

Abstract
Recent decades have been filled with groundbreaking research in the field of endocrine hormone signaling. Pivotal events like the isolation and purification of the estrogen receptor, the cloning of glucocorticoid receptor cDNA, or dissemination of nuclear hormone receptor (NHR) DNA binding sequences are well recognized for their contributions. However, the novel genome-wide and gene-specific information obtained over the last decade describing NHR association with chromatin, cofactors, and epigenetic modifications, as well as their role in gene regulation, has been largely facilitated by the adaptation of the chromatin immunoprecipitation (ChIP) technique. Use of ChIP-based technologies has taken the field of hormone signaling from speculating about the transcription-enabling properties of acetylated chromatin and putative transcription (co-)factor genomic occupancy to demonstrating the detailed, stepwise mechanisms of factor binding and transcriptional initiation; from treating hormone-induced transcription as a steady-state event to understanding its dynamic and cyclic nature; from looking at the DNA sequences recognized by various DNA-binding domains in vitro to analyzing the cell-specific genome-wide pattern of nuclear receptor binding and interpreting its physiological implications. Not only have these events propelled hormone research, but, as some of the pioneering studies, have also contributed tremendously to the field of molecular endocrinology as a whole. In this review, we give a brief summary of some of the most important discoveries in hormone signaling using ChIP and other derivative techniques and speculate on what the future may hold.
Introduction
Hormones are important signaling molecules that facilitate intercellular communication and synchronization not only between adjacent cells (paracrine signaling) but also between distant organs in the body through endocrine signaling. Endocrine messengers include a vast assortment of molecules that can be categorized into four groups based on their core constituents: protein/peptide hormones, steroid hormones (derived from cholesterol), tyrosine/tryptophan-derived hormones, and (phospho-) lipid hormones (Nussey & Whitehead 2001).
Steroid hormone signaling constitutes a major endocrine pathway that primarily functions by regulating transcription of target genes. Steroid and thyroid hormones are water insoluble and are transported by carrier proteins. However, due to their hydrophobic nature, these hormones can easily diffuse across the plasma membrane into the cell, where they interact with nuclear hormone receptors (NHRs) such as estrogen receptor (ER) and thyroid hormone receptor (THR). NHRs are, therefore, hormone-responsive transcription factors that directly interact with DNA and regulate transcription by recruiting cofactor proteins, namely, coactivator and corepressor complexes, which function by establishing epigenetic marks on target genes (Rosenfeld et al. 2006, Perissi et al. 2010). Conversely, protein/peptide hormones, like insulin or growth hormone (GH), are water soluble, cannot freely diffuse across the plasma membrane, and require membrane-bound receptors to initiate signaling pathways within their target cells. Nevertheless, in select cases, these signaling cascades eventually result in activation of transcription factors and modulation of transcription. Lipid-derived hormones, like prostaglandins and leukotrienes, use both cell-surface and cytoplasmic receptors to invoke cellular responses, which include de novo protein synthesis. Thus, directly or indirectly, transcriptional regulation plays a central role in most endocrine signaling pathways, and its regulation, particularly the temporal and spatial dynamics of transcription factors and cofactors, has been the subject of numerous investigations over the last four decades.
Our understanding of the mechanism of transcriptional regulation is both enabled and limited by the tools available for analysis of this process. In this review, we summarize how the use of chromatin immuno-precipitation (ChIP)-based assays advanced our knowledge of nuclear hormone signaling, while also revisiting some of the earlier methods that were instrumental in our current understanding of hormone action. We discuss several techniques for investigating protein–DNA interactions that fueled early research of gene regulation and explore the advancements that have been facilitated by ChIP, all in the context of molecular endocrinology. While peptide hormone signaling systems are the most numerous, the direct role that NHRs play in modulating transcription has made them the paradigm for analyzing mechanisms of gene regulation in endocrine biology. Since in this context, the most comprehensive information is available for NHR signaling, we focus on the discoveries made with steroid and other NHRs to illustrate the significance of ChIP in advancing hormone research. While other endocrine signaling systems are equally important and fascinating, the relatively short nature of this review limits us from exploring the influence of ChIP in those areas. We conclude the review by presenting some shortcomings of ChIP and forecasting where this powerful technique may take us in the near future.
Early studies of NHR–DNA interaction
Pioneering studies hinting at NHR interaction with DNA started to appear in the late 1960s. Radioactively labeled ligands and cellular fractionation experiments were instrumental in determining NHR association with chromatin (Jensen et al. 1968, Fang et al. 1969, Beato et al. 1970, O’Malley et al. 1970, Swaneck et al. 1970, Harris 1971, Baxter et al. 1972). However, direct evidence for NHR interaction with DNA was difficult to come by and the hypothesis that proteins were the immediate downstream effectors prevailed for another decade. Major breakthroughs in the NHR field were facilitated by the adaptation of nitrocellulose filter binding assays and the availability of highly purified receptors. Use of these developments presented convincing evidence for direct receptor–DNA interaction and enabled elucidation of the DNA recognition sequences specific to many receptors (reviewed in Glass (1994)).
Nitrocellulose filter binding assays are no longer common for assessing protein–DNA interactions and have been largely replaced by electrophoretic mobility shift assays (EMSAs; Fig. 1A). EMSAs not only offer reduced background but also allow more simplified investigations of direct DNA interactions with single or multiple proteins and protein complexes in vitro (Hellman & Fried 2007). Several other DNA binding assays have been developed and offer some distinct advantages. DNA footprinting assays can detect sequences of DNA that are protected by bound proteins from physical shearing or enzymatic digestion both in vitro and in vivo (Fig. 1B). In vivo assays are advantageous due to the maintenance of proper chromatin structure; however, the presence of multiple cellular proteins makes it impossible to know which protein is directly binding and protecting the DNA. In vitro assays can be used with purified proteins, but, just like EMSAs and filter binding assays, are performed on naked DNA. In a similar assay – the DNase I hypersensitivity assay – enzymatic digestion of chromatin is used to detect nucleosome-free regions, which are indicative of transcription factor binding. This assay is performed in vivo, but, as with DNA footprinting, identification of transcription factors binding to the identified regions is difficult.
