51). We found that mutation P334L in Munc18-2 did not affect Stx11 binding but enhanced the propensity of the protein to form dimers in solution, which in the cellular context may disrupt interactions between Munc18-2 and SNARE complexes or other binding partners.
One of the most puzzling aspects of FHL4 and FHL5 has been the finding that although CTL and NK assayed directly ex vivo are unable to kill target cells, this genetic defect can often be corrected upon activation with IL-2. Our results reveal a possible molecular basis for this compensation. We show that upon activation with IL-2 NK express increased levels of Stx3 (Fig. 5A), which is able to bind Munc18-2 with a similar affinity as Stx11 (Fig. 6), and would therefore be able to compensate for loss of Stx11. This finding is supported by previous reports, demonstrating that both Stx3 and Stx11 are capable of forming a SNARE complex with SNAP23 and VAMP8 (52–54). Many other changes take place with IL-2 activation of cells, including clustering of granules (55). However, our studies reveal changes that could compensate for lost SNARE interactions, which are critical for NK and CTL function.
Although binding between the full-length Stx3 and Stx11 to Munc18-2 is of comparable strength, we find that the binding affinity of their N peptides for Munc18-2 differ significantly. Our Munc18-2 structure shows a larger hydrophobicity around the pocket into which L8 of the syntaxin peptide is proposed to insert (Fig. S3A). This finding may explain why Stx11, with its leucine at position 9, binds more strongly to Munc18-2 than Stx3, which bears a hydrophilic lysine at that site, thus supporting the model that the N peptide interaction serves as an initial contact site, which allows Munc18 proteins to attach to and then probe the syntaxin for further binding (31). Because the Stx11 N peptide binds Munc18-2 ∼20-times better than the Stx3 N peptide, this result suggests that, when both syntaxins are present, Stx11 will be selected by Munc18-2. Therefore, our data suggest that the N peptide plays a role in selective binding of competing syntaxins to the same Munc18 protein. In addition, our findings support the idea that Munc18-2 acts as a chaperone, stabilizing Stx11 (5), which is also reflected by the 70% decrease in Stx11 protein levels in CTL lacking Munc18-2, but Stx3 levels were unaffected (Fig. 7D). Moreover, we could show that in the absence of Stx11, Stx3 is trafficked in a Munc18-2–dependent manner to the plasma membrane in CTL (where we found Stx11 localized). These observations support a model in which Munc18-2 selectively traffics Stx11 to the plasma membrane, but can traffic Stx3 when Stx11 is absent. Interestingly, a homozygous mutation that creates a stop codon at residue 268 of Stx11, preventing membrane attachment, also causes FHL4, highlighting the requirement for membrane attachment of Stx11 for normal CTL and NK cell function (6).
Not only do our data point toward a functional redundancy between Stx3 and Stx11 in immune cells, but also between Munc18-2 and Munc18-1. Although Munc18-1 was originally thought to be neuron-specific, its expression in other cell types, including mast cells, has been noted (41, 56). In neurons, Munc18-1 is crucial for many of the steps leading to exocytosis (57), some but not all of which can be rescued by Munc18-2 expression (58), suggesting that although Munc18 homologs show some redundancy a degree of specificity is maintained. Here we find that not only is Munc18-1 also expressed in activated CTL (Fig. 7 A and B) and up-regulated upon IL-2 activation (Fig. S5A), it also binds to Stx11 (Fig. 7C). This finding raises the interesting possibility that Munc18-1 might not only stabilize the residual Stx11 protein detected in FHL5 patient cells (5), but may also provide the molecular basis for the rescue of cytotoxicity seen upon IL-2 activation of FHL5 CTL and NK.
By solving the crystal structure of human Munc18-2 and mapping 18 disease-causing mutations, we have been able to provide a clear picture of how these mutations in Munc18-2 are likely to give rise to disease. Furthermore, by analyzing a known disease-causing mutation, our analysis shows that the syntaxin N peptide binding site on Munc18-2 is crucial for function and suggests that the N peptide plays an important part in selecting the binding of Stx11 over other syntaxins in the cell. Our data suggest a mechanism whereby NK and CTL might overcome loss of Stx11 (FHL4) or Munc18-2 (FHL5) upon IL-2 activation, with Stx3 and Munc18-1 being able to act as surrogates for Stx11 or Munc18-2, respectively.
Materials and Methods
Antibodies.
