

Abstract
PUMA is a BCL-2 family protein that transmits stress signals to promote apoptosis. Upon DNA damage, a unique binding determinant within PUMA triggers partial unfolding of BCL-XL, resulting in the release of sequestered p53 and commitment to p53-dependent cell death.
The BCL-2 family of apoptotic proteins arbitrates the cellular response to internal and external stress through a complex network of protein interactions that ultimately block or activate mitochondrial poration. Specialized stress sensors within the family communicate with downstream members via a conserved motif called the BCL-2 homology 3 (BH3) domain, which engages its targets as a single α-helix. These so-called ‘BH3-only proteins’ are otherwise heterogeneous, subserving discrete cellular functions and subject to distinct transcriptional and post-translational modes of regulation. PUMA, or ‘p53 upregulated modulator of apoptosis’, was first discovered as a p53 target that contributed to the apoptotic response at the level of the mitochondria1,2. PUMA has since been shown to engage the broad range of antiapoptotic BCL-2 family proteins through the now-classic interaction between its BH3 helix and a hydrophobic groove on the antiapoptotic surface3,4. Targeting antiapoptotic BCL-2 family proteins in this fashion lowers the apoptotic threshold by preventing sequestration of proapoptotic members and displacing them when already bound. In this issue, Follis et al.5 uncover a unique BCL-XL–binding feature of the PUMA BH3 domain (PUMABH3) and its mechanistic role in activating p53-dependent apoptotic signaling through induced unfolding of BCL-XL and p53 release.
As guardian of the human genome, p53 has an essential tumor suppressive role in regulating the cell cycle and cell death in response to DNA damage. Whereas the transcriptional basis for p53 activity has been thoroughly defined in this context, transcription-independent signaling has also emerged as a contributory mechanism6,7. The localization of p53 in complex with BCL-XL at the mitochondria of apoptotic cells implicated direct communication between the p53 and BCL-2 family pathways in regulating the cell’s life-death rheostat7. Interestingly, the same basic domain within p53 that confers DNA binding functionality was shown to interact with an acidic surface formed by residues of the BCL-XL α1 C terminus and the α3-α4 and α5-α6 loops8. That is, p53 engaged BCL-XL at an interaction site distinct from that previously defined for the amphipathic BH3 helices, which bind a surface hydrophobic groove formed by the confluence of residues from BCL-XL α-helices 2, 3, 4, 5 and 8. Here, Follis et al.5 apply a comprehensive battery of structural, biochemical and cellular analyses to investigate the mechanism by which PUMA dissociates the p53–BCL-XL complex to drive p53-dependent apoptosis.
The first clue was derived from NMR experiments examining the influence of PUMABH3 titration on the resonances of a 15N-labeled BCL-XL construct termed BCL-XLΔLΔC, which lacked the unstructured α1-α2 loop (whose resonances overlapped or interfered with those of the structured portions of the protein) and the C-terminal helix. Whereas PUMABH3 binding induced chemical shift changes in the canonical BH3-binding pocket of BCL-XLΔLΔC, additional residues within α2, α3 and α4 were likewise perturbed (Fig. 1a), a phenomenon not observed, for example, upon titration with another BH3-pocket binder such as BADBH3. Structural analysis of the PUMABH3–BCL-XL complex by X-ray crystallography further showed that Pro116 of the α3-α4 loop underwent a 5-Å displacement relative to its position in the uncomplexed and BADBH3–BCL-XL structures. Taken together, these data suggested that, in addition to the expected interactions between PUMABH3 and BCL-XL, unique allosteric changes in the structure flanked by α2 and α4 occurred that map to a portion of the region previously identified as the p53-binding site on BCL-XL8(Fig. 1a). Could PUMABH3 binding to BCL-XL partially unfold and thereby alter the p53 binding site such that p53 dissociation ensues? Correlative biochemical studies indeed showed that preincubation of BCL-XLΔC with PUMABH3 or exposure of preformed p53–BCL-XLΔC complex to PUMABH3 abrogated the capacity of p53 to bind BCL-XLΔC. Notably, no other BH3 domains tested recapitulated these effects, suggesting that PUMABH3 harbors a unique binding determinant that mediates its distinct functionality.