Figure 1.
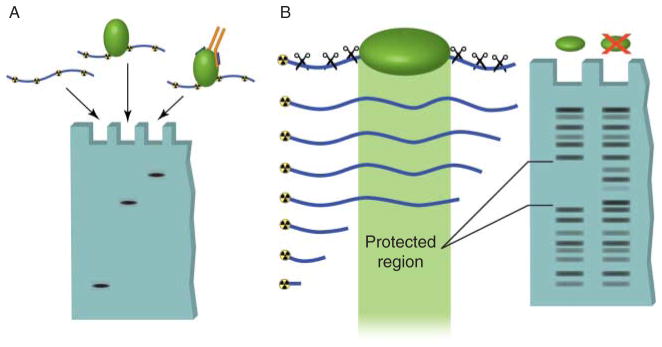
In vitro techniques used to detect DNA–protein interactions. (A) Electromobility shift assays can be used to determine direct binding between a specific sequence of radioactively labeled DNA and a purified protein. Unbound DNA, termed free probe, migrates at a relatively low molecular weight in the agarose gel. Binding of protein to this sequence results in the DNA band shifting to a high molecular weight region. Addition of an antibody that recognizes the bound protein causes an even greater shift in mobility, called supershifting. This assay can also be used with protein complexes to detect indirect protein–DNA interactions. (B) DNase footprinting assays allow identification of regions of DNA bound by proteins. A DNA oligomer is radioactively labeled on one end and mixed with the protein of interest. The DNA is then digested by a DNA endonuclease (DNase). The regions of DNA that are bound by proteins are protected from digestion. When the DNA is run out on a gel, the protected region shows up as a break in the laddering produced by DNase digestion.
While the binding assays described above are invaluable tools, the functional significance of transcription factor binding cannot be determined using these methods. Chloramphenicol acetyltransferase, green fluorescent protein (GFP), or, more commonly, luciferase reporter assays have been employed to answer these types of questions. In these techniques, a genomic region of interest, typically containing a transcription factor binding site, is cloned upstream of a reporter gene and transcriptional activity is measured. Mutations of the transcription factor (TF) binding site or the transcription factor itself allow analysis of sequences and residues important for gene regulation. A different reporter assay-based technique – the yeast one-hybrid screen – has been used to screen libraries of proteins that can functionally interact with a particular sequence of DNA and activate gene transcription. However, as is the case with binding assays, these functional assays are performed on DNA placed out of context and may not take into account nucleosome positioning effects (for a more detailed review of these techniques, see Carey & Smale (1999)). Nevertheless, such assays were instrumental in identifying and determining the regulatory properties of hormone response elements (HREs) to which NHRs bind, and, thereby, advanced our understanding of the mechanism of NHR-mediated gene regulation in the pre-genomic era.
Removal of DNA and protein from their native environment can produce substantial artifacts and result in deviations from physiological significance. Thus, it is essential to analyze protein–DNA interactions in whole cells, where DNA is properly chromatinized and transcription factors are complexed with appropriate cofactors. Furthermore, binding and reporter assays make it very difficult to detect indirect or nonspecific protein–DNA associations, like those apparent between cofactors or histones and DNA. Perhaps one of the most significant shortcomings of many of these pre-genomic techniques is the difficulty of adapting them for genome-wide analysis. The development of ChIP-based assays has made it possible to avoid such drawbacks and has opened up the field for gene-specific and genome-wide in vivo analysis in an unprecedented manner. Importantly, integration of pre-genomic techniques (i.e. reporter and EMSA assays) with the now widely used ChIP-based methodologies allows for analysis of functional DNA–protein interactions in the post-genomic time (Table 1). For example, DNA binding and reporter assays are still frequently used today for NHRs and other transcription factors to characterize newly identified binding sequences originating from genome-wide studies.
Table 1.
Various techniques used to analyze transcription factor interactions with DNA, along with their benefits and shortcomings. This list is not meant to be exhaustive
| Technique
|
Detects...