Rabbit polyclonal α-Stx3 (Western blot: SynapticSystems; ICC: Calbiochem), α-Stx11 (gift from R. Prekeris, University of Colorado Denver, Denver), α-hMunc18-2 (produced in the laboratory) (Fig. S6B), α-Calnexin (Sigma), α-actin (Sigma), and anti-CD8 (Abcam); mouse monoclonal α-Stx4 (BD Bioscience; clone 49), α-hMunc18-1 (BD Transduction Laboratories; clone 31), α-CD8 (Sigma; clone UCHT-4), α-LAMP2 (Development studies hybridoma bank; clone H4B4), α-β-actin (Sigma; clone AC-15), α-HA1.1 (Covance; clone 16B12), and α-myc tag (Millipore; clone 4A6).
Cell Culture, Purification, and Lysis.
Peripheral blood lymphocytes were purified using a Ficoll (GE Healthcare) gradient according to the manufacturer’s manual and NK were subsequently extracted by negative selection using a human NK cell purification kit (Miltenyi Biotec). Seventy-percent of cells were harvested, pelleted, and stored frozen at −80 °C after purification; the remaining 30% were cultured for 4 d in RPMI supplemented with 50 μM β-mercaptoethanol, 5% human serum (SeraLab), 2 mM l-glutamine (Invitrogen), 2% (vol/vol) recombinant IL-2 (produced in house), 0.9 M NaHCO3 (Invitrogen), and 1 mM sodium pyruvate (Invitrogen) at 37 °C in a humidified atmosphere with 8% CO2. NK were lysed in 50 mM Tris⋅HCl, 150 mM NaCl, 1 mM MgCl2, 2% (vol/vol) Nonidet P-40, and EDTA-free Protease Inhibitor mixture (1 tablet/50 mL buffer; Roche) at pH 8. Cleared lysate proteins were denatured at 95 °C in 2× sample buffer [0.1 M Tris⋅HCl, 4% (wt/vol) SDS, 0.2% (wt/vol) Bromophenol Blue, 20% (vol/vol) glycerol, 10% (vol/vol) β-mercaptoethanol (Invitrogen), pH 6.8] and subjected to SDS/PAGE.
CTL from a healthy individual, FHL5 patient 1976-2 (P477L) (5) and FHL4 patient 847-1850 (homozygous AL135917:g.25561–44749_del; provided by S.E.) were cultured as previously described (59) and harvested 9 d following restimulation. Whole-cell proteins were separated by SDS/PAGE, transferred onto nitrocellulose membrane, and probed with protein-specific antibodies.
OT-I transgenic CTL were purified and cultured as previously described (60). Cells were infected 1 d after stimulation with Stx11-HA–expressing retrovirus produced in HEK293 cells, sorted 4 d post infection and used for immunocytochemistry 6 d following infection (detailed protocol in SI Materials and Methods).
HEK293 cells were maintained in DMEM supplemented with 10% FCS.
Spodoptera frugiperda (Sf9) insect cells were grown in TNM-FH insect medium (Sigma), 10% (vol/vol) FCS, 10% (vol/vol) EX-CELL TiterHigh medium (SAFC Biosciences), and 1% (vol/vol) CD lipid concentrate (Gibco) in a shaking incubator at 27 °C and 125 rpm.
Purification of Munc18-2 and Syntaxins.
Frozen Sf9 pellets were thawed and resuspended in buffer 1 [20 mM Tris⋅HCl, 200 mM NaCl, 1 mM CaCl2, 1 mM dithiothreitol (DTT), 25 μg/mL 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), pH 8], supplemented with 500 U/L DNaseI type II (Sigma). Cells were lysed by passing through a G21 and G23 needle and the lysate was cleared by centrifugation (70,000 × g, 4 °C, 1 h), applied to glutathione Sepharose (GE Healthcare), and mixed for 1 h at 4 °C. The beads were washed with 30 column volumes of 20 mM Tris⋅HCl (pH 8), 200 mM NaCl, 1 mM CaCl2, 1 mM DTT followed by eight column volumes with 20 mM Tris⋅HCl (pH 8), 200 mM NaCl, 1 mM CaCl2. Munc18-2 was cleaved off the beads by overnight incubation at room temperature with 50 U/L thrombin (relative to Sf9 culture volume; Serva). Thrombin was inactivated with 250 μg/mL AEBSF before concentrating soluble Munc18-2 and applying it to a Superdex 200 10/30 GL gel-filtration column (GE Healthcare) preequilibrated with 20 mM Tris⋅HCl (pH 8), 200 mM NaCl, 1 mM DTT. Fractions containing pure Munc18-2 were pooled and concentrated to ∼8.85 mg/mL.