Figure 1.
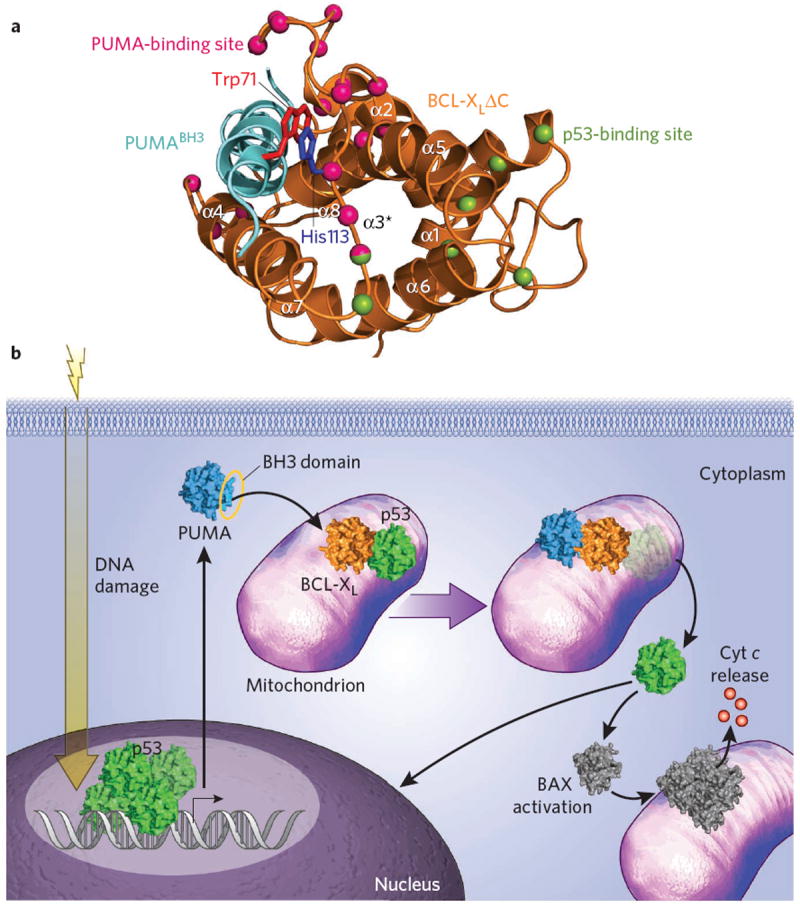
Induction of p53-dependent apoptosis by selective PUMABH3-mediated dissociation of the mitochondrial p53–BCL-XL complex. (a) A unique π-stacking interaction between PUMABH3 Trp71 (red) and BCL-XL His113 (blue) triggers partial unfolding of the BCL-XL α2-α3 region (α3*), a component of the previously defined p53 binding site on BCL-XL. BCL-XL residues that undergo the greatest chemical shift change upon PUMABH3 titration are depicted as pink balls, whereas those residues affected by p53 DNA-binding domain engagement, as defined by Petros et al.8, are represented by green balls. Thr115 is dual-colored, reflecting an exemplary residue that is affected both by PUMABH3 (ref. 5) and p53 (ref. 8) binding. The ribbon diagram of BCL-XLΔC protein and the PUMABH3 helix are colored orange and cyan, respectively. (b) Irradiation-induced DNA damage triggers the p53 transcriptional response, resulting in the upregulation of PUMA, among other targets. PUMABH3 engages BCL-XL at the canonical binding site, resulting in structural changes that promote dissociation of p53. This liberation of cytosolic p53 is believed to contribute to p53-dependent apoptosis through amplification of the transcriptional response and direct activation of proapoptotic BAX. Cyt c, cytochrome c.