|
Benefits
|
Drawbacks
|
References
|
|---|---|---|---|---|
| Nitrocellulose membrane binding assay Electrophoretic mobility shift assay |
In vitro interaction between protein and ‘naked’ DNA | Simple and convenient Can discriminate between direct and indirect interactions |
Not conclusive of in vivo interactions Low throughput |
Damm et al. (1989) and Umesono et al. (1991) |
| DNase footprinting | Protein interaction with DNA at high resolution | Nucleotide level resolution allows precise identification of binding sites or nucleosome occupancy |
In vitro assays are low throughput and are not conclusive of in vivo functionality In vivo approaches cannot identify bound protein |
Payvar et al. (1983) and Gross & Garrard (1988) |
| Reporter assay | Ability of specific DNA sequence to activate, enhance, or repress transcription | Indicates transcriptional functionality of the DNA sequence in question | Sequence is placed out of context and may not reflect in vivo properties | Umesono et al. (1991) |
| Chromatin immunoprecipitation (ChIP) | Presence of specific protein at a specific location in vivo |
In vivo system Allows observation of highly dynamic events |
Requires a population of cells, so observations are not necessarily concurrent in a single cell Cannot directly indicate functional significance Interactions may be indirect |
Orlando (2000) and Collas (2010) |
| ChIP–reChIP | Co-localization of two proteins at a specific genomic locus | Highly suggestive of functional relationship between two proteins at the chromatin level | Reduced signal to noise ratio Requires high quality antibodies |
Furlan-Magaril et al. (2009) |
| μChIP | Presence of a specific protein and a specific location in vivo with as few as 100 cells | Low amount of starting material | Requires high-quality antibodies and high abundance protein | Collas (2010) |
| ChIP-chip ChIP-seq |
Presence of a specific protein throughout a large portion or the entire genome | High throughput | Considerable variation between experiments Highly dependent on the quality of antibody Cannot directly indicate functional significance |
Collas (2010) |
| ChIA–PET | Chromatin looping and interaction of distal loci of the genome due to a protein of interest | Identifies chromatin looping associated with protein of interest | Cannot demonstrate whether looping depends on the protein of interest Low signal to noise ratio |
de Wit & de Laat (2012) |
Chromatin immunoprecipitation
ChIP is a technique used to map protein–DNA interactions in vivo to a specific region of the genome. Briefly, DNA and proteins are cross-linked together, commonly using formaldehyde. The chromatin is then sheared by sonication or enzymatic digestion and subjected to precipitation with antibodies. After the cross-links are reversed, DNA undergoes PCR amplification using primers targeting a particular genomic locus. When the target protein interacts with this locus, more DNA is pulled down and amplified, resulting in a higher signal (Fig. 2). Several reviews provide detailed descriptions and historical perspectives on the ChIP protocol (e.g. see Kuo & Allis (1999) and Orlando (2000)).
Figure 2.

Chromatin immunoprecipitation assay. DNA and proteins are covalently cross-linked together using formaldehyde and the chromatin is fragmented using either enzymatic digestion or physical shearing by sonication. Antibodies against various proteins associated with DNA, like histones, transcription factors, and cofactors, can be used to immunoprecipitate any associated fragments of DNA. Once the cross-links are reversed, the DNA can be subjected to a number of downstream analysis techniques, including targeted approaches, like semiquantitative PCR and quantitative PCR, and genome-wide analyses using microarrays (ChIP-chip) and deep sequencing (ChIP-seq).
The ability to freeze in vivo protein–DNA interactions through cross-linking makes ChIP a very powerful technique. Not only does this allow investigations of whether and where on chromatin transcription factors and cofactors bind in their native environment, but also makes it possible to examine the dynamic nature of such interactions. More recently, ChIP has been adapted to answer questions on a genome-wide scale via ChIP-chip or ChIP-seq techniques. It has also been incorporated into more elaborate methods like chromatin interaction analysis by paired-end tag sequencing (ChIA–PET), which can analyze long-range chromatin interactions. All of these aspects have made ChIP an indispensable tool in modern molecular endocrinology research (Collas 2010). Below we present some notable examples of ChIP applications in nuclear hormone signaling that provided expected and unexpected information and helped move the field forward.
Use of ChIP in targeted and genome-wide in vivo analyses
Detecting histone modifications
The general effects that histones have on transcription have been observed for a long time. Electron microscopy and biochemical studies provided evidence that transcriptionally active genes are either depleted of nucleosomes (Scheer 1978, Karpov et al. 1984, Widmer et al. 1984) or the nucleosome conformation is altered (Lohr 1983, Prior et al. 1983, Weischet et al. 1983, Benezra et al. 1986). Additional biochemical in vitro and in vivo analyses indicated that histone hyper-acetylation is highly correlated with transcription, while silent genes lack such histone marks (Pogo et al. 1966, Gorovsky et al. 1973; reviewed in Brownell & Allis (1996)). Nevertheless, direct in vivo evidence for these observations was lacking and data demonstrating a causative relationship between histone acetylation and transcriptional activation was very limited (Ura et al. 1997). Some of the earliest experiments using ChIP were aimed at assessing histone occupancy and acetylation status upon gene activation. Solomon et al. (1988) demonstrated that, contrary to previous observations (Karpov et al. 1984), transcriptionally active hsp70 locus in Drosophila melanogaster was not completely deprived of histones but retained at least histone H4. Similarly, histone acetylation status was probed by various groups at other genes and was found to increase coordinately with and be required for transcription (Hebbes et al. 1988, Kuo et al. 1998, Parekh & Maniatis 1999). Notably, ChIP offered finer resolution than previous techniques, allowing for discovery of promoter-specific histone acetylation (Parekh & Maniatis 1999).
One of the earliest studies of histone modifications in endocrine signaling using ChIP was carried out in MCF7 and HL-60 cells and analyzed the status of ER and retinoic acid receptor (RAR) target gene promoters. Hormone stimulation resulted in rapid histone hyperacetylation of these regions in a localized and NHR cofactor and histone acetyltransferase CREBBP/EP300-dependent manner. Interestingly and unexpectedly, histone acetylation was not maintained with sustained hormone treatment. The decline of acetylation after the initial peak at 1 h correlated closely with a decrease in mRNA expression (Chen et al. 1999). These results complemented previous observations of hormone-mediated transcription that is attenuated after an initial burst (Sasaki et al. 1984, Cavailles et al. 1988, Dubik & Shiu 1988) and provided a mechanistic understanding for this phenomenon. More recently, using ChIP–DNA selection and ligation (DSL) technology, Kwon et al. (2007) analyzed ER binding, along with histone acetylation and methylation, upon hormone treatment of MCF7 cells on promoter and enhancer tiling arrays. As expected, hyperacetylation of histone H3 lysine 9 occurred in both promoters and enhancers of ER target genes TFF1 and GREB1, while H3 lysine 4 trimethylation was observed exclusively in the promoters. Analyses of additional histone methylation marks established a gene- and chromatin domain-specific modification pattern, suggesting that each individual gene is uniquely marked with histone modifications (Kwon et al. 2007). Thus, it appears that a combinatorial modification pattern that is expected to be different for each gene would define activated and repressed states, thereby making it difficult to assign a single set of histone markings to the transcriptional status of a gene. Therefore, while some evidence was already available for the effects of histones, and their modifications, on transcription, use of ChIP provided more direct and physiologically accurate data to support such hypotheses. With the discovery of more histone modifications, like ubiquitination, and sumoylation, ChIP has become an indispensable tool for studying their temporal and spatial organization and physiological significance.