For purification of syntaxins, frozen E. coli pellets were thawed and resuspended in buffer 1, supplemented with 500 units DNaseI per 6 L of culture, and lysed by single passage through a Type Z cell disruptor (Constant Systems) at 30 kpsi. Recombinant fusion proteins were purified on glutathione Sepharose as outlined above. Vps33A was purified as described previously (19).
Interaction Studies.
For pull-downs using recombinant proteins expressed in bacteria (syntaxins, Vps33A) and insect cell (Munc18-2) 0.5 nmol bait (Stx11ΔC-GST, GST-Stx11-NHABC, GST-Stx11-SNARE, Stx3ΔTM-GST, or GST) were incubated with 0.5 nmol recombinant hMunc18-2 or recombinant Vps33A in the presence of MagneGST Glutathione Particles (Promega) in PD buffer [20 mM Tris⋅HCl, 200 mM NaCl, 1 mM CaCl2, 0.2% Nonidet P-40, and 1× EDTA-free Protease Inhibitor mixture (Roche), pH 7.4] for 1 h rotating at 4 °C. Particles were washed five times in PD buffer and bound material was eluted in SDS sample buffer at 95 °C and analyzed by SDS/PAGE and Coomassie staining.
Pull-downs of myc-tagged Munc18 proteins produced in HEK293 were performed with 1 nmol of bait immobilized on MagneGST Glutathione Particles (Promega) in PD buffer [20 mM Tris⋅HCL, 200 mM NaCl, 1 mM DTT, 0.1% (vol/vol) Nonidet P-40; 1× EDTA-free protease inhibitor mixture; pH 7.4] for 10 min at 4 °C. Beads were washed before incubation with HEK293 lysate, prepared from cells expressing myc-tagged Munc18 proteins, for 1 h rotating at 4 °C. Bound material was washed four times in PD buffer and eluted in 1× sample buffer at 95 °C. Samples were analyzed by SDS/PAGE and Western blot against the myc tag.
SDS/PAGE and Western Blotting.
For NK lysates, SDS/PAGE was performed using NuPAGE 4–12% Bis⋅Tris precast gels (Invitrogen). Proteins were separated at 170 V (constant voltage) and transferred onto Hybond-C Extra nitrocellulose membrane (Amersham). Membranes were blocked in PBS, 5% (vol/vol) skimmed milk (Marvel), 0.05% Tween 20 (Sigma), and incubated with primary antibody for 1 h at room temperature. Membranes were washed, incubated with the appropriate HRP-conjugated secondary antibody, developed using UptiLight HS Western blot reagents (Uptima) and exposed onto X-ray film. Quantitation of protein bands was performed in ImageJ. Protein samples from pull-down experiments were subjected to SDS/PAGE as described above and stained with Coomassie brilliant blue R-250 (Sigma) in 10% acetic acid and 50% methanol.
Crystallization and Data Collection.
Diffraction-quality crystals of WT Munc18-2 (8.85 mg/mL) were grown in sitting drops (200 nL protein + 200 nL reservoir) equilibrated at 20 °C against 80-μL reservoirs containing 0.1 M MES (pH 6) and 10% (vol/vol) 2-methyl-2,4-pentanediol. Crystals were cryoprotected by diluting the mother liquor with 0.5 μL of reservoir solution supplemented with 25% (vol/vol) ethylene glycol immediately before snap-cryocooling by plunging into liquid nitrogen. Diffraction data were recorded at 100 K at Diamond Light Source beam line I03 on a Pilatus 6M-F detector. All datasets were carefully indexed and integrated using MOSFLM (61) and scaled with AIMLESS (62). Resolution cutoff was chosen to ensure <I/σI> > 2 and CC1/2 > 0.5 (61, 63).
Structure Solution, Refinement, Validation, and Interpretation.
Molecular replacement was performed in PHASER (64) using a single molecule of rat Munc18-1 [PDB ID code 3C98; chain A (22)] as a search model. Manual building was performed in COOT (65) using the structure of rat Munc18-1 as a starting model and maps generated in phenix.refine (66). Refinement was performed in phenix.refine in consultation with the validation statistics provided by COOT and MolProbity (67). Structural superpositions were performed using the SSM (68) utility in COOT. Molecular graphics were prepared using PyMOL (DeLano Scientific). Structure factors and final refined coordinates are deposited in the PDB.