Alignment of BH3 sequences and inspection of both the NMR and crystal structures of the PUMABH3–BCL-XL complexes provided a second mechanistic clue underlying the activity and specificity of PUMABH3-mediated partial unfolding of BCL-XL. The PUMABH3 sequence contains a uniquely positioned tryptophan, N-terminal to the core BH3 consensus sequence, that was observed to engage in π-stacking interactions with His113 of BCL-XL (Fig. 1a). To determine whether this unique interaction pair influenced the observed structural and biochemical effects of PUMABH3 on BCL-XL and its association with p53, studies with the mutant constructs PUMABH3 W71A and BCL-XLΔCH113A were undertaken. In each case, elimination of the π-stacking interaction by alanine mutagenesis completely eliminated the selective PUMABH3 activity, mechanistically linking the induced partial unfolding to a single complementary aromatic interaction. Importantly, the authors vetted their mechanistic conclusions in a cellular context, demonstrating that reconstitution of Puma−/− mouse embryonic fibroblasts with wild-type PUMA, but not PUMAW71A, restored UV-induced apoptosis. Correspondingly, the UV-induced p53–BCL-XL complex was effectively dissociated upon engagement by reintroduced wild-type PUMA but not by PUMAW71A, as elegantly demonstrated by coimmunoprecipitation experiments. As even further evidence of mechanism-based specificity, both PUMA constructs fully restored TNF-induced apoptosis and were equally ineffective in triggering UV-induced apoptosis in the absence of p53.
The p53 and BCL-2 family pathways represent critical control points in the regulation of cellular life and death, and thus their intersection through direct interactions at the mitochondria seems logical, albeit noncanonical. BH3 domains are well known to be discriminating and, however homologous, encode sequence-based selectivity that dictates their unique and common functions. Here, one such determinant, a tryptophan within PUMABH3, confers a unique PUMABH3–BCL-XL binding mode that selectively induces allosteric unfolding within a portion of the p53-binding site, rendering BCL-XL incompatible for p53 interaction. As a consequence, cytosolic p53 is released to compound transcriptional activation and promote apoptosis through direct protein interaction with mitochondrial proapoptotic effectors, such as BAX6 (Fig. 1b). Interestingly, a previous NMR study demonstrated that preincubation of BCL-XLΔC with BADBH3 precluded p53 interaction8; inspection of the BADBH3 sequence reveals an N-terminal tryptophan localized one residue upstream of PUMABH3 Trp71. Given the flexibility of the α3-α4 loop, it is plausible that in discrete contexts, the regulatory mechanism elucidated here for PUMABH3–BCL-XL–p53 may translate to other multiprotein BCL-2 family complexes. Indeed, noncanonical BH3 interaction modes and their unique effects on target structure and function are emerging as important themes in BCL-2 family biology9,10. Rigorous multidisciplinary analyses, as showcased by the current study, are essential for both validating and integrating such discoveries, however unexpected, into the evolving and complex canon that defines the signaling mechanisms of life and death.
Footnotes
Competing financial interests
The author declares no competing financial interests.
References
- 1.Nakano K, Vousden KH. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 3.Day CL, et al. J Mol Biol. 2008;380:958–971. doi: 10.1016/j.jmb.2008.05.071. [DOI] [PubMed] [Google Scholar]
- 4.Sattler M, et al. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 5.Follis AV, et al. Nat Chem Biol. 2013;9:163–168. doi: 10.1038/nchembio.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chipuk JE, et al. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 7.Mihara M, et al. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 8.Petros AM, Gunasekera A, Xu N, Olejniczak ET, Fesik SW. FEBS Lett. 2004;559:171–174. doi: 10.1016/S0014-5793(04)00059-6. [DOI] [PubMed] [Google Scholar]
- 9.Danial NN, et al. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]