Investigating NHR, cofactor, and chromatin dynamics
ChIP makes it possible to perform meticulous time-course analyses of transcription (co-)factor binding in vivo, which has significantly enhanced our understanding of transcriptional initiation as well as its maintenance. In addition to demonstrating the dynamic nature of histone acetylation in response to hormone (see above), Chen et al. (1999) used ChIP to investigate the recruitment of ER, along with some of its cofactors, to target gene promoters throughout the course of estradiol (E2) treatment. While the presence of ER on chromatin appeared unaltered, the downregulation of target gene c-Myc (MYC) and CTD (CTSD) transcription, after an initial burst, was accompanied by decreased recruitment of ACTR (NCOA3), CBP (CREBBP), and p300 (EP300) acetyl-transferases. More detailed studies by Shang et al. (2000) and Métivier et al. (2003) using the same model system revealed a slightly different mechanism. Because ChIP allows fixing of proteins in place at any point in time, these groups were able to obtain impressive temporal resolutions of 15 and 5 min respectively and capture, in great detail, the cyclic nature of ER and pre-initiation complex assembly over 3 h of hormone treatment. These experiments demonstrated an ordered and sequential recruitment of coactivators, including histone acetyltransferases (HATs), histone methyltransferases, and nucleosome remodelers, leading to transcriptional initiation and gene upregulation. Furthermore, they showed that the entire ER complex, not just select cofactors, periodically associates with and dissociates from chromatin. Huang et al. (2003) expanded on these studies and demonstrated that the recruitment of CREBBP/EP300 along with the acetylation of histones is a required step for further assembly of the pre-initiation complex. Similar observations of cyclic transcription have also been made with androgen receptor (AR; Kang et al. 2002), THR (Sharma & Fondell 2002), and vitamin D receptor (Väisänen et al. 2005). The findings presented above laid down the basis for our current model of transcriptional dynamics (Hager et al. 2009). Additionally, the observations of ordered recruitment of histone modifying and remodeling complexes, followed by the assembly of the Mediator complex and the RNA polymerase II holoenzyme, remain the dogma of hormone-induced transcriptional activation (Chen & Roeder 2011).
Genome-wide approaches
The ability of different members of the NHR superfamily to recognize and regulate specific sets of genes had been well established before the use of ChIP (Yamamoto 1985, Beato 1989, Glass 1994). Nitrocellulose membrane binding, EMSA, and DNase footprinting assays (Payvar et al. 1981, 1983, Scheidereit et al. 1983) coupled with reporter assays (Klock et al. 1987, Strähle et al. 1987, Umesono et al. 1991) were instrumental in elucidating the binding sequences, termed HREs, for many members of this large family of transcription factors (reviewed in Beato (1989) and Glass (1994)).
In vivo NHR binding sites
With the emergence of ChIP, one major aim of research in the field of nuclear hormone signaling has been to determine the functional in vivo binding sites for various NHRs. ChIP certainly provides the means for investigating functionality of individual response elements (e.g. Shang et al. (2000) and Métivier et al. (2003)), but such a targeted approach is impractical when attempting to understand the global characteristics of hormone signaling. By coupling ChIP with genome-wide assays (i.e. microarrays and deep sequencing), a more comprehensive analysis of nuclear receptor action can be undertaken. Numerous experiments have been performed using ChIP-chip or ChIP-seq in several cell lines and tissues investigating many NHRs. Collectively, the findings of these studies demonstrate that the number of NHR binding sites ranges anywhere from 1000 to numbers in the tens of thousands depending on cell type and the conditions of the experiment (Jagannathan & Robinson-Rechavi 2011). Furthermore, these assays have uncovered an unexpected result – the majority of NHR binding sites are not found within the proximal promoter of the target genes but are located several to over 100 kb both upstream and downstream of the transcription start site (Carroll et al. 2005, 2006, Wang et al. 2007). ChIP-chip experiments in MCF7 cells found that <4% of all ER binding sites occur within the 1 kb promoter–proximal regions (Carroll et al. 2006). These results challenge the historical bias toward promoter–proximal TF binding as the major driving force for transcriptional activation. Genome-wide analysis of AR binding in prostate cancer LNCaP cell lines essentially established a similar phenomenon: most of the AR binding sites lie 20–50 kb upstream of AR target genes (Wang et al. 2007). Additionally, noncanonical AREs were identified in the enhancer regions and AR binding to these sites was found to be dependent on cooperating factor FoxA1 (Wang et al. 2009). The discovery of distal enhancers raises the question of their impact on transcription: can nuclear receptors regulate transcription over such large distances, and if so, which enhancers are involved in transcriptional control of which genes? Using another recently developed technique termed chromosome conformation capture (3C; reviewed in de Wit & de Laat (2012)), which can be employed to identify interactions between distant regions of the genome, some of the distal HREs have been shown to physically contact the promoters of the genes they regulate (Deschênes et al. 2007, Barnett et al. 2008, Bretschneider et al. 2008). Of note is a recent study using ChIA–PET, a technique that combines ChIP, 3C, and deep sequencing. Fullwood et al. (2009) used ChIA–PET to analyze chromatin interactions created by the ERα (ESR1) on a global scale. The findings show that most of the high-confidence ERα binding sites determined through ChIP-seq interact with promoters and suggest that they are likely to be functionally important. The role that the distal enhancers play on gene regulation, however, is still being investigated. While physical interaction may hint at functional relevance, such claims need to be evaluated directly through deletion or site-directed mutagenesis studies. Recent development of zinc finger nuclease (ZFN)- and transcription activator-like effector nuclease (TALEN)-based technologies makes it possible to perform in vivo mutations of distal binding sites and evaluate their functional relevance in the native environment (Wood et al. 2011). The genome-wide studies also indicate that there are more ER binding sites throughout the genome than the number of ER-regulated genes, suggesting that either some of the binding sites are nonfunctional in any given cell or binding of ER at multiple sites is a prerequisite for target gene regulation (Carroll et al. 2006). The presence of multiple ER binding sites for a single gene may also enable differential transcriptional regulation in various cell types. Discoveries of distal enhancers, although confounding and unintuitive at first glance, provide excellent examples of the dynamic and flexible nature of chromatin, as well as the complexity of transcriptional regulation. The collective genome-wide data from all these experiments provide an invaluable database of information that will stimulate even more in-depth analysis of nuclear receptor mechanisms of action.