Supplementary Material
Acknowledgments
We thank Edmund Jackson for advice and helpful discussion regarding statistical analysis, Helen Kent for help with the expression of human Munc18-2 in insect cells, Janet Deane for assistance with size-exclusion chromatography-multiangle light scattering, Phil Evans for assistance with modeling Fig. 2A, and the beamline scientists at Diamond Light Source. This work was funded by Wellcome Trust Principal Research Fellowships 075880 (to G.M.G.), 090909 (to D.J.O.); Strategic Award 100140 (to the Cambridge Institute for Medical Research); a Daimler Benz scholarship (to Y.H.); the Kendal-Dixon Fund (Y.H.); the Cambridge European Trust (Y.H.); King’s College (Cambridge) (Y.H.); the National Institute for Health Research Biomedical Research Center (Y.H.); a Royal Commission for the Exhibition of 1851 Research Fellowship (to S.C.G.); Federal Ministry of Education and Research (BMBF) Grant 01-EO-0803 (to S.E.); German Science Foundation Grant SFB 635, TP A3 (to S.H.); Antonio Pinzino – Associazione per la Ricerca sulle Sindromi Emofagocitiche (M.A.); Associazione Italiana Ricerca istiocitosi (M.A.); and Italian Ministry of Health, Progetti dioe ricerca finalizzata 2008, Bando Malattie Rare RF-TOS-2008-1219488 (to M.A.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4CCA).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313474110/-/DCSupplemental.
References
- 1.Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140(3):221–230. doi: 10.1007/BF00443367. [DOI] [PubMed] [Google Scholar]
- 2.Henter JI, et al. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78(11):2918–2922. [PubMed] [Google Scholar]
- 3.Stepp SE, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 4.Feldmann J, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3) Cell. 2003;115(4):461–473. doi: 10.1016/s0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 5.zur Stadt U, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–492. doi: 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.zur Stadt U, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–834. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 7.Côte M, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119(12):3765–3773. doi: 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryceson YT, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119(12):2754–2763. doi: 10.1182/blood-2011-08-374199. [DOI] [PubMed] [Google Scholar]
- 9.Südhof TC, Rothman JE. Membrane fusion: Grappling with SNARE and SM proteins. Science. 2009;323(5913):474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carr CM, Rizo J. At the junction of SNARE and SM protein function. Curr Opin Cell Biol. 2010;22(4):488–495. doi: 10.1016/j.ceb.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hata Y, Slaughter CA, Südhof TC. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature. 1993;366(6453):347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- 12.Pevsner J, Hsu SC, Scheller RH. n-Sec1: A neural-specific syntaxin-binding protein. Proc Natl Acad Sci USA. 1994;91(4):1445–1449. doi: 10.1073/pnas.91.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thurmond DC, et al. Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J Biol Chem. 1998;273(50):33876–33883. doi: 10.1074/jbc.273.50.33876. [DOI] [PubMed] [Google Scholar]
- 14.Tang BL, Low DY, Hong W. Syntaxin 11: A member of the syntaxin family without a carboxyl terminal transmembrane domain. Biochem Biophys Res Commun. 1998;245(2):627–632. doi: 10.1006/bbrc.1998.8490. [DOI] [PubMed] [Google Scholar]
- 15.Bracher A, Weissenhorn W. Crystal structures of neuronal squid Sec1 implicate inter-domain hinge movement in the release of t-SNAREs. J Mol Biol. 2001;306(1):7–13. doi: 10.1006/jmbi.2000.4347. [DOI] [PubMed] [Google Scholar]
- 16.Hu SH, Latham CF, Gee CL, James DE, Martin JL. Structure of the Munc18c/Syntaxin4 N-peptide complex defines universal features of the N-peptide binding mode of Sec1/Munc18 proteins. Proc Natl Acad Sci USA. 2007;104(21):8773–8778. doi: 10.1073/pnas.0701124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404(6776):355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 18.Baker RW, Jeffrey PD, Hughson FM. Crystal structures of the Sec1/Munc18 (SM) protein Vps33, alone and bound to the homotypic fusion and vacuolar protein sorting (HOPS) subunit Vps16. PLoS ONE. 2013;8(6):e67409. doi: 10.1371/journal.pone.0067409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham SC, et al. Structural basis of Vps33A recruitment to the human HOPS complex by Vps16. Proc Natl Acad Sci USA. 2013;110(33):13345–13350. doi: 10.1073/pnas.1307074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang B, Steegmaier M, Gonzalez LC, Jr, Scheller RH. nSec1 binds a closed conformation of syntaxin1A. J Cell Biol. 2000;148(2):247–252. doi: 10.1083/jcb.148.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bracher A, Weissenhorn W. Structural basis for the Golgi membrane recruitment of Sly1p by Sed5p. EMBO J. 2002;21(22):6114–6124. doi: 10.1093/emboj/cdf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J. 2008;27(7):923–933. doi: 10.1038/emboj.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deák F, et al. Munc18-1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J Cell Biol. 2009;184(5):751–764. doi: 10.1083/jcb.200812026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128(1):183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 25.Xu Y, Su L, Rizo J. Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry. 2010;49(8):1568–1576. doi: 10.1021/bi9021878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi L, Kümmel D, Coleman J, Melia TJ, Giraudo CG. Dual roles of Munc18-1 rely on distinct binding modes of the central cavity with Stx1A and SNARE complex. Mol Biol Cell. 2011;22(21):4150–4160. doi: 10.1091/mbc.E11-02-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carr CM, Grote E, Munson M, Hughson FM, Novick PJ. Sec1p binds to SNARE complexes and concentrates at sites of secretion. J Cell Biol. 1999;146(2):333–344. doi: 10.1083/jcb.146.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Togneri J, Cheng YS, Munson M, Hughson FM, Carr CM. Specific SNARE complex binding mode of the Sec1/Munc-18 protein, Sec1p. Proc Natl Acad Sci USA. 2006;103(47):17730–17735. doi: 10.1073/pnas.0605448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latham CF, et al. Molecular dissection of the Munc18c/syntaxin4 interaction: Implications for regulation of membrane trafficking. Traffic. 2006;7(10):1408–1419. doi: 10.1111/j.1600-0854.2006.00474.x. [DOI] [PubMed] [Google Scholar]
- 30.Peng RW, Guetg C, Abellan E, Fussenegger M. Munc18b regulates core SNARE complex assembly and constitutive exocytosis by interacting with the N-peptide and the closed-conformation C-terminus of syntaxin 3. Biochem J. 2010;431(3):353–361. doi: 10.1042/BJ20100145. [DOI] [PubMed] [Google Scholar]
- 31.Hu SH, et al. Possible roles for Munc18-1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc Natl Acad Sci USA. 2011;108(3):1040–1045. doi: 10.1073/pnas.0914906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ter Beest MB, Chapin SJ, Avrahami D, Mostov KE. The role of syntaxins in the specificity of vesicle targeting in polarized epithelial cells. Mol Biol Cell. 2005;16(12):5784–5792. doi: 10.1091/mbc.E05-07-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen J, Rathore SS, Khandan L, Rothman JE. SNARE bundle and syntaxin N-peptide constitute a minimal complement for Munc18-1 activation of membrane fusion. J Cell Biol. 2010;190(1):55–63. doi: 10.1083/jcb.201003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rathore SS, et al. Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci USA. 2010;107(52):22399–22406. doi: 10.1073/pnas.1012997108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christie MP, et al. Low-resolution solution structures of Munc18:Syntaxin protein complexes indicate an open binding mode driven by the Syntaxin N-peptide. Proc Natl Acad Sci USA. 2012;109(25):9816–9821. doi: 10.1073/pnas.1116975109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou P, et al. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J. 2013;32(1):159–171. doi: 10.1038/emboj.2012.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson JR, et al. Binding of UNC-18 to the N-terminus of syntaxin is essential for neurotransmission in Caenorhabditis elegans. Biochem J. 2009;418(1):73–80. doi: 10.1042/BJ20081956. [DOI] [PubMed] [Google Scholar]
- 38.McEwen JM, Kaplan JM. UNC-18 promotes both the anterograde trafficking and synaptic function of syntaxin. Mol Biol Cell. 2008;19(9):3836–3846. doi: 10.1091/mbc.E08-02-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malintan NT, et al. Abrogating Munc18-1-SNARE complex interaction has limited impact on exocytosis in PC12 cells. J Biol Chem. 2009;284(32):21637–21646. doi: 10.1074/jbc.M109.013508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meijer M, et al. Munc18-1 mutations that strongly impair SNARE-complex binding support normal synaptic transmission. EMBO J. 2012;31(9):2156–2168. doi: 10.1038/emboj.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bin NR, Jung CH, Piggott C, Sugita S. Crucial role of the hydrophobic pocket region of Munc18 protein in mast cell degranulation. Proc Natl Acad Sci USA. 2013;110(12):4610–4615. doi: 10.1073/pnas.1214887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saltzman RW, et al. Novel mutation in STXBP2 prevents IL-2-induced natural killer cell cytotoxicity. J Allergy Clin Immunol. 