NHR interplay with other transcription factors
The importance of mapping global nuclear receptor binding sites using ChIP-seq has recently been exemplified with the discovery of the role that the forkhead DNA-binding protein (FOXA1) plays in hormone signaling (reviewed in Zaret & Carroll (2011)). Analysis of the DNA sequences surrounding the genome-wide ERα binding sites unveiled FOXA1 binding motifs at a large portion of the surveyed regions. Additional ChIP experiments revealed that the binding of the FOXA1 transcription factor in the proximity of ERα response elements (EREs), accompanied by the remodeling of chromatin structure, is an essential pioneering event in establishing stable recruitment of ER to many of its functional EREs (Carroll et al. 2005, Laganière et al. 2005, Lupien et al. 2008, Hurtado et al. 2011). A strong positive correlation was also observed between ER, C/EBP, and Oct occupancy, while a negative correlation between ER and AP-1 response elements was noted (Carroll et al. 2006). Furthermore, these studies suggest that the presence or absence of FOXA1 expression in different cell types could account for the cell type-specific gene regulation by the ER. Similar genome-wide AR binding site sequence analysis identified enrichment of GATA2 and OCT1 response elements, also suggesting their role as collaborating/pioneering factors for AR (Wang et al. 2007, 2009). Use of the genome-wide data will likely lead to the discovery of similar pioneering factors for other NHRs in the future.
Recent global analysis of ER and RAR binding in MCF7 cells has uncovered an intricate and somewhat controversial interplay between these two groups of receptors. Hua et al. (2009), through ChIP-seq analysis, discovered that a large portion of ERα binding sites overlap with RARα and RARγ sites throughout the genome. Further targeted experiments demonstrated that this overlap is functionally significant and results in ER and RAR receptors competing for binding, which leads to antagonistic regulation of a subset of target genes. At around the same time, Ross-Innes et al. (2010) performed a similar ChIP-seq experiment, confirming the large overlap of binding sites seen in the Hua et al. (2009) study but, contrary to previous results, did not observe competitive binding between ERα and RARα. Nevertheless, antagonistic gene regulation was confirmed and may be due to differential recruitment of cofactors by the two different receptors. Additionally, the group uncovered cooperative interactions between these two groups of NHRs at a subset of target genes. It was determined that ERα and RARα form a complex together and that RARα significantly enhances the ability of ERα to activate gene expression in an estrogen-dependent manner. The differential observations from these two studies may have been due to varying experimental conditions (e.g. endogenous vs exogenous, epitope-tagged RARα; or pre-treatment of MCF7 cells with hormone-free media vs no hormone depletion) and reveal the susceptibility of ChIP to such variations. Rather than a shortcoming of the ChIP technique, this example demonstrates the ability of ChIP to detect such in vivo alterations, which may be overlooked in other assays. Further experiments will be necessary to elucidate the true nature of ER/RAR dynamics.
Advantages, limitations, and remedies
The impact that ChIP has had on the field of nuclear hormone signaling is unquestionable. It has tremendously advanced our understanding of the role of NHR in transcriptional regulation and provided more physiologically relevant data that facilitate translational research. In some instances, however, ChIP assays played a more confirmatory role demonstrating validity of in vitro DNA binding data. On the other hand, ChIP-based genome-wide binding studies on nuclear receptors, cofactors, and histone modifications provided new information that surprised us and allowed us to predict and test new hypotheses. For example, the astounding number of ER binding sites and their preponderance at the distal enhancers were not predicted by previous studies but have gained a lot of interest in recent years.
ChIP has some clear advantages over techniques like nitrocellulose membrane binding, DNase footprinting, EMSA, and reporter assays. The ability to observe native protein–DNA interactions in vivo is extremely valuable and physiologically significant. However, because formaldehyde cross-linking creates covalent bonds between proteins as well as DNA, ChIP results need to be supplemented with in vitro binding assays to determine whether the observed protein–DNA interactions are in fact direct. One of the most influential aspects of ChIP is that it allowed transition of protein–DNA interaction assays into genome-wide techniques, which are becoming the de facto standards for performing research.
Nevertheless, ChIP is not an ideal assay for all purposes and has some shortcomings. The quality of the results obtained through ChIP is greatly dependent on the quality of the antibodies used. The relatively low signal-to-noise ratio of the ChIP assay makes it susceptible to PCR artifacts and places much importance on highly specific and strong antibodies. While more and more commercial antibodies are marketed as suitable for ChIP, finding ones that produce good results is still a challenge. This issue is particularly true for less commonly studied proteins. A common way to overcome this limitation is through the use of epitope-tagged or fusion constructs, like FLAG, His, and GFP. Although this provides much better means of pulling down the protein of interest, overexpression of an exogenous protein shifts the assay away from its physiological relevance. As ChIP-suitable antibodies become more available, this particular issue should become less relevant in the future.
ChIP assays may capture transient DNA–protein interactions, which may or may not be functionally relevant. Thus, other approaches, like in vivo deletion or site-directed mutagenesis discussed earlier, are necessary to confirm true functionality of transcription factor binding in regulating gene expression. The spatial resolution of ChIP is also limited. For targeted assays, it may be difficult to design quality primers at a desired locus due to repetitive sequences or, more commonly, high GC content of CpG islands found in the promoters of most genes. Even with ChIP-seq, the resolution is limited by the heterogeneity of the fragments that are obtained through sonication. Recently, this limitation has been bypassed using a ChIP-exo assay (Rhee & Pugh 2011). In this technique, the DNA is sonicated and immunoprecipitated as previously, but before it is sequenced, each strand is digested up to the bound protein using an exonuclease. The 5′-end of the remaining fragment marks the exact position of transcription factor binding and can be sequenced after adapter ligation. Variations of the ChIP assay offering better performance are constantly being developed, providing even more proof to the power and flexibility of this technique.
Current methods for ChIP require a starting amount of chromatin that can only be obtained from a large population of cells. Thus, the events observed in ChIP experiments are not necessarily representative of those inside a single cell but are rather the average of a population. Binding of two different proteins at a single location as observed by ChIP may in fact be temporally exclusive. The current approach to disambiguate such results is through ChIP–reChIP, where the chromatin that is pulled down with a single protein is subjected to a consecutive pull-down by a different antibody (Furlan-Magaril et al. 2009). Nevertheless, the efficiency of the pull down is not always sufficient to retain the signal after a second immunoprecipitation. Furthermore, extending this method to more than two proteins, while desirable, is often not practical. Advancements in ChIP have allowed scaling down if the amount of starting material to as little as 100 cells for probing a single protein using the mChIP technique (Collas 2010), which should reduce the heterogeneity of the cell population. As more advancements are made in the coming years, we can expect to see ChIP assays reach single-cell levels.
Summary and future directions
In this review, we highlighted major advancements in genome-wide analysis of NHR signaling using ChIP-based technologies. To put these new advancements in perspective, it was equally important to revisit some of the pre-genomic era techniques that yielded much useful information. We predict that a mix of both pre-and post-genomic technologies will be used in the future to analyze genome-wide and gene-specific regulation by hormone signaling. Extrapolating from the last 20 years, the future of endocrine hormone signaling research is likely to see even more exciting and groundbreaking discoveries based on the ChIP technique. Of particular interest will continue to be genome-wide studies probing yet-unexplored proteins and analyzing minor variations of binding sites across different cell types. Additionally, we hope to begin seeing single cell-based genome-wide analyses used more frequently in the future, particularly in the context of human diseases, including cancer. For example, using single cells isolated from different regions of a breast cancer sample, we may see differential NHR binding and gene expression patterns. These observations then can be harnessed for better understanding and treatment of endocrine-linked diseases. Finally, although the new in vivo techniques are exciting and offer unprecedented means of investigation, the significance of older in vitro assays, like EMSA, should not be undermined, as they are still commonly used and offer confirmation of the results observed in vivo. In the coming years, we expect newer genome-wide ChIP-utilizing assays to lead the technological advancement of exciting hormone research and uncover even more wonders of NHR transcriptional regulation.
Acknowledgments
The authors would like to thank Dr James Brandon Parker for helpful discussions during the writing of this review.
Funding
Research in the Chakravarti laboratory was supported in part by NIH grants R01 CA133755 and P01 HD57877.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review reported. Owing to space restrictions, additional relevant references could not be included.
References
- Barnett DH, Sheng S, Howe Charn T, Waheed A, Sly WS, Lin C-Y, Liu ET, Katzenellenbogen BS. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Research. 2008;68:3505–3515. doi: 10.1158/0008-5472.CAN-07-6151. [DOI] [PubMed] [Google Scholar]
- Baxter JD, Rousseau GG, Benson MC, Garcea RL, Ito J, Tomkins GM. Role of DNA andspecific cytoplasmic receptors in glucocorticoid action. PNAS. 1972;69:1892–1896. doi: 10.1073/pnas.69.7.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M. Gene regulation by steroid hormones. Cell. 1989;56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- Beato M, Brändle W, Biesewig D, Sekeris CE. On the mechanism of hormone action. XVI. Transfer of (1,2-3H2)cortisol from the cytoplasm to the nucleus of rat-liver cells. Biochimica et Biophysica Acta. 1970;208:125–136. doi: 10.1016/0304-4165(70)90055-3. [DOI] [PubMed] [Google Scholar]
- Benezra R, Cantor CR, Axel R. Nucleosomes are phased along the mouse β-major globin gene in erythroid and nonerythroid cells. Cell. 1986;44:697–704. doi: 10.1016/0092-8674(86)90835-4. [DOI] [PubMed] [Google Scholar]
- Bretschneider N, Kangaspeska S, Seifert M, Reid G, Gannon F, Denger S. E2-mediated cathepsin D (CTSD) activation involves looping of distal enhancer elements. Molecular Oncology. 2008;2:182–190. doi: 10.1016/j.molonc.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell JE, Allis CD. Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Current Opinion in Genetics & Development. 1996;6:176–184. doi: 10.1016/S0959-437X(96)80048-7. [DOI] [PubMed] [Google Scholar]