2012;129(6):1666–1668. doi: 10.1016/j.jaci.2011.12.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cetica V, et al. STXBP2 mutations in children with familial haemophagocytic lymphohistiocytosis type 5. J Med Genet. 2010;47(9):595–600. doi: 10.1136/jmg.2009.075341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 45.Hata Y, Südhof TC. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins. Use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J Biol Chem. 1995;270(22):13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- 46.Al Hawas R, et al. Munc18b/STXBP2 is required for platelet secretion. Blood. 2012;120(12):2493–2500. doi: 10.1182/blood-2012-05-430629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meeths M, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood. 2010;116(15):2635–2643. doi: 10.1182/blood-2010-05-282541. [DOI] [PubMed] [Google Scholar]
- 48.Khvotchev M, et al. Dual modes of Munc18-1/SNARE interactions are coupled by functionally critical binding to syntaxin-1 N terminus. J Neurosci. 2007;27(45):12147–12155. doi: 10.1523/JNEUROSCI.3655-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schollmeier Y, Krause JM, Kreye S, Malsam J, Söllner TH. Resolving the function of distinct Munc18-1/SNARE protein interaction modes in a reconstituted membrane fusion assay. J Biol Chem. 2011;286(35):30582–30590. doi: 10.1074/jbc.M111.269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.D’Orlando O, et al. Syntaxin 11 is required for NK and CD8⁺ T-cell cytotoxicity and neutrophil degranulation. Eur J Immunol. 2013;43(1):194–208. doi: 10.1002/eji.201142343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyd A, et al. A random mutagenesis approach to isolate dominant-negative yeast sec1 mutants reveals a functional role for domain 3a in yeast and mammalian Sec1/Munc18 proteins. Genetics. 2008;180(1):165–178. doi: 10.1534/genetics.108.090423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ravichandran V, Chawla A, Roche PA. Identification of a novel syntaxin- and synaptobrevin/VAMP-binding protein, SNAP-23, expressed in non-neuronal tissues. J Biol Chem. 1996;271(23):13300–13303. doi: 10.1074/jbc.271.23.13300. [DOI] [PubMed] [Google Scholar]
- 53.Valdez AC, Cabaniols JP, Brown MJ, Roche PA. Syntaxin 11 is associated with SNAP-23 on late endosomes and the trans-Golgi network. J Cell Sci. 1999;112(Pt 6):845–854. doi: 10.1242/jcs.112.6.845. [DOI] [PubMed] [Google Scholar]
- 54.Ye S, et al. Syntaxin-11, but not syntaxin-2 or syntaxin-4, is required for platelet secretion. Blood. 2012;120(12):2484–2492. doi: 10.1182/blood-2012-05-430603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.James AM, et al. Rapid activation receptor- or IL-2–induced lytic granule convergence in human natural killer cells requires Src, but not downstream signaling. Blood. 2013;121(14):2627–2637. doi: 10.1182/blood-2012-06-437012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh E, Kalwat MA, Kim MJ, Verhage M, Thurmond DC. Munc18-1 regulates first-phase insulin release by promoting granule docking to multiple syntaxin isoforms. J Biol Chem. 2012;287(31):25821–25833. doi: 10.1074/jbc.M112.361501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verhage M, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287(5454):864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 58.Gulyas-Kovacs A, et al. Munc18-1: Sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci. 2007;27(32):8676–8686. doi: 10.1523/JNEUROSCI.0658-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holt O, et al. Slp1 and Slp2-a localize to the plasma membrane of CTL and contribute to secretion from the immunological synapse. Traffic. 2008;9(4):446–457. doi: 10.1111/j.1600-0854.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jenkins MR, Tsun A, Stinchcombe JC, Griffiths GM. The strength of T cell receptor signal controls the polarization of cytotoxic machinery to the immunological synapse. Immunity. 2009;31(4):621–631. doi: 10.1016/j.immuni.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leslie AGW, Powell HR. 2007. Processing diffraction data with mosflm. Evolving Methods for Macromolecular Chrystallography, eds Read RJ, Sussman JL, NATO Science Series vol 245 (Springer, New York), pp. 41–51.
- 62.Evans PR. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 69.Bond CS, Schüttelkopf AW. ALINE: A WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 5):510–512. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.