- Carey M, Smale ST. Transcriptional Regulation in Eukaryote: Concepts, Strategies, and Techniques. Woodbury, NY, USA: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nature Genetics. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Cavailles V, Augereau P, Garcia M, Rochefort H. Estrogens and growth factors induce the mRNA of the 52K-pro-cathepsin-D secreted by breast cancer cells. Nucleic Acids Research. 1988;16:1903–1919. doi: 10.1093/nar/16.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Roeder RG. Mediator-dependent nuclear receptor function. Seminars in Cell & Developmental Biology. 2011;22:749–758. doi: 10.1016/j.semcdb.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/S0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- Collas P. The current state of chromatin immunoprecipitation. Molecular Biotechnology. 2010;45:87–100. doi: 10.1007/s12033-009-9239-8. [DOI] [PubMed] [Google Scholar]
- Damm K, Thompson CC, Evans RM. Protein encoded by v-erbA functions as a thyroid-hormone receptor antagonist. Nature. 1989;339:593–597. doi: 10.1038/339593a0. [DOI] [PubMed] [Google Scholar]
- Deschênes J, Bourdeau V, White JH, Mader S. Regulation of GREB1 transcription by estrogen receptor α through a multipartite enhancer spread over 20 kb of upstream flanking sequences. Journal of Biological Chemistry. 2007;282:17335–17339. doi: 10.1074/jbc.C700030200. [DOI] [PubMed] [Google Scholar]
- Dubik D, Shiu RP. Transcriptional regulation of c-myc oncogene expression by estrogen in hormone-responsive human breast cancer cells. Journal of Biological Chemistry. 1988;263:12705–12708. [PubMed] [Google Scholar]
- Fang S, Anderson KM, Liao S. Receptor proteins for androgens: on the role of specific proteins in selective retention of 17β-hydroxy-5α-androstan-3-one by rat ventral prostate in vivo and in vitro. Journal of Biological Chemistry. 1969;244:6584–6595. [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-α-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan-Magaril M, Rincón-Arano H, Recillas-Targa F. Sequential chromatin immunoprecipitation protocol: ChIP–reChIP. In: Leblanc B, Moss T, editors. DNA–Protein Interactions. Totowa, NJ: Humana Press; 2009. pp. 253–266. [DOI] [PubMed] [Google Scholar]
- Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocrine Reviews. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- Gorovsky MA, Pleger GL, Keevert JB, Johmann CA. Studies on histone fraction F2A1 in macro- and micronuclei of Tetrahymena pyriformis. Journal of Cell Biology. 1973;57:773–781. doi: 10.1083/jcb.57.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annual Review of Biochemistry. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- Hager GL, McNally JG, Misteli T. Transcription dynamics. Molecular Cell. 2009;35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GS. Nature of oestrogen specific binding sites in the nuclei of mouse uteri. Nature. 1971;231:246–248. doi: 10.1038/231296a0. [DOI] [PubMed] [Google Scholar]
- Hebbes TR, Thorne AW, Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO Journal. 1988;7:1395–1402. doi: 10.1002/j.1460-2075.1988.tb02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nature Protocols. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S, Kittler R, White KP. Genomic antagonism between retinoic acid and estrogen signaling in breast cancer. Cell. 2009;137:1259–1271. doi: 10.1016/j.cell.2009.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z-Q, Li J, Sachs LM, Cole PA, Wong J. A role for cofactor–cofactor and cofactor–histone interactions in targeting p300, SWI/SNF and mediator for transcription. EMBO Journal. 2003;22:2146–2155. doi: 10.1093/emboj/cdg219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nature Genetics. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan V, Robinson-Rechavi M. The challenge of modeling nuclear receptor regulatory networks in mammalian cells. Molecular and Cellular Endocrinology. 2011;334:91–97. doi: 10.1016/j.mce.2010.06.012. [DOI] [PubMed] [Google Scholar]
- Jensen EV, Suzuki T, Kawashima T, Stumpf WE, Jungblut PW, DeSombre ER. A two-step mechanism for the interaction of estradiol with rat uterus. PNAS. 1968;59:632–638. doi: 10.1073/pnas.59.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Pirskanen A, Jänne OA, Palvimo JJ. Involvement of proteasome in the dynamic assembly of the androgen receptor transcription complex. Journal of Biological Chemistry. 2002;277:48366–48371. doi: 10.1074/jbc.M209074200. [DOI] [PubMed] [Google Scholar]
- Karpov VL, Preobrazhenskaya OV, Mirzabekov AD. Chromatin structure of hsp 70 genes, activated by heat shock: selective removal of histones from the coding region and their absence from the 5′ region. Cell. 1984;36:423–431. doi: 10.1016/0092-8674(84)90235-6. [DOI] [PubMed] [Google Scholar]
- Klock G, Strahle U, Schutz G. Oestrogen and glucocorticoid responsive elements are closely related but distinct. Nature. 1987;329:734–736. doi: 10.1038/329734a0. [DOI] [PubMed] [Google Scholar]
- Kuo M-H, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods. 1999;19:425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- Kuo M-H, Zhou J, Jambeck P, Churchill MEA, Allis CD. Histone acetyltransferase activity of yeast Gcn5p is required for the activation of target genes in vivo. Genes and Development. 1998;12:627–639. doi: 10.1101/gad.12.5.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y-S, Garcia-Bassets I, Hutt KR, Cheng CS, Jin M, Liu D, Benner C, Wang D, Ye Z, Bibikova M, et al. Sensitive ChIP–DSL technology reveals an extensive estrogen receptor α-binding program on human gene promoters. PNAS. 2007;104:4852–4857. doi: 10.1073/pnas.0700715104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganière J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguère V. Location analysis of estrogen receptor α target promoters reveals that FOXA1 defines a domain of the estrogen response. PNAS. 2005;102:11651–11656. doi: 10.1073/pnas.0505575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr D. The chromatin structure of an actively expressed, single copy yeast gene. Nucleic Acids Research. 1983;11:6755–6773. doi: 10.1093/nar/11.19.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métivier R, Penot G, Hübner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/S0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Nussey S, Whitehead S. Endocrinology: an Integrated Approach. Oxford: BIOS Scientific Publishers; 2001. Principles of Endocrinology. [PubMed] [Google Scholar]
- O’Malley BW, Sherman MR, Toft DO. Progesterone ‘receptors’ in the cytoplasm and nucleus of chick oviduct target tissue. PNAS. 1970;67:501–508. doi: 10.1073/pnas.67.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends in Biochemical Sciences. 2000;25:99–104. doi: 10.1016/S0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- Parekh BS, Maniatis T. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-β promoter. Molecular Cell. 1999;3:125–129. doi: 10.1016/S1097-2765(00)80181-1. [DOI] [PubMed] [Google Scholar]
- Payvar F, Wrange O, Carlstedt-Duke J, Okret S, Gustafsson JA, Yamamoto KR. Purified glucocorticoid receptors bind selectively in vitro to a cloned DNA fragment whose transcription is regulated by glucocorticoids in vivo. PNAS. 1981;78:6628–6632. doi: 10.1073/pnas.78.11.6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payvar F, DeFranco D, Firestone GL, Edgar B, Wrange O, Okret S, Gustafsson JA, Yamamoto KR. Sequence-specific binding of glucocorticoid receptor to MTV DNA at sites within and upstream of the transcribed region. Cell. 1983;35:381–392. doi: 10.1016/0092-8674(83)90171-X. [DOI] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nature Reviews. Genetics. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- Pogo BG, Allfrey VG, Mirsky AE. RNA synthesis and histone acetylation during the course of gene activation in lymphocytes. PNAS. 1966;55:805–812. doi: 10.1073/pnas.55.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior CP, Cantor CR, Johnson EM, Littau VC, Allfrey VG. Reversible changes in nucleosome structure and histone H3 accessibility in transcriptionally active and inactive states of rDNA chromatin. Cell. 1983;34:1033–1042. doi: 10.1016/0092-8674(83)90561-5. [DOI] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. Comprehensive genome-wide protein–DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes and Development. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, Massie CE, Vowler SL, Eldridge M, Carroll JS. Cooperative interaction between retinoic acid receptor-α and estrogen receptor in breast cancer. Genes and Development. 2010;24:171–182. doi: 10.1101/gad.552910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Cripe TP, Koch SR, Andreone TL, Petersen DD, Beale EG, Granner DK. Multihormonal regulation of phosphoenolpyruvate carboxykinase gene transcription. The dominant role of insulin. Journal of Biological Chemistry. 1984;259:15242–15251. [PubMed] [Google Scholar]
- Scheer U. Changes of nucleosome frequency in nucleolar and non-nucleolar chromatin as a function of transcription: an electron microscopic study. Cell. 1978;13:535–549. doi: 10.1016/0092-8674(78)90327-6. [DOI] [PubMed] [Google Scholar]
- Scheidereit C, Geisse S, Westphal HM, Beato M. The glucocorticoid receptor binds to defined nucleotide sequences near the promoter of mouse mammary tumour virus. Nature. 1983;304:749–752. doi: 10.1038/304749a0. [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/S0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- Sharma D, Fondell JD. Ordered recruitment of histone acetyltransferases and the TRAP/mediator complex to thyroid hormone-responsive promoters in vivo. PNAS. 2002;99:7934–7939. doi: 10.1073/pnas.122004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon MJ, Larsen PL, Varshavsky A. Mapping protein–DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 1988;53:937–947. doi: 10.1016/S0092-8674(88)90469-2. [DOI] [PubMed] [Google Scholar]
- Strähle U, Klock G, Schütz G. A DNA sequence of 15 base pairs is sufficient to mediate both glucocorticoid and progesterone induction of gene expression. PNAS. 1987;84:7871–7875. doi: 10.1073/pnas.84.22.7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaneck GE, Chu LLH, Edelman IS. Stereospecific binding of aldosterone to renal chromatin. Journal of Biological Chemistry. 1970;245:5382–5389. [PubMed] [Google Scholar]
- Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ura K, Kurumizaka H, Dimitrov S, Almouzni G, Wolffe AP. Histone acetylation: influence on transcription, nucleosome mobility and positioning, and linker histone-dependent transcriptional repression. EMBO Journal. 1997;16:2096–2107. doi: 10.1093/emboj/16.8.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väisänen S, Dunlop TW, Sinkkonen L, Frank C, Carlberg C. Spatio-temporal activation of chromatin on the human CYP24 gene promoter in the presence of 1α,25-dihydroxyvitamin D3. Journal of Molecular Biology. 2005;350:65–77. doi: 10.1016/j.jmb.2005.04.057. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li W, Liu XS, Carroll JS, Jänne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Molecular Cell. 2007;27:380–392. doi: 10.1016/j.molcel.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischet WO, Glotov BO, Zachau HG. Protection of expressed immunoglobulin genes against nuclease cleavage. Nucleic Acids Research. 1983;11:3593–3612. doi: 10.1093/nar/11.11.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer RM, Lucchini R, Lezzi M, Meyer B, Sogo JM, Edström JE, Koller T. Chromatin structure of a hyperactive secretory protein gene (in Balbiani ring 2) of chironomus. EMBO Journal. 1984;3:1635–1641. doi: 10.1002/j.1460-2075.1984.tb02022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes and Development. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Lo T-W, Zeitler B, Pickle CS, Ralston EJ, Lee AH, Amora R, Miller JC, Leung E, Meng X, et al. Targeted genome editing across species using ZFNs and TALENs. Science. 2011;333:307. doi: 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annual Review of Genetics. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes and Development